

Abstract
The first total syntheses of the Lycopodium alkaloids (+)-nankakurine A (2), (+)-nankakurine B (3) and the originally purported structure 1 of nankakurine A were accomplished. The syntheses of 2 and 3 feature a demanding intramolecular azomethine imine cycloaddition as the key step for generating the octahydro-3,5-ethanoquinoline moiety and installing the correct relative configuration at the spiro piperidine ring juncture. The cyclization precursor was prepared from octahydronaphthalene ketone 50, which was assembled from enone (+)-9 and diene 48 by a cationic Diels–Alder reaction. The Diels–Alder reactants were synthesized from 5-hexyn-1-ol (16) and (+)-pulegone (49), respectively. The tetracyclic ring system of 1 was generated using an unprecedented nitrogen-terminated aza-Prins cyclization cascade. The enantioselective total syntheses of (+)-nankakurine A (2) and (+)-nankakurine B (3) establish the relative and absolute configuration of these alkaloids, and are sufficiently concise that substantial quantities of 2 and 3 were prepared for biological studies. (+)-Nankakurine A and (+)-nankakurine B showed no effect on neurite outgrowth in rat hippocampal H-19 cells over a concentration range of 0.3–10 μM.
Keywords: nankakurine, Lycopodium alkaloids, intramolecular azomethine imine dipolar cycloaddition, nitrogen-terminated aza-Prins reaction
Introduction
Because of their diverse architectures and biological activities, Lycopodium alkaloids1 have attracted significant synthetic interest for many years. In 2004, Kobayashi and co-workers reported the isolation of a structurally unique Lycopodium alkaloid, (+)-nankakurine A,2 from the club moss Lycopodium hamiltonii collected at the southwest tip of Kyushu Island, Japan. On the basis of two-dimensional NMR data and mass spectrometric analysis, structure 1 was proposed (Figure 1).2 The relative configuration at the spiro stereocenter of nankakurine A was assigned on the basis of 1H NMR NOE experiments, and concurred with the structure of spirolucidine (4), whose relative configuration had been secured earlier by single-crystal X-ray analysis of a derivative.3
FIGURE 1.

Nankakurines A and B and two structurally related Lycopodium alkaloids.
Two years later, the Kobayashi laboratory reported that further purification of this club moss extract provided a less abundant alkaloid, (+)-nankakurine B, for which structure 3 was suggested.4 In this case, 1H NMR NOE data were interpreted to support a configuration at the spiro stereocenter opposite to that originally proposed for nankakurine A (1). Reductive methylation of natural nankakurine A gave nankakurine B, thus, the structure of nankakurine A was revised to 2.4
Neurotrophic factors play an important role in mediating neuronal growth and can counteract neuronal atrophy and death in the adult nervous system.5 Because of the poor pharmacokinetics documented by naturally occurring polypeptidyl-neurotrophic factors,6 small molecules that exhibit neurotrophic effects are attracting considerable attention as potential therapeutics.7 Thus, Kobayashi’s disclosure in 2006 that nankakurine A (2) induced secretion of neurotrophic factors in human astrocytoma cells at a concentration of 1 μM heightened our interest in this area.4 Similar examination of nankakurine B was not possible at the time as a result of the low abundance of these alkaloids in their natural source.8 It was particularly provocative that nankakurines A and B bore no obvious structural relationship to other small molecule neurotrophic agents reported at that time,7 and, moreover, that their structures were devoid of polar functional groups known to negatively affect central nervous system exposure of small-molecule drugs.9
Stimulated initially by the opportunity to explore a new construction of polycyclic diamines (vide infra) and by the prospect of obtaining sufficient quantities of nankakurines A (2) and B (3) to evaluate their potential as leads for developing new agents for combating neurodegenerative disorders, we initiated a total synthesis program in this area.10 Herein, we report the development of the chemistry that allowed the first total syntheses of (+)-nankakurine A, (+)-nankakurine B, and the originally proposed structure of nankakurine A (1) to be accomplished, syntheses that confirmed the relative configuration and established the absolute configuration of these rare alkaloids.11 Moreover, these syntheses allowed the neurotrophic activities of both 2 and 3 to be examined. In this exploration, neurotrophic activity was not observed in rat hippocampal H-19 cells over a concentration range of 0.3–10 μM.
Results and Discussion
Initial Retrosynthetic Analysis
The focus of our initial effort was the originally purported structure 1 (5-epi-nankakurine A) of nankakurine A (Scheme 1). We envisioned tetracyclic diamine 6 arising in one step from bis-homoallylic amine 8 by an amino-terminated aza-Prins cyclization of formaldiminium intermediate 7 (Scheme 1). Although carboxylate-terminated aza-Prins reactions of amino acids had been described,12 amino-terminated aza-Prins cyclizations were unknown. This lack of precedent provided extra motivation for evaluating this approach, as we envisioned amino-terminated aza-Prins cyclizations potentially having considerable utility in heterocyclic synthesis. Cyclization of the tethered nitrogen nucleophile from the sterically less-hindered-convex face of intermediate 7 was expected to set the spiropiperidine stereocenter of 1 with high selectivity. We foresaw the central challenge in orchestrating a successful amino-terminated aza-Prins reaction as co-generating and properly matching the iminium ion electrophile and the nitrogen nucleophile. Cis-octahydronaphthalene amine 8 was seen deriving from the Diels–Alder reaction of (R)-5-methyl-2-cyclohexen-1-one (9) and diene 10, a strategy that drew ample precedent from Oppolzer’s pioneering early synthesis of (+)-luciduline.13
SCHEME 1.

Retrosynthetic Analysis of 5-epi-Nankakurine (1)
Synthesis of cis-Octahydronaphthalene Amine Intermediates
Our early exploratory studies were carried out in the racemic series, and required convenient access to 5-methyl-2-cyclohexen-1-one ((±)-9) and 1,3-diene 19 (Scheme 2). After examining several of the known methods for preparing cyclohexenone (±)-9,14 we developed an alternate synthesis from 5-methyl-1,3-cyclohexanedione (13), which is commercially available or readily prepared from inexpensive orcinol (11) by methylation15 followed by Birch reduction and acidic hydrolysis.16 Reaction of dione 13 with oxalyl bromide to form β-bromoenone 14,17 followed by reduction with zinc in THF gave 5-methyl-2-cyclohexen-1-one ((±)-9) in 55% overall yield from orcinol. As noted by Cossy,18 attempts to prepare ethylene ketal 15 from (±)-9 by reaction with ethylene glycol in the presence of a variety of Brønsted acids under Dean–Stark conditions were complicated by migration of the double bond to provide significant amounts of 9-methyl-1,4-dioxaspiro[4.5]dec-7-ene. Although the use of the weak acid pyridinium p-toluenesulfonate (PPTS) allowed ketal 15 to be prepared in 29% yield (55% based on consumed (±)-9),18 this ketalization was more efficient (73% yield) when carried out by reaction of (±)-9 with 1,2-bis-(trimethylsiloxy)ethane and catalytic trimethylsilyl triflate (TMSOTf) at −78 °C.19
SCHEME 2.

Synthesis of cis-Octahydronaphthalene Amine 22.
Using a Mitsunobu variant of the Gabriel amine synthesis, diene 19 was prepared in a three-step sequence from 5-hexyn-1-ol (16). Alcohol 16 was transformed first into amine hydrochloride salt 17, which was protected as a sulfonamide (18) in 83% yield for the two step sequence.20,21 Reaction of alkyne 18 with ethylene using Grubbs’ 2nd generation catalyst22 afforded diene 19 in 90% yield.23 The TMSOTf-catalyzed ionic Diels–Alder reaction24 of cyclohexenone (±)-9 with diene 19 took place in 50–60% yield using a stoichiometric quantity of 1,2-bis-(trimethylsiloxy)ethane. During attempts to optimize this conversion, it became apparent that the moderate yield arose from reaction inhibition resulting from the buildup of (TMS)2O. When ketal 15 was employed, the ionic Diels–Alder reaction proceeded in high yield with excellent cis-diastereoselectivity.25 Subsequent cleavage of the ketal using FeCl3 adsorbed on silica gel26,27 provided ketone 20 in 90% yield from diene 19.28 Ketone 20 was quantitatively converted to cis-oxime 21 upon reaction at room temperature with aqueous hydroxylamine in methanol; the use of acidic or basic conditions for oxime formation resulted in significant epimerization. The robust sequence summarized in Scheme 2 reproducibly provided oxime 21 on scales of >10 g. Reduction of this intermediate with MoO3 and NaBH4 provided cis-octahydronaphthalene amine 22 in excellent yield.29
Exploration of Amino-Terminated aza-Prins Cyclizations and the Synthesis of (±)-5-epi-Nankakurine (1)
In our initial attempts to develop an amino-terminated aza-Prins cyclization, we employed a cyanomethyl amine as a formaldiminium ion precursor to allow the iminium cation to be generated under mild conditions in various solvents.30 Cyanomethylamine derivative 23 was prepared in 78% yield by alkylation of amine 22 with chloroacetonitrile. Reaction of this formaldiminium precursor with AgO2CCF3 in CHCl3 at room temperature gave the monocyclized product, octahydro-3,5-ethanoquinoline 24, in nearly quantitative yield (Scheme 3). Variation of the solvent or changing the initiating reagent to Cu(O2CCF3) led in no case to the formation of detectable tetracyclic product. We also investigated related reactions of congeners of cyanomethylamine 23 in which the tethered nitrogen substituent was NH2, NHBn, or NHC6H4p-OMe instead of NHTs; however, Ag-mediated cyclization reactions of these substrates also produced simple aza-Prins products analogous to 24. In an attempt to see if a potential tricyclic tertiary carbenium precursor of azatricyclic 24 could be trapped by an external nitrogen nucleophile, the silver-promoted cyclization of 23 was carried out in the presence of 10 equiv of sodium azide. This reaction also led to the formation of octahydro-3,5-ethanoquinoline 24 in high yield.
SCHEME 3.

Attempted Nitrogen-Terminated Aza-Prins Reaction of Cyanomethyl Amine 23.
Although it would be less desirable than a cascade sequence, we investigated briefly whether the spiropiperidine ring could be formed in a subsequent step from aza-Prins product 24 (Scheme 4). As intramolecular hydroamination of unsaturated N-tosylamines to form pyrrolidine rings in the presence of strong acids is well established,31 tricyclic sulfonamide 24 was allowed to react with trifluoromethanesulfonic acid (TfOH) or trifluoroacetic acid (TFA, neat or in toluene, rt–80 °C). These reactions generated double bond isomers of 24, but no trace of tetracyclic product 26. In order to minimize protonation at N1, aza-Prins product 24 was protected as a methyl carbamate. Reaction of carbamate 25 with TfOH or TFA (neat or in CHCl3, MeCN, or PhMe, rt–100 °C) resulted in the formation of multiple products, including those that resulted from loss of the carbamate and isomerization of the double bond; the desired tetracyclic product 27 was not produced. In order to further reduce the basicity of the protected amine and increase the stability of this substituent, p-nitrophenylsulfonyl (Ns) sulfonamide 28 was prepared. Reaction of 28 with TFA or p-toluenesulfonic acid (TsOH, 25–80 °C, various solvents), returned only starting material, whereas exposure of 28 to TfOH at 120 °C delivered tetralin derivative 29. This extensive degradation presumably results from protonation of N1, followed by the cleavage of two C–N bonds to eliminate p-nitrophenylsulfonamide, and finally double bond isomerization to produce tetralin 29 (30→31→32).
SCHEME 4.

Attempted Hydroamination of Tricyclic Unsaturated Sulfonamides 24, 25, and 28.
A metal-catalyzed hydroamination reaction was considered as an alternative approach for preparing tetracyclic products 26 or 27 from tricyclic intermediates 24 or 25,32,33 however, existing literature precedent focuses largely on substrates that contain only one nitrogen substituent and typically result in the formation of pyrrolidine rings. There are only a few examples that result in the formation of piperidine rings or that utilize tri-substituted alkenes.34 Intramolecular hydroamination of 24 and 25 utilizing FeCl3 was evaluated.35 Extended exposure of unsaturated sulfonamides 24 or 25 to FeCl3 at 80 °C in 1,2-dichloroethane resulted only in slow conversion of the starting material to mixtures of alkene-migrated products.
Being sensitized at this point to the ready formation of alkene regioisomers in reactions of azatricyclic alkenes 24 and 25 with acids, we reconsidered the observation that product 24 was produced as a single double-bond isomer in the original aza-Prins cyclization. This high regioselectivity suggested the possibility that the nitrogen atom of the hydro-3,5-ethanoquinoline fragment of 23 was participating in the generation of the tricyclic aza-Prins product (Scheme 5). For stereoelectronic reasons,36 aza-Prins cyclization of formaldiminium ion 33 should generate initially tricyclic carbenium ion 34, which would be expected to undergo rapid nitrogen inversion37 to isomer 35, in which the nitrogen non-bonded electron pair would be positioned ideally to assist in deprotonation to form the conjugate acid 36 of octahydro-3,5-ethanoquinoline product 24.38
SCHEME 5.

Potential Nitrogen Participation in the Aza-Prins Cyclization to form Octahydro-3,5-ethanoquinoline 24.
To enhance the lifetime of carbenium ion intermediate 35 and potentially favor a nitrogen-terminated aza-Prins process, we investigated related reactions of N-acyloxy-formaldiminium ion intermediates. We initially examined initiating the cyclization from N-cyanomethyl and N-benzotriazolmethyl39 carbamates 37 and 39 (Scheme 6). At temperatures up to 120 °C, cyanomethyl derivative 37 was unchanged when exposed to 2 equiv of AgO2CCF3, AgBF4, or TsOH in acetonitrile or toluene. Exposure of benzotriazole derivative 39 to 0.9 equiv of ZnBr2 in dichloromethane at 60 °C initiated aza-Prins cyclization to provide alkylidene decahydro-3,5-ethanoquinoline products 40 in 63% yield. Other modes of initiating the cyclization were also pursued;39 however, in no case was any tetracyclic product detected.
SCHEME 6.

Attempted Sulfonamide-Terminated Aza-Prins Reaction of Carbamates 37 and 39.
We next turned to initiate the cyclization by acid-promoted reaction of carbamate 38 with formaldehyde (Scheme 7). Exposure of carbamate 38 to an excess of paraformaldehyde in TFA/CHCl3 at room temperature12b resulted in formation of two inseparable tetracyclic products 41 whose molecular formulas (C26H36N2O4S, MS analysis) indicated that 2 equiv of formaldehyde were involved in their genesis. As these products were also produced upon exposure of alkylidene decahydro-3,5-ethanoquinoline products 40 to identical conditions, they were assigned as the tetracyclic alkene products of aza-Prins cyclization of N-sulfonylformaldiminium ion intermediate 42. Using one equiv of paraformaldehyde, the tetracyclic product 27 resulting from sulfonamide-terminated aza-Prins cyclization was formed, albeit in low yield (10–20%). In this reaction, 27 was produced in a complex mixture of products that contained the already characterized aza-Prins product 40 and tetracyclic products 41. Much effort was expended in trying to optimize the formation of spirocyclic product 27. Numerous variations in solvent, protic or Lewis acid, and formaldehyde precursor were surveyed, but no conditions were identified under which 27 was formed in more than 20% yield.40,41
SCHEME 7.

Sufonamide-terminated Aza-Prins Cyclization to Form Spirotetracyclic Product 27.
Although the yield of the sulfonamide-terminated aza-Prins reaction of bicyclic precursor 38 was low, this reaction did provide sufficient quantities of tetracyclic diamine 27 to allow the originally proposed structure of nankakurine A (1) to be prepared in two additional steps (Scheme 8). Cleavage of the sulfonamide of 27 by reaction with Na/NH3 provided amino carbamate 43 in excellent yield. Lithium aluminum hydride reduction of the methyl carbamate of this product afforded (±)-5-epi-nankakurine A (1) in 65% yield.
SCHEME 8.

Elaboration of 27 to (±)-5-epi-Nankakurine A (1).
The NMR data for synthetic (±)-1 did not match data reported for nankakurine A.2,42 We considered it secure that the tethered sulfonamide would undergo C–N bond formation to the cup-shaped decahydro-3,5-ethanoquinoline intermediate (7, Scheme 1) from the less-hindered convex face in the sulfonamide-terminated aza-Prins reaction. With the relative configuration at the spirocyclic stereocenter of product 27 established, we hypothesized that the originally proposed structure 1 for nankakurine A was incorrect. At approximately the same time, Kobayashi and coworkers reported that the relative configuration of nankakurine A at the spiropiperidine stereocenter C5 should be reversed.4 As discussed earlier, this conclusion was founded on 1H NMR NOE studies of the conjugate acid of nankakurine B (3).
A serious complication in assigning the relative configuration of the spiro stereocenter of nankakurines A and B involved the existence of two conformational isomers of the spiropiperidine ring, which made NOE measurements difficult to interpret. Kobayashi and co-workers suggested that this complication was responsible for the original misassignment of the relative configuration of nankakurine A (2).4 This issue was proposed to be less serious for the disalt of nankakurine B (3), which was suggested to exist predominantly in conformer B on the basis of molecular mechanics calculations (Figure 2).4 However, molecular modeling carried out in our laboratory at the time did not support this conclusion (B3-LYP/6-311G*), nor do more recent DFT calculations using two functionals and higher-level basis sets.43 We concluded that a definitive answer to the relative and absolute configuration of the nankakurines would require their total syntheses.
FIGURE 2.

Molecular models of low-energy conformations of the most stable disalts of nankakurine B (2); ΔE = E(conf B) – E(conf A) in kcal/mol; energies marked with an asterisk refer to DFT calculations using the COSMO model for methanol solvent.
Synthesis of (+)-Nankakurine A and (+)-Nankakurine B
A strategy for construction of structures (+)-2 and (+)-3 requires that the spiropiperidine nitrogen be located on the concave face of the tricyclic decahydro-3,5-ethanoquinoline moiety. We envisaged tetracyclic pyrazolidine 44, which possesses the proper relative configuration at C5, as a logical precursor to nankakurines A and B (Scheme 9). Pyrazolidine 44 was seen arising by an intramolecular dipolar cycloaddition of azomethine imine 45.44 High regioselection in this step was anticipated based on the prospect that C–C bond-formation would be more advanced than C–N bond-formation,44a an expectation bolstered by the high regioselectivity observed in Oppolzer’s construction of (+)-luciduline by intramolecular cycloaddition of an analogous nitrone intermediate.13 Hydrazine 6 would readily derive from octahydronaphthalene ketone 47.
SCHEME 9.

Retrosynthetic Analysis of (+)-Nankakurines A (2) and B (3)
In a sequence analogous to that employed for the synthesis of 5-epi-nankakurine A (1), octahydronaphthalenone 50 was constructed in enantioenriched form by the cationic Diels–Alder reaction of (R)-5-methyl-2-cyclohexen-1-one ((+)-9) and 2-substituted-1,3-butadiene 48 (Scheme 10). Diene 48 was available in two high-yielding steps from 5-hexyn-1-ol (16): benzyl-protection,45 followed by enyne cross metathesis with ethylene using Grubbs’ 2nd generation catalyst.22,23 Cyclohexenone (+)-9 (>99% ee) was prepared from (+)-pulegone (49) in four steps by a known, efficient sequence.46 Allowing diene 48, cyclohexenone (+)-9, and 1,2-bis(trimethylsiloxy)ethane to react in CH2Cl2 at −78 °C in the presence of 10 mol % of TMSOTf,25 followed by cleavage of the dioxolane with FeCl3 adsorbed on silica gel,26 gave cis-octahydroquinolone 50 in 67% yield as a single stereoisomer.47
SCHEME 10.

Synthesis of cis-Octahydronaphthalene Hydrazine Derivatives 51–53.
Intramolecular or bimolecular dipolar cycloadditions of azomethine imines derived from formaldehyde with unactivated trisubstituted alkenes were to our knowledge unknown. As a result, we anticipated that the electron-withdrawing substituent on the dipole might need to be finely tuned for the projected cycloaddition to succeed.44a,g Hydrazine derivatives carrying carbomethoxy, p-toluenesulfonyl, and benzoyl groups were prepared in varying yields by hydrazone formation, followed by reaction with sodium cyanoborohydride, the reduction occurring exclusively from the convex β-face to deliver 51–53.48 In order to avoid partial epimerization adjacent to the ketone, it was essential that the hydrazone intermediate was prepared under neutral conditions.
The pivotal intramolecular dipolar cycloaddition was surveyed under both thermal and acidic conditions. In preliminary studies, hydrazines 51–53 were allowed to react with excess (CH2O)n either in refluxing toluene in the presence of 4Å molecular sieves or at room temperature in CHCl3 in the presence of 20 equiv of TFA (Table 1). With the carbamate and sulfonamide precursors, only the octahydro-3,5-ethanoquinoline aza-Prins products 55 were produced (entries 1–4). Although acidic conditions were not successful (entry 5), reaction of benzoylhydrazide 53 with (CH2O)n and 4Å molecular sieves in refluxing toluene provided the tetracyclic pyrazolidine product 54 in 40% yield together with aza-Prins products 55 (entry 6).49 Carrying out this reaction in the presence of basic additives provided dipolar cycloaddition product 54 in 80–85% yield, with the reaction being slightly faster in the presence of diisopropylethylamine than triethylamine (entries 7–8).
TABLE 1.
Dipolar Cycloaddition of Unsaturated Hydrazine Derivatives 51–53.
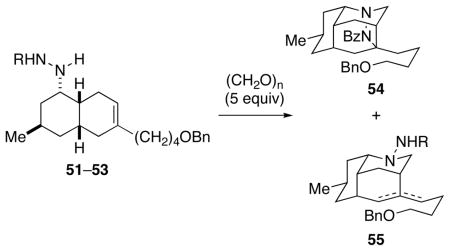 | ||||
|---|---|---|---|---|
| entry | R | solventa | additive | yield 54 |
| 1 | CO2Me | CHCl3 | TFA (20 equiv) | 0% |
| 2 | CO2Me | PhMe | 4Å mol. sieves | 0% |
| 3 | Ts | CHCl3 | TFA (20 equiv) | 0% |
| 4 | Ts | PhMe | 4Å mol. sieves | 0% |
| 5 | Bz | CHCl3 | TFA (20 equiv) | 0% |
| 6 | Bz | PhMe | 4Å mol. sieves | 40% |
| 7 | Bz | PhMe | Et3N 4Å mol. sieves |
80%b |
| 8 | Bz | PhMe |
i-Pr2NEt 4Å mol. sieves |
85%c |
Reactions run in CHCl3 at rt or PhMe at 120 °C.
24 h.
5 h.
Our interpretation of the positive effect of basic additives on the efficiency of the pivotal intramolecular dipolar cycloaddition is outlined in Scheme 11. Dehydrative condensation of benzoylhydrazide 53 with paraformaldehyde generates azomethine imine 56, which undergoes intramolecular cycloaddition to yield cycloadduct 54. In the absence of a basic additive, the efficiency of this reaction is undermined by protonation of 56 by adventitious acid (likely formic acid) to generate N-benzamidoformaldiminium ion 57, which undergoes aza-Prins cyclization to give octahydro-3,5-ethanoquinoline 55 via tertiary carbenium ion intermediate 58. In the presence of base, iminium electrophile 57 is not generated.50
SCHEME 11.

Competition Between Intramolecular Azomethine Imine Dipolar Cycloaddition and Aza-Prins Cyclization.
The total syntheses of (+)-nankakurine A (2) and (+)-nankakurine B (3) were concluded in five and six steps, respectively, from cycloadduct 54 (Scheme 12). Cleavage of the N–N bond with SmI251 and selective reductive methylation of the secondary amine product afforded diamine 59 in 80% overall yield in this one-pot sequence. Hydrogenolytic cleavage of the O-benzyl protecting group, followed by reduction of the amide with aluminum hydride,52 provided diamine alcohol 60 in 72% yield. Selective O-mesylation of this intermediate at −40 °C, followed by warming the primary mesylate to ambient temperature generated the spiropiperidine ring and furnished N-benzylnankakurine A (61) in 96% yield.53 Hydrogenolysis of this intermediate in acidic methanol then gave (+)-nankakurine A (2), [α]24D +13 (c 0.4, MeOH), in 99% yield. Standard reductive methylation of nankakurine A (2) delivered (+)-nankakurine B (3), [α]24D +12 (c 1.5, MeOH), in 80% yield.54
SCHEME 12.

Completion of the Enantioselective Total Syntheses of (+)-Nankakurine A (2) and (+)-Nankakurine B (3)
The 1H NMR spectra of synthetic 2 and 3 did not initially match those reported for the natural products (Figure 3). Because basic nitrogen atoms readily protonate, we were able to reproducibly obtain 1H and 13C NMR spectra of the free base forms of nankakurines A (2) and B (3) only in CD3OD containing a trace of NaOCD3. These spectra were not identical to those reported for natural 2 and 3 in CD3OD.4 We surmised that the natural isolates contained unknown amounts of their conjugate acids. Thus, we examined the 1H NMR spectra of synthetic (+)-nankakurine A (2) and (+)-nankakurine B (3) by titrating samples of the free bases with TFA. As reproduced in Figure 3, 1H NMR spectra identical to those of the natural products were obtained. These data are consistent with the notion that the natural samples contained an undetermined amount of the conjugate acids.
FIGURE 3.

1H NMR Spectra of the Titration of Synthetic (+)-2 and (+)-3 with TFA. a Matches 1H NMR of 2 reported in 2004.2 b Matches 1H NMR of 3 reported in 2006.4
Evaluation of the Neurotrophic Properties of (+)-Nankakurine A (2) and (+)-Nankakurine B (3)
The purported neurotrophic properties of (+)-nankakurine A (2) and (+)-nankakurine B (3) were investigated in the following fashion. Immortalized embryonic rat hippocampus H19-7 cells were grown in the presence of N-2 supplement medium. After 24 h, cells were treated with either 2 or 3. After an additional 24 h, the cells were fixed, and incubated successively with an antibody against neuronal class III β-tubulin and a secondary Alexa Fluor® 546 goat anti-mouse antibody. After staining, the length of neurites and the cell count were quantified using fluorescence microscopy.55,56 Neither (+)-nankakurine A nor (+)-nankakurine B increased neurite outgrowth over a concentration range of 0.3–10 μM (Figure 4), nor did they display any toxicity or change in cell number. Under the same conditions, a positive reference compound increased neurite outgrowth by 60% at 1.0 μM.
FIGURE 4.

Absence of neurotrophic properties of (+)-nankakurine A (2) and (+)-nankakurine B (3) in rat hippocampus cells; data are expressed as the mean of two separate experiments; control = DMSO; reference = positive reference compound.
Conclusions
In summary, the first total syntheses of (+)-nankakurine A (2), (+)-nankakurine B (3), and the originally proposed structure of nankakurine A (1, 5-epi-nankakurine A) were accomplished. Pivotal steps in these syntheses include the first example of a dipolar cycloadditions of an azomethine imine derived from formaldehyde with an unactivated trisubstituted alkene, and a nitrogen-terminated aza-Prins cyclization. The total syntheses of (+)-2 and (+)-3, together with the synthesis of the originally purported structure 1 of nankakurine A, rigorously establish the relative and absolute configuration of these Lycopodium alkaloids. These syntheses were sufficiently concise that substantial quantities of these alkaloids could be prepared for biological evaluation. (+)-Nankakurine A (2) and (+)-nankakurine B (3) showed no effect on neurite outgrowth in rat hippocampus H-19 cells over a concentration range of 0.3–10 μM, which stands in contrast to the report of neurotrophic activity of the former in human astrocytoma cells.2
Experimental Section
Materials and Methods
Unless stated otherwise, reactions were conducted in flame-dried glassware under an atmosphere of nitrogen using anhydrous solvents. THF, DMF, CH2Cl2, PhMe, MeOH, MeCN, Et2O, NEt3 and i-Pr2NEt were passed through activated alumina columns.57 CHCl3 was distilled from Na2SO4 and stored over 4 Å molecular sieves.58 EtOH (200 proof) and acetone (ACS grade) were purchased from commercial sources and used without further purification. FeCl3/silica26 and AlH352 were prepared as previously described. SmI251,59 was freshly prepared prior to each usage. Trimethylsilyl triflate was distilled from CaH2.58 Methyl chloroformate was distilled from CaCl2.58 All other commercially obtained reagents were used as received. Reaction temperatures were controlled using a temperature modulator, and unless stated otherwise, reactions were performed at room temperature (rt, approximately 25 °C).
Thin-layer chromatography (TLC) was conducted with silica gel 60 F254 pre-coated plates, (0.25 mm) and visualized by exposure to UV light (254 nm) or stained with anisaldehyde, ceric ammonium molybdate, potassium permanganate or iodide. Flash column chromatography was performed using normal phase silica gel (60 Å, 230–240 mesh).
1H NMR spectra were recorded on 500 or 600 MHz spectrometers and are reported relative to deuterated solvent signals. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz) and integration. 13C NMR spectra were recorded at 125 or 150 MHz. Data for 13C NMR spectra are reported in terms of chemical shift (δ ppm). IR spectra were recorded on a spectrometer and are reported in terms of frequency of absorption (cm−1). Optical rotations were measured with a polarimeter. High resolution mass spectra were obtained using the electrospray ionization method.
Experimental Procedures
1,3-Dimethoxy-5-methylbenzene (12)
Orcinol (11, 25.0 g, 0.202 mol) was heated at reflux with MeI (62 mL, 1.0 mol) and anhydrous K2CO3 (140 g, 1.0 mol) in acetone (500 mL) overnight. After cooling to rt, the reaction mixture was filtered through diatomaceous earth, and the filtrate was concentrated in vacuo. Chromatographic purification of the residue (hexanes:EtOAc 19:1) provided 1260 as a clear oil (27 g, 89%): 1H NMR (500 MHz, CDCl3) δ 6.35 (d, J = 2.1 Hz, 2H), 6.31 (t, J = 2.2 Hz, 1H), 3.80, (s, 6H), 2.32 (s, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 160.9, 140.4, 107.3, 97.7, 66.1, 55.4, 22.0 ppm.
1,5-Dimethoxy-3-methylcyclohexa-1,4-diene (S1)
A solution of 12 (760 mg, 5.0 mmol) in anhydrous EtOH (5.0 mL) was added dropwise over 10 min to a solution of Li0 (350 mg, 0.050 mol) and NH3 (50 mL) at −50 °C. The reaction mixture was stirred at −50 °C for 1 h, then quenched by the addition of saturated aqueous NH4Cl (10 mL) and warmed to rt. The aqueous mixture was extracted with hexanes (3 × 10 mL). The combined extracts were dried with MgSO4, and concentrated in vacuo to provide S161 as a clear oil (650 mg, 85%): 1H NMR (500 MHz, CDCl3) δ 4.58 (dt, J = 1.2, 3.5 Hz, 2H), 3.57 (s, 6H), 3.05 (1H, m), 2.77 (dq, J = 1.1, 6.7 Hz, 2H), 1.09 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 151.4, 98.0, 54.3, 31.3, 30.9, 24.7 ppm.
5-Methylcyclohexane-1,3-dione (13)
Cyclohexadiene S1 (650 mg, 4.2 mmol) was stirred in 1 M HCl (5 mL) and THF (50 mL) overnight at rt, and then concentrated in vacuo. The aqueous layer was extracted with CH2Cl2 (3 × 10 mL), and the combined organic extracts were dried with MgSO4. The solvent was removed in vacuo to provide 1362 as a colorless solid (440 mg, 83%, a 5:1 mixture of hydroxyenone–diketone tautomers): 1H NMR (500 MHz, CDCl3, 3-hydroxyenone tautomer) δ 11.30, (bs, 1H), 5.47 (s, 1H), 2.43 (dd, J = 4.3, 17.0 Hz, 2H), 2.29–2.20 (m, 1H), 2.10 (dd, J = 11.1, 17.0 Hz, 2H), 1.08 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 204.2, 192.6, 104.0, 48.2, 40.7, 29.0, 21.0 ppm.
3-Bromo-5-methylcyclohex-2-enone (14)
Following a known procedure,17 oxalyl bromide (22 mL, 240 mmol) was added dropwise over 20 min (CAUTION gas evolution) to a solution of diketone 13 (25.0 g, 198 mmol), DMF (20 mL, 260 mmol), and CH2Cl2 (500 mL) at 0 °C. The reaction was stirred at 0 °C for 15 min, allowed to warm to rt, and stirred for 1 h at rt, after which H2O (500 mL) was added, and the biphasic mixture was stirred for 1 h. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (500 mL). The combined organic layers were washed with brine (500 mL), dried with MgSO4, and concentrated in vacuo. The residue was purified by flash chromatography (hexanes:EtOAc 1:0→3:1) to provide 1417a as a yellow oil (35.2 g, 94%): 1H NMR (500 MHz, CDCl3) δ 6.46 (s, 1H), 2.86 (dd, J = 4.7, 18.5 Hz, 1H), 2.51 (m, 1H), 2.46 (d, J = 4.0 Hz, 1H), 2.37–2.30 (m, 1H), 2.10 (dd, 1H, J = 12.0, 16.3 Hz), 1.10 (d, 3H, J = 6.7 Hz) ppm; 13C NMR (125 MHz, CDCl3) δ 196.7, 149.6, 132.4, 44.8, 44.5, 30.8, 20.8 ppm.
5-Methylcyclohex-2-enone [(±)-9]
A 1L 3-neck round-bottom flask was charged with Zn0 (37 g, 580 mmol, washed with AcOH and dried), 1,2-dibromoethane (6.2 mL, 72 mmol) and THF (200 mL), and the slurry was stirred and heated with a heat gun until ethylene gas evolved (CAUTION gas evolution). The slurry was allowed to cool to rt. The heating/cooling cycle was repeated two additional times. A solution of 14 (27.2 g, 144 mmol) in THF (200 mL) was added to the reaction mixture at a rate that maintained the reaction temperature <35 °C. The reaction was stirred at rt for 24 h, and then quenched by the addition of saturated aqueous NH4Cl (300 mL). The aqueous layer was extracted with CH2Cl2 (3 × 100 mL), and the combined organic extracts were dried with MgSO4, and concentrated. Purification by flash chromatography (hexanes:EtOAc 0:1→1:1) provided enone (±)-963 as a pale yellow oil (14.2 g, 94%): 1H NMR (500 MHz, CDCl3) δ 6.95 (ddd, J = 2.6, 5.5, 10.0 Hz, 1H), 6.01 (dd, J = 1.4, 10.1 Hz, 1H), 2.48 (dd, J = 2.6, 16.0 Hz, 1H), 2.42 (dt, J = 5.4, 18.6 Hz, 1H), 2.29–2.17 (m, 1H), 2.12 (dd, J = 12.3, 15.8 Hz, 1H), 2.04 (ddt, J = 2.7, 9.8, 18.5 Hz, 1H), 1.07 (d, J = 6.6 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 200.3, 150.1, 129.8, 46.5, 34.2, 30.5, 21.4 ppm; IR (thin film) 1681, 1457, 1390, 879, 734 cm−1.
9-Methyl-1,4-dioxaspiro[4.5]dec-6-ene (15)
An oven-dried Schlenk tube was charged with (±)-9 (12.1 g, 110 mmol) and 1,2-bis(trimethylsiloxy)ethane (27 mL, 0.10 mmol), evacuated, and back-filled with N2. Dichloromethane (8.0 mL) was added, the solution was cooled to −78 °C, and TMSOTf (360 μL, 2.0 mmol) was added dropwise. The reaction vessel was sealed, and the reaction mixture was stirred at −78 °C for 23 h. The reaction mixture was quenched by the addition of NEt3 (2 mL), warmed to rt, and filtered through a plug of basic Al2O3. The plug was rinsed with excess Et2O (50 mL), and the volatiles were removed in vacuo. Purification of the residue by flash chromatography (hexanes:Et2O:NEt3 99:0:1→90:10:1) provided ketal 1518 as a pale yellow oil (12.3 g, 73%) along with recovered (±)-9 (3.5 g, 30%); ketal 15: 1H NMR (500 MHz, CDCl3) δ 5.95 (ddd, J = 2.2, 5.2, 10.0 Hz, 1H), 5.60–5.57 (m, 1H), 4.07–3.83 (m, 4H), 2.14 (dt, J = 5.1, 17.8 Hz, 1H), 2.01–1.93 (m, 1H), 1.88–1.84 (m, 1H), 1.65 (ddt, J = 2.5, 10.7, 17.8 Hz, 1H), 1.48 (t, J = 13.0 Hz, 1H), 1.00 (d, J = 6.6 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 132.5, 127.3, 106.7, 64.8, 64.5, 42.2, 38.8, 28.1, 21.8 ppm.
Hex-5-yn-1-amine Hydrochloride (16)
A solution of 17 (25.0 g, 255 mmol) and diisopropyl azodicarboxylate (51 mL, 260 mmol) in THF (160 mL) was added by cannula at 0 °C to a solution of PPh3 (66.8 g, 255 mmol) and phthalimide (37.5 g, 255 mmol) in THF (330 mL) over 20 min. The clear yellow solution was allowed to warm to rt overnight. Hydrazine hydrate (20 mL, 51% wt, 306 mmol) was added, and the resulting solution was heated at reflux for 6 h. The solution was allowed to cool to rt and concentrated HCl (50 mL) was slowly added. The solution was refluxed for 2 h, then allowed to cool to rt, and stirred overnight. The colorless precipitate (PPh3O) was removed by filtration, and the eluent was removed in vacuo. The residual colorless solid was redissolved in H2O (500 mL), and extracted with CH2Cl2 (3 × 500 mL). The aqueous layer was concentrated to provide 1664 as an off-white solid (32.8 g, 97%): 1H NMR (500 MHz, CD3OD) δ 2.96 (t, J = 7.6 Hz, 2H), 2.29–2.24 (m, 3H), 1.79 (m, 2H), 1.64–1.57 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 84.2, 70.5, 40.5, 27.8, 26.5, 18.7 ppm. This ammonium salt was used in the subsequent step without further purification.
N-(Hex-5-ynyl)-4-methylbenzenesulfonamide (18)
p-Toluenesulfonyl chloride (12.6 g, 65.9 mmol) was added in four portions over 30 min to a solution of ammonium salt 17 (8.00 59.9 mmol), NEt3 (25 mL, 180 mmol) and CH2Cl2 (120 mL) at 0 °C. The reaction was allowed to warm to rt and stirred overnight. The reaction was quenched with saturated aqueous NH4Cl, and extracted with CH2Cl2 (3 × 150 mL). The combined organic layers were dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexanes:EtOAc 1:0→3:2) to afford sulfonamide 1865 as a colorless solid (12.6 g, 85%): mp 63–65 °C; 1H NMR (500 MHz, CDCl3) δ 7.75 (d, J = 8.5 Hz, 2H), 7.30 (d, J = 8 Hz, 2H), 4.95–4.88 (m, 1H), 2.94 (q, J = 6.5 Hz, 2H), 2.42 (s, 3H), 2.15–2.12 (m, 2H), 1.91 (t, J = 2.5 Hz, 1H), 1.61–1.55 (m, 2H), 1.51–1.47 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 143.5, 137.1, 129.9, 127.2, 83.9, 69.0, 42.8, 28.6, 25.4, 21.7, 18.0 ppm; IR (thin film) 3294, 2944, 2870, 1598, 1426, 1322, 1154, 1092 cm−1; HRMS (ESI) m/z 274.0871 (274.0878 calcd for C13H17NO2SNa+ [MNa]+).
4-Methyl-N-(5-methylenehept-6-enyl) Benzenesulfonamide (19)
A solution of alkyne 18 (1.5 g, 5.9 mmol) and CH2Cl2 (100 mL) was degassed by sparging with nitrogen for 30 min. Benzylidene[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(tricyclohexyl-phosphine)ruthenium (0.18 g, 0.21 mmol)22 was added to the degassed solution, and the mixture was charged to a high pressure bomb. The reaction bomb was filled with ethylene gas (300 psi), vented and re-pressurized with ethylene (300 psi). This process was repeated a total of three times. The reaction was stirred at rt under high pressure (300 psi) for 4–5 h. After venting unreacted ethylene, the reaction mixture was transferred to a round bottom flask containing silica gel and the solvent was removed in vacuo. The residue was purified by column chromatography (dry load, hexanes:EtOAc 4:1) to give diene 19 (1.5 g, 90%) as a tan oil: 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.5 Hz, 2H), 7.28 (d, J = 8 Hz, 2H), 6.30 (dd, J = 17.5, 10.5 Hz, 1H), 5.25 (t, J = 6 Hz, 1H), 5.14 (d, J = 18 Hz, 1H), 5.00 (d, J = 11 Hz, 1H), 4.95 (s, 1H), 4.88 (s, 1H), 2.92 (q, J = 6 Hz, 2H), 2.40 (s, 3H), 2.11 (t, J = 7 Hz, 2H), 1.49–1.43 (m, 4H) ppm; 13C NMR (125 MHz, CDCl3) δ 145.8, 143.3, 138.7, 137.1, 129.7, 127.1, 115.8, 113.3, 43.1, 30.7, 29.3, 25.0, 21.5 ppm; IR (thin film) 3299, 2929, 1325, 1160, 1094 cm−1; HRMS (ESI) m/z 302.1186 (302.1191 calcd for C15H21NO2SNa+ [MNa]+).
4-Methyl-N-(4-(7-methyl-5-oxo-1,4,4a,5,6,7,8,8a-octahydronaphthalen-2-yl)butyl) benzenesulfonamide (20)
An oven-dried Schlenk tube was charged with 15 (5.7 g, 37 mmol) and 19 (6.9 g, 25 mmol), evacuated and back-filled with Ar. Dichloromethane (25 mL) was added, the reaction mixture was cooled to −78 °C, and TMSOTf (420 μL, 2.3 mmol) was added dropwise. The tube was sealed and stirred at −78 °C for 20 h. The reaction mixture was quenched at −78 °C by the addition of NEt3 (2 mL), and warmed to rt. The mixture was diluted with 20 mL Et2O, filtered through a short silica gel column, and the eluent was concentrated in vacuo to afford S2 as a yellow oil.
Ketal S2 was dissolved in 80 mL acetone (80 mL) and stirred at rt for 4 h with FeCl3/SiO2 (1.3 g).26 Silica gel (50 g) was added and the solvent was removed in vacuo. Purification of the residue by flash chromatography (dry packed, hexanes:Et2O 9:2→1:2) provided 20 as a yellow oil (9.07 g, 93%): 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.5 Hz, 2H), 5.19 (bs, 1H), 5.14 (t, J = 6.0 Hz, 1H), 2.83 (q, J = 6.5 Hz, 2H), 2.61 (t, J = 5.5 Hz, 1H), 2.44–2.39 (m, 1H), 2.37 (s, 3H), 2.34–2.29 (m, 1H), 2.16–2.12 (m, 1H), 2.09–2.04 (m, 2H), 1,97–1.90 (m, 2H), 1.87–1.74 (m, 3H), 1.68–1.59 (m, 2H), 1.40–1.35 (m, 2H), 1.34–1.26 (m, 2H), 0.97 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 211.9, 143.1, 137.1, 134.9, 129.6, 127.1, 118.8, 49.6, 47.0, 43.1, 38.4, 36.9, 35.6, 30.7, 29.8, 28.7, 24.2, 23.6, 22.5, 21.5 ppm; IR (thin film) 3286, 2915, 1702, 1599, 1325, 1160, 1095, 905 cm−1; HRMS (ESI) m/z 412.1929 (412.1922 calcd for C22H31NO3SNa+ [MNa]+).
(E)-N-(4-(5-(Hydroxyimino)-7-methyl-1,4,4a,5,6,7,8,8a-octahydronaphthalen-2-yl)butyl)-4-methylbenzenesulfonamide (21)
Aqueous H2NOH (6.2 mL, 94 mmol, 50% wt.) was added over 5 min to a stirred solution of ketone 20 (9.07 g, 23.2 mmol) in MeOH (250 mL) at rt. The reaction was stirred for 4 h, and the solvent was removed in vacuo. The residue was dissolved in CH2Cl2 (150 mL), and saturated aqueous NH4Cl (100 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 100 mL), the combined organic layers were dried with MgSO4, and concentrated to provide the cis-oxime as a colorless solid (9.24 g, 99%): 1H NMR (500 MHz, CDCl3) δ 8.52 (bs, 1 H), 7.77 (d, J = 8 Hz, 2H), 7.31 (d, J = 7.5 Hz, 2H), 5.23 (s, 1H), 4.75 (bs, 1H), 3.21 (dd, J = 13, 3.5 Hz, 1H), 2.93–2.91 (m, 2H), 2.51–2.50 (m, 1H), 2.44 (s, 3H), 2.42–2.38 (m, 1H), 2.22–2.18 (m, 1H), 2.09–2.05 (m, 1H), 2.01–1.96 (m, 1H), 1.88–1.84 (m, 2H), 1.83–1.79 (m, 1H), 1.71–1.68 (m, 2H), 1.52 (dd, J = 13.5, 11 Hz, 1H), 1.44–1.39 (m, 2H), 1.38–1.35 (m, 3H), 1.01 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 160.2, 143.4, 137.4, 135.4, 129.8, 127.3, 119.2, 42.9, 39.6, 38.4, 36.9, 34.5, 32.0, 30.1, 28.9, 28.6, 25.8, 23.8, 22.3, 21.7 ppm; IR (thin film) 3286, 2907, 1597, 1458, 1446, 1323, 1156, 1092 cm−1; HRMS (ESI) m/z 427.2025 (427.2031 calcd for C22H32N2O3SNa+ [MNa]+).
N-(4-(-5-Amino-7-methyl-1,4,4a,5,6,7,8,8a-octahydronaphthalen-2-yl)butyl)-4-methylbenzenesulfonamide (22)
cis-Oxime 21 (4.04 g, 10.0 mmol) was dissolved in EtOH (250 mL) and stirred at 30–35 °C. MoO3 (1.9 g, 13 mmol) was added followed by NaBH4 (1.9 g, 50 mmol; CAUTION, gas evolution), and the reaction was stirred for 1 h, during which the solution turned brown. A second portion of MoO3 (1.9 g, 13 mmol) and NaBH4 (1.9 g, 50 mmol) were added, and the reaction was stirred for an additional 1 h. A third portion of MoO3 (1.9 g, 13 mmol) and NaBH4 (1.9 g, 50 mmol) was added, and the reaction was stirred for an additional 1 h. The reaction mixture was cooled to rt, and a solution of KOH(aq) (0.2 M, 50 mL) was added. The slurry was stirred for 2 h. The brown precipitate was filtered with diatomaceous earth, and the filter cake was washed with excess methanol (300 mL). The filtrate was concentrated in vacuo. The residue was dissolved in saturated aqueous NH4Cl (150 mL), and extracted with CH2Cl2 (4 × 100 mL). The combined organic layers were dried with MgSO4 and concentrated to provide the amine as a colorless solid that was used without further purification (3.52 g, 90%): 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 8.0 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 5.23 (s, 1H), 3.61 (bs, 2H), 3.13 (bs, 1H), 2.85 (t, J = 6.5, Hz, 2H), 2.38 (s, 3H), 2.10–2.06 (m, 2H), 1.99–1.85 (m, 4H), 1.81 (t, J = 7.5 Hz, 2H), 1.58–1.29 (m, 8H), 0.96 (d, J = 7.5 Hz, 3H), 0.86–0.83 (m, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 143.1, 137.3, 135.5, 129.6, 127.1, 118.7, 49.1, 43.1, 37.7, 37.1, 35.8, 34.1, 33.0, 29.7, 29.1, 28.9, 26.6, 24.6, 21.5, 19.9 ppm; IR (thin film) 2907, 1599, 1458, 1324, 1159, 1095 cm−1; HRMS (ESI) m/z 391.2417 (391.2419 calcd for C22H35N2O2S [MH]+).
N-(4-((4a,8a)-5-(Cyanomethylamino)-7-methyl-1,4,4a,5,6,7,8,8a-octahydronaphthalen-2-yl)butyl)-4-methylbenzenesulfonamide (23)
A mixture of amine 22 (484 mg, 1.2 mmol), chloroacetonitrile (0.16 mL, 2.5 mmol), NEt3 (0.52 mL, 3.7 mmol), and tetrabutylammonium iodide (114 mg, 0.31 mmol) was stirred in THF (50 mL) at 70 °C for 30 h. The reaction was cooled to rt, and concentrated in vacuo. The residue was redissolved in CH2Cl2 (40 mL), washed with saturated aqueous NaHCO3 (30 mL). The organic layer was dried with MgSO4, and concentrated. The residue was purified by column chromatography (hexanes:EtOAc 4:1→3:2) to give cyanomethylamine 23 as a tan oil (418 mg, 78%): 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 8.0 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 5.29 (s, 1H), 4.63 (bs, 1H), 3.62–3.61 (m, 2H), 3.07–3.05 (m, 1H), 2.93 (q, J = 6.5 Hz, 2H), 2.44 (s, 3H), 2.03–2.00 (m, 2H), 1.96–1.93 (m, 3H), 1.88 (t, J = 7.5 Hz, 2H), 1.77 (bs, 1H), 1.65–1.52 (m, 3H), 1.46–1.41 (m, 2H), 1.37–1.32 (m, 3H), 1.25–1.24 (m, 1H), 1.12–1.05 (m, 1H), 0.94 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 143.3, 137.1, 129.7, 127.2, 126.5, 119.1, 118.3, 60.5, 48.0, 43.2, 37.2, 37.1, 34.9, 33.8, 29.8, 29.2, 24.6, 21.6, 21.1, 20.5, 14.2, 12.6 ppm; IR (thin film) 3267, 2917, 1600, 1466, 1459, 1380, 1328, 1161, 1095 cm−1; HRMS (ESI) m/z 452.2333 (452.2348 calcd for C24H35N3O2SNa+ [MNa]+).
Tricyclic Amine 24
A solution of cyanomethyl amine 23 (420 mg, 0.97 mmol) in dry CHCl3 (90 mL) was added to AgO2CCF3 (709 mg, 3.21 mmol). The suspension was stirred at rt for 2 h, after which time the reaction mixture was filtered with diatomaceous earth. The filter cake was washed with CHCl3 (50 mL), and the filtrate was washed with saturated aqueous NaHCO3 (50 mL), dried with MgSO4, and concentrated to give 24 as a tan oil (350 mg, 90%): 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8 Hz, 2H), 5.49 (s, 1H), 2.95 (t, J = 6.5 Hz, 3H), 2.80 (dd, J = 13.5, 2 Hz, 1H), 2.67 (d, J = 13.5 Hz, 1H), 2.46 (m, 4H), 2.46 (m, 1H), 2.10 (bs, 1H) 2.01 (bs, 1H), 1.88–1.86 (m, 2H), 1.81 (bs, 2H), 1.76–1.72 (m, 2H), 1.60–1.54 (m, 1H), 1.53–1.50 (m, 1H), 1.49–1.44 (m, 3H), 1.40–1.36 (m, 1H), 1.24–1.19 (m, 1H), 1.19–1.12 (m, 1H), 0.90 (d, J = 6.0 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 143.3, 138.2, 137.3, 130.6, 129.8, 127.2, 55.0, 47.9, 43.2, 40.6, 40.4, 36.5, 35.0, 33.2, 33.0, 32.4, 29.5, 24.9, 22.7, 22.0, 21.7 ppm; IR (thin film) 2928, 2873, 1600, 1466, 1457, 1326, 1305, 1160, 1095 cm−1; HRMS (ESI) m/z 403.2412 (403.2419 calcd for C23H35N2O2S+ [MH]+).
Tricyclic Carbamate 25
Triethylamine (0.10 mL, 0.75 mmol) was added to a stirred solution of amine 24 (150 mg, 0.37 mmol) and methylchloroformate (58 μL, 0.75 mmol) in CH2Cl2 (4.2 mL), and the reaction was stirred at rt for 12 h. The reaction mixture was concentrated, and the residue was purified by chromatography (hexanes:EtOAc 4:1→3:2) to give carbamate 25 as a colorless oil (114 mg, 67%): 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8 Hz, 2H), 5.22 (s, 1H), 5.14 (t, J = 6 Hz, 1H), 3.89–3.86 (m, 1H), 3.63 (s, 3H), 3.55 (q, J = 3 Hz, 1H), 3.03–3.00 (m, 1H), 2.94 (q, J = 6.5 Hz, 2H), 2.91 (dd, J = 12.5, 3.5 Hz, 1H), 2.45 (m, 4H), 2.10 (bs, 1H), 1.94–1.92 (m, 2H), 1.83–1.77 (m, 2H), 1.73–1.70 (m, 1H), 1.68–1.65 (m, 1H), 1.53–1.49 (m, 3H), 1.41–1.36 (m, 2H), 1.26–1.20 (m, 1H), 0.98–0.92 (m, 1H), 0.85 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 158.7, 143.3, 137.4, 137.2, 129.7, 128.9, 127.2, 55.9, 52.2, 49.6, 43.2, 41.0, 39.4, 36.9, 34.7, 33.7, 31.9, 31.8, 29.2, 24.7, 22.3, 22.0, 21.6 ppm; IR (thin film) 3269, 2945, 2917, 2850, 1702, 1685, 1442, 1326, 1307, 1294, 1268, 1243, 1230, 1201, 1157, 1094 cm−1; HRMS (ESI) m/z 483.2290 (483.2293 calcd for C25H36N2O4SNa+ [MNa]+).
Tricyclic Sulfonamide 28
p-Nitrophenylsulfonyl chloride (96 mg, 0.43 mmol) was added to a stirred solution of amine 24 (175 mg, 0.434 mmol) and NEt3 (90 μL, 0.65 mmol) in CH2Cl2 (20 mL), and the yellow solution was stirred overnight at rt. The reaction was quenched with 2 M HCl (10 mL), the layers were separated, and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed with brine (20 mL), dried with MgSO4, and concentrated. The resulting residue was purified by chromatography (hexanes:EtOAc, 1:1) to give sulfonamide 28 as yellow foam/oil (216 mg, 88%): 1H NMR (500 MHz, CDCl3) δ 8.00 (m, 1H), 7.77–7.67 (m, 5H), 7.32 (d, J =8.0 Hz, 2H), 5.22 (s, 1H), 4.62 (t, J = 6.0 Hz, 1H), 3.72 (d, J = 11.2 Hz, 1H), 3.57 (d, J = 2.9 Hz, 1H), 3.00–2.93 (m, 3H), 2.43 (s, 3H), 2.21–2.10 (m, 3H), 1.96 (m, 1H), 1.81 (m, 2H), 1.76–1.36 (m, 6H), 1.22–1.07 (m, 3H), 0.90 (dt, J = 14.3, 2.3 Hz, 1H), 0.28 (d, J = 6.1 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 147.7, 143.2, 136.9, 135.0, 133.4, 132.1, 130.9, 129.6, 128.4, 127.0, 124.9, 60.3, 58.0, 50.6, 42.9, 40.2, 38.5, 36.6, 34.8, 34.2, 31.9, 29.1, 23.9, 21.6, 21.4, 21.0, 14.1 ppm.
N-(4-(3,7-Dimethyl-5,6,7,8-tetrahydronaphthalen-2-yl)butyl)-4-methylbenzenesulfonamide (29)
Trifluoromethanesulfonic acid (13 mg, 0.085 mmol) was added to a solution of 28 (50 mg, 0.085 mmol) in toluene (5.0 mL). The reaction mixture was heated to reflux for 2 h, and then cooled to rt, and quenched with saturated aqueous NaHCO3 (10 mL). The layers were separated, and the organic layer was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were dried with MgSO4, concentrated in vacuo, and the resulting residue was purified by column chromatography (hexanes:EtOAc 4:1) to give 29 in low yield: 1H NMR (500 MHz, CDCl3) δ 7.74 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 6.84 (s, 1H), 6.74 (s, 1H), 4.26 (t, J = 6.1 Hz, 1H), 2.86 (q, J = 14.5, 7.9 Hz, 2H), 2.73–2.70 (m, 2H), 2.48–2.39 (m, 2H), 2.42 (s, 3H), 2.33–2.28 (m, 2H), 2.17 (s, 3H), 1.86–1.80 (m, 2H), 1.55–1.50 (m, 3H), 1.36–1.25 (m, 2H), 1.04 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 143.4, 137.1, 136.9, 134.3, 134.2, 132.8, 130.5, 129.7, 129.3, 127.1, 43.2, 37.7, 32.3, 31.6, 29.5, 29.4, 28.7, 27.3, 22.0, 21.5, 18.8 ppm; IR (thin film) 3282, 2932, 1455, 1326, 1158, 1094, 814 cm−1; HRMS (ESI) m/z 408.1964 (408.1973 calcd for C23H31NO2SNa [(MNa)+]).
Methyl Cyanomethyl(3-methyl-6-(4-(4-methylphenylsulfonamido)butyl)-1,2,3,4,4a,5,8,8a-octahydronaphthalen-1-yl)carbamate (37)
Methyl chloroformate (26 μL, 0.340 mmol) was added to a mixture of 23 (73 mg, 0.017 mmol) and K2CO3 (0.14 g, 1.0 mmol) in THF (3.0 mL), and the yellow mixture was stirred overnight at rt. The reaction mixture was directly purified by column chromatography (hexanes/EtOAc 4:1→3:2) to provide carbamate 37 as yellow oil (43 mg, 52%): 1H NMR (500 MHz, CDCl3) δ 7.24 (d, J = 8.2 Hz, 2H), 7.29 (d, J = 8.1 Hz, 2H), 5.24 (s, 1H), 4.35 (t, J = 6.1 Hz, 1H), 4.26 (bs, 1H), 3.92 (d, J = 17.7 Hz, 1H), 3.79 (s, 3H), 2.92 (q, J = 13.3, 6.6 Hz, 2H), 2.43 (s, 3H), 2.26–2.08 (m, 4H), 1.97 (ddd, J = 12.9, 12.9, 5.5 Hz, 1H), 1.85 (t, J = 7.1 Hz, 2H), 1.71 (d, J = 15.7 Hz, 1H), 1.52–1.45 (m, 5H), 1.36 (m, 2H), 1.28 (m, 2H), 1.07 (dd, J = 7.3 Hz, 3H), 0.98 (m, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 143.42, 136.9, 135.2, 129.7, 127.1, 117.7, 116.9, 53.8, 53.4, 43.1 36.9, 34.0, 32.4, 31.4, 29.3, 29.1, 28.4, 27.8, 24.4, 21.5, 20.3, 18.9, 14.6 ppm.66
Methyl 3-Methyl-6-(4-(4-methylphenylsulfonamido)butyl)-1,2,3,4,4a,5,8,8a-octahydro-naphthalen-1-yl carbamate (38)
Methyl chloroformate (81 μL, 1.0 mmol) was added dropwise over 5 min to a stirred mixture of amine 22 (340 mg, 0.87 mmol) and Na2CO3 (120 mg, 1.1 mmol) in H2O–CH2Cl2 (1:1, 2 mL) at rt. The reaction was stirred vigorously for 3 h, after which time the layers were separated. The organic layer was dried with MgSO4, and the solvent was removed in vacuo. The resulting residue was purified by column chromatography (hexanes:EtOAc 1:0→1:1) to provide carbamate 38 as a colorless solid (310 mg, 80%): 1H NMR (500 MHz, DMSO-d6, 298.0 K, peak broadening caused by dynamic NMR processes) δ 7.65 (d, J = 7.8 Hz, 2H), 7.45 (t, J = 4.9 Hz, 1H), 7.37 (d, J = 7.8 Hz, 2H), 6.98 (bs, 1H), 5.21 (bs, 1H), 3.71 (bs, 1H), 3.49 (s, 3H), 2.69 (bs, 2H), 2.37 (s, 3H), 2.10 (d, J = 17.0 Hz, 1H), 2.05–1.86 (m, 4H), 1.85–1.57 (m, 4H), 1.48 (m, 1H), 1.40 (t, J = 9.6 Hz, 1H), 1.36–1.19 (m, 5H), 1.00 (d, J = 7.0 Hz, 3H), 0.93 (d, J = 12.7 Hz, 1H) ppm; 13C NMR (125 MHz, DMSO-d6, 323.0 K, peak broadening caused by dynamic NMR processes) δ 155.7, 144.2, 137.9, 134.5, 129.3, 126.3, 118.3, 50.8, 48.0, 42.3, 39.9, 36.4, 35.3, 33.6, 31.6, 28.3, 27.6, 26.3, 23.9, 20.7, 19.8, 19.0 ppm; IR (thin film) 3267, 2908, 1698, 1528, 1158 cm−1; HRMS (ESI) m/z 471.2282 (471.2293 calcd for C24H36N2O4SNa [MNa]+).
Methyl (1H-Benzo[d][1,2,3]triazol-1-yl)methyl(3-methyl-6-(4-(4-methylphenylsulfonamido)-butyl)-1,2,3,4,4a,5,8,8a-octahydronaphthalen-1-yl)carbamate (39)
Trifluoroacetic acid (4–5 drops) was added to a solution of 38 (50 mg, 0.11 mmol) and (1H-benzo[d][1,2,3]triazol-1-yl)methanol (0.050 mL, 0.33 mmol) in dry acetonitrile (2.5 mL). The resulting solution was stirred at rt overnight, and the solvent was removed in vacuo. The residue was dissolved in Et2O and unreacted benzotriazole was removed by filtration. The remaining material was purified by column chromatography (hexanes:EtOAc 4:1→1:1) to afford benzotriazole 39 (45 mg, 70%). An analytical sample was further purified using preparatory HPLC (hexanes:EtOAc 19:1→3:2) 1H NMR (500 MHz, CDCl3) δ 8.05 (d, J = 8.3 Hz, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.57–7.54 (m, 3H), 7.42 (t, J = 7.6 Hz, 1H), 7.20 (d, J = 8.0 Hz, 2H), 6.12 (s, 2H), 5.23 (s, 1H), 4.70 (bs, 1H), 3.88 (bs, 1H), 3.63 (s, 3H), 3.25 (t, J = 7.5 Hz, 2H), 2.39 (s, 3H), 2.05–1.93 (m, 6H), 1.78 (t, J = 7.2 Hz, 2H), 1.71–1.26 (m, 8H), 1.15 (m, 1H), 1.03 (d, J = 7.0 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.7, 146.3, 144.2, 136.5, 135.7, 132.8, 130.0, 128.4, 127.1, 124.7, 120.0, 119.4, 111.0, 66.1, 59.3, 52.0, 50.0, 47.7, 37.2, 36.8, 35.2, 33.6, 29.9, 28.9, 27.5, 24.6, 21.7, 20.3, 15.5 ppm; IR (thin film) 3386, 3332, 1712, 1520, 1454, 1342, 1261, 1234, 1157, 1115, 1034, 933, 748 cm−1; HRMS (ESI) m/z 602.2777 (602.2776 calcd for C31H41N5O4SNa [MNa]+).
Sulfonamide-Terminated Aza-Prins Cyclization to Form Tetracyclic Sulfonamide 27
A stock solution of paraformaldehyde (20 mg), TFA (1 mL) and CHCl3 (10 mL) was prepared in a flame-dried round-bottom flask under nitrogen. An aliquot of the stock formaldehyde solution (0.30 mL, ~1 equiv of paraformaldehyde and 20 equiv of TFA) was added to a solution of amide 38 (0.10 g, 0.022 mmol) in CHCl3 (1 mL), and the resulting mixture was stirred at rt until complete consumption of the starting material was observed by electrospray MS analysis (~3 h). The reaction was quenched with saturated aqueous NaHCO3 (3 mL) and extracted with CHCl3 (3 × 3 mL). The combined organic layers were dried with MgSO4, and concentrated in vacuo. The crude residue was purified by preparative HPLC (Alltima silica 5 micron, length 250 mm, ID 22 mm; hexanes:EtOAc 7:3; 16 mL/min; retention time 9.8–11 min) to give tetracyclic product 27 (20 mg, 20%) as a colorless solid: 1H NMR (500 MHz, CDCl3) δ 7.66 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 3.72 (m, 1H), 3.62 (s, 3H), 3.48 (ddd, J = 14.6, 4.0, 4.0 Hz, 1H), 3.32 (t, J = 12.7 Hz, 1H), 2.74 (m, 1H), 2.50 (dd, J = 13.2, 5.0 Hz, 1H), 2.42 (s, 3H), 2.40–2.39 (m, 1H), 2.18–2.12 (m, 2H), 1.95–1.91 (m, 3H), 1.82 (ddd, J = 13.5, 10.8, 2.7 Hz, 1H), 1.85–1.73 (m, 2H), 1.63–1.45 (m, 4H), 1.01 (ddd, J = 12.8, 12.8, 3.0 Hz, 1H), 0.97 (ddd, J = 13.9, 3.5, 3.5 Hz, 1H), 0.88 (dd, J = 6.5 Hz, 3H), 0.83 (ddd, J = 14.4, 14.4, 3.0 Hz, 1H) ppm; 13C NMR (125 MHz, CDCl3) δ 156.2, 143.3, 135.2, 129.8, 127.4, 59.4, 56.7, 54.3, 51.9, 49.3, 40.1, 38.9, 36.1, 36.0, 35.1, 34.1, 32.1, 30.9, 29.3, 24.9, 22.0, 21.7, 21.4 ppm; IR (thin film) 1713 cm−1; HRMS (ESI) m/z 483.2284 (483.2293 calcd for C25H36N2O4SNa [MNa]+).
Tetracyclic Carbamate 43
A flame dried three-neck flask was equipped with a stir bar, two rubber septa, and a cold-finger condenser under nitrogen. The condenser and the round bottom flask were cooled to −78 °C and ammonia gas was condensed into the three-neck round bottom flask (~10 mL). To this solution at −78 °C was added Na0 (0.010 g, 0.46 mmol, washed with HPLC grade pentane three times). The resulting deep blue reaction was stirred at −78 °C for 5 min, and then a solution of tetracyclic aza-Prins product 27 (7.0 mg, 0.015 mmol) in THF (3 mL) was added dropwise. The reaction was stirred at −78 °C for 15 min and quenched by the slow addition of methanol (MeOH added dropwise until reaction becomes clear, ~2 mL) followed by the addition of saturated aqueous NH4Cl (3 mL). The resulting mixture was allowed to warm to rt overnight open to the atmosphere. The aqueous solution was extracted with CH2Cl2 (3 × 10 mL), and the combined organic solutions were dried with MgSO4, and concentrated in vacuo to provide an orange oil. This residue was purified by column chromatography (MeOH:CHCl3:NH4OH 10:90:1) to afford carbamate 43 (5.0 mg, 98%) as a colorless oil. NMR spectra are reported as the free base generated by the addition of NaOCD3 to the NMR sample: 1H NMR (500 MHz, CD3OD) δ 3.74 (bs, 1H), 3.60 (s, 3H), 2.88 (m, 1H), 2.76–2.66 (m, 2H), 2.57 (dd, J = 11.0, 3.6 Hz, 1H), 2.33 (m, 1H), 2.12 (d, J = 14.0 Hz, 1H), 1.99 (dd, J = 11.6, 2.8 Hz, 1H), 1.92 (ddd, J = 13.8, 11.1, 3.2 Hz, 1H), 1.88–1.71 (m, 3H), 1.70–1.50 (m, 4H), 1.48–1.42 (m, 2H), 1.40–1.29 (m, 2H), 1.06 (ddd, J = 12.8, 12.8, 3.1 Hz, 1H), 0.96 (ddd, J = 16.6, 4.6, 4.6 Hz, 1H), 0.87 (d, J = 6.5 Hz, 3H), 0.86 (m, 1H) ppm; 13C NMR (125 MHz, CD3OD) δ 158.1, 60.9, 57.8, 56.6, 52.5, 41.5, 40.0, 38.4, 37.8, 37.1, 35.5, 34.1, 32.2, 31.3, 24.8, 22.6, 22.5 ppm;66 IR (thin film) 3363, 1706 cm−1; HRMS (ESI) m/z 307.2386 (307.2393 calcd for C18H31N2O2 [MH]+).
5-epi-Nankakurine A (1)
An Et2O solution of LiAlH4 (0.30 mL, 0.29 mmol, 1.0 M) was added dropwise to a solution of N-methyl carbamate 43 (9.0 mg, 0.029 mmol) in THF (3.0 mL) at 0 °C. The reaction mixture was allowed to warm to rt, stirred overnight at rt, quenched with H2O (2.0 mL), and Rochelle salt (20 mL), and stirred for an additional 2 h. The mixture was transferred to a separatory funnel, diluted with Et2O (20 mL), agitated and the two solutions were separated. The aqueous solution was extracted with CH2Cl2 (5 × 20 mL), the combined organic solutions were dried over MgSO4, filtered and then concentrated in vacuo to yield orange oil residue. The residue was purified by column chromatography (MeOH:CHCl3:NH4OH 10:90:1) to afford diamine (±)-1 (5.0 mg, 65%) as a yellow oil: IR (thin film) 3458, 1679 cm−1; HRMS (ESI) m/z 263.2490 (263.2487 calcd for C17H31N2+ [MH]+).
A portion of this sample was dissolved in 0.60 mL of CD3OD for NMR analysis and 150 μL of NaOCD3 (~0.1 M in CD3OD) was added to assure that only the free base was present. Aliquots of a solution of TFA in CD3OD (15–25 μL portions of a 0.15 M solution) were added to this sample and 1H NMR spectra were obtained after each addition (Figure S1). Under no conditions were spectra obtained that matched the 1H NMR data reported for nankakurine A.2 1H NMR (500 MHz, CD3OD/CD3ONa) δ 2.80 (m, 1H), 2.73 (dd, J = 14.6, 3.6 Hz, 1H), 2.54 (dd, J = 14.5, 10.9 Hz, 1H), 2.36 (bs, 1H), 2.28 (s, 3H), 2.28 (m, 1H), 2.02 (m, 1H), 1.95–1.82 (m, 3H), 1.75–1.66 (m, 4H), 1.54–1.43 (m, 4H), 1.19 (t, J = 2.9 Hz, 1H), 1.06 (m, 1H), 1.00 (ddd, J = 12.8, 12.8, 3.2 Hz, 1H), 0.86 (m, 1H), 0.86 (dd, J = 6.7 Hz, 3H), 0.74 (t, J = 12.8 Hz, 1H) ppm; 13C NMR (125 MHz, CD3OD) δ 63.3, 56.5, 56.1, 42.0, 40.7, 39.0, 37.8, 35.4, 34.4, 32.5, 32.2, 30.9, 23.0, 22.9, 22.6 ppm.67
((Hex-5-ynyloxy)methyl)benzene (S3)45
A slurry of KH (35 g, 260 mmol) in THF (100 mL) was added to a mixture of alcohol 16 (28 mL, 250 mmol), tetra-n-butylammonium iodide (470 mg, 1.3 mmol) and benzyl bromide (33 mL, 280 mmol) in THF (500 mL) at 0 °C. The suspension was warmed to rt, and stirred overnight. The reaction was quenched by the addition of H2O (400 mL), and the layers were separated. The aqueous layer was extracted with Et2O (2 × 200 mL). The combined organic extracts were dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexanes:EtOAc 99:1) to give benzyl ether S3 as a yellow oil (46.5 g, 97%): 1H NMR (500 MHz, CDCl3) δ 7.43–7.33 (m, 4H), 7.31 (m, 1H), 4.53 (s, 2H), 3.52 (t, J = 6.3 Hz, 2H), 2.24 (ddd, J = 7.0, 7.0, 2.5 Hz, 2H), 1.97 (t, J = 2.5 Hz, 1H), 1.84–1.72 (m, 2H), 1.71–1.60 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 138.7, 128.5, 127.7, 127.6, 84.5, 73.0, 69.8, 68.6, 28.9, 25.4, 18.4 ppm; IR (thin film) 3298, 2941, 2860, 1455, 1362, 1206, 1106 cm−1; HRMS (ESI) m/z 211.1100 (211.1099 calcd for C13H16ONa+ [MNa]+).
((5-Methylenehept-6-enyloxy)methyl)benzene (48)
Benzylidene[1,3-bis(2,4,6-trimethylphenyl)-2-imidazolidinylidene]dichloro(tricyclohexylphosphine)ruthenium22 (1.12 g, 1.33 mmol) was added to a solution of alkyne S3 (5.0 g, 27 mmol) and CH2Cl2 (400 mL), which was degassed with nitrogen for 60 min. The solution was charged to a high-pressure bomb. The reaction bomb was filled with ethylene gas (300 psi), vented and re-pressurized with ethylene (300 psi). This process was repeated a total of three times. The reaction was stirred under pressure (300 psi) for 4–5 h. After venting unreacted ethylene, the reaction mixture was transferred to a round bottom flask containing silica gel and the solvent was removed in vacuo. The residue was dry loaded and purified by column chromatography (hexanes:EtOAc, 19:1) to give diene 48 (5.2 g, 90%) as a tan oil: 1H NMR (500 MHz, CDCl3) δ 7.39 (d, J = 4.5 Hz, 4H), 7.35–7.28 (m, 1H), 6.42 (dd, J = 17.6, 10.8 Hz, 1H), 5.27 (d, J = 17.6 Hz, 1H), 5.10 (d, J = 10.9 Hz, 1H), 5.05 (d, J = 11.5 Hz, 2H), 4.55 (s, 2H), 3.54 (t, J = 6.4 Hz, 2H), 2.27 (t, J = 7.6 Hz, 2H), 1.80–1.68 (m, 2H), 1.67–1.59 (m, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 146.3, 139.1, 138.8, 128.5, 127.8, 127.6, 115.9, 113.3, 73.1, 70.4, 31.3, 29.8, 24.9 ppm; IR (thin film) 2939, 2858, 1596, 1455, 1362, 1104, 895 cm−1; HRMS (ESI) m/z 217.1600 (217.1592 calcd for C15H20O+ [MH]+).
(3′R,4a′S,8a′R)-6′-(4-(Benzyloxy)butyl)-3′-methyl-3′,4′,4a′,5′,8′,8a′-hexahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalene] (S4)
Trimethylsilyl trifluoromethanesulfonate (0.20 mL, 1.1 mmol) was added dropwise to a solution of enone (+)-946 (1.31 g, 11.9 mmol, >99% ee as determined by chiral HPLC) in CH2Cl2 (0.75 mL) at −78 °C. 1,2-Bis(trimethylsiloxy)ethane (2.93 mL, 11.9 mmol) was added, and the resulting solution was stirred at −78 °C for 5 min. Diene 48 (2.35 g, 10.9 mmol) was added, and the reaction was stirred at −78 °C for 22 h. The reaction was then quenched with NEt3 (0.25 mL), and the reaction was warmed to rt. The solution was passed through a small plug of neutral alumina and eluted with 50 mL of CH2Cl2, and the eluent was concentrated in vacuo. The residue was purified by column chromatography (hexanes:Et2O 49:1) to give ketal S4 (2.53 g, 64%) as a clear oil: [α]D23 − 1.7 (c = 1.25, CH3Cl); 1H NMR (500 MHz, CDCl3) δ 7.35 (d, J = 4.4 Hz, 4H), 7.29 (m, 1H), 5.27 (s, 1H), 4.52 (s, 2H), 3.95–3.82 (m, 4H), 3.48 (t, J = 6.6 Hz, 2H), 2.18–2.02 (m, 4H), 1.97 (t, J = 7.3 Hz, 2H), 1.97 (m, 1H), 1.93–1.86 (m, 1H), 1.80 (dd, J = 13.7, 3.9 Hz, 2H), 1.66–1.55 (m, 3H), 1.53–1.40 (m, 2H), 1.22–1.12 (m, 2H), 0.98 (d, J = 6.9 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 138.9, 136.7, 128.5, 127.7, 127.5, 118.8, 111.7, 73.0, 70.6, 65.2, 64.4, 41.6, 39.7, 37.8, 37.2, 32.0, 31.1, 29.3, 26.2, 24.2, 24.2, 21.8 ppm; IR (thin film) 2916, 2871, 1455, 1102, 1048, 949 cm−1; HRMS (ESI) m/z 388.2852 (388.2852 calcd for C24H34O3NH4+ [MNH4]+).
(3R,4aS,8aR)-6-(4-(Benzyloxy)butyl)-3-methyl-3,4,4a,5,8,8a-hexahydronaphthalen-1(2H)-one (50)
A preformed mixture of FeCl3/SiO226 (0.375 g) was added to a solution of ketal S4 (2.0 g, 5.4 mmol) in acetone (50 mL, ACS grade), and the resulting suspension was stirred at rt for 2 h. The solvent was removed in vacuo, and the residue was purified by column chromatography (hexanes:Et2O, 19:1) to provide the ketone 50 (1.75 g, 99%) as a colorless oil: [α]D23 + 55 (c = 2.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.31 (d, J = 4.4 Hz, 4H), 7.28–7.19 (m, 1H), 5.31 (s, 1H), 4.47 (s, 2H), 3.43 (t, J = 6.5 Hz, 2H), 2.64 (t, J = 5.8 Hz, 1H), 2.54 (dd, J = 17.5, 2.9 Hz, 1H), 2.46–2.38 (m, 1H), 2.35 (dd, J = 13.1, 4.3 Hz, 1H), 2.24–2.10 (m, 1H), 1.96 (t, J = 12.6 Hz, 2H), 1.89 (td, J = 13.2, 6.4 Hz, 2H), 1.88–1.78 (m, 3H), 1.63 (ddd, J = 16, 11.9, 4.3 Hz, 1H), 1.54 (dd, J = 14.1, 6.9 Hz, 2H), 1.48–1.36 (m, 2H), 1.00 (d, J = 6.4 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 211.6, 138.9, 135.6, 128.5, 127.7, 127.6, 118.6, 72.9, 70.4, 49.7, 47.2, 38.7, 37.6, 35.7, 30.8, 30.1, 29.5, 24.2, 23.8, 22.6 ppm; IR (thin film) 3397, 2913, 2868, 1708, 1454, 1104 cm−1; HRMS (ESI) m/z 349.2143 (349.2144 calcd for C22H30O2Na+ [MNa]+).
Methyl N′-6-(4-(Benzyloxy)butyl)-3-methyl-1,2,3,4,4a,5,8,8a-octahydronaphthalen-1-ylmethanehydrazonoperoxoate (51)
A solution of methyl hydrazidocarbamate (0.010 g, 0.11 mmol) and decalone 50 (33 g, 0.010 mmol) was stirred at rt for 3 days in MeOH (1.0 mL), and NaCNBH3 (6.9 mg, 0.11 mmol) and bromocresol green indicator (~1 mg) were added to the solution. A MeOH/HCl solution then was added dropwise over 30 min at a rate to maintain the pH ~3–5. (A green/blue color was maintained for 30 minutes and the reaction was complete when a yellow color persisted.) The reaction mixture was quenched with 6 N KOH (1.0 mL), and stirred for 1 h. This mixture was diluted with saturated aqueous NH4Cl (5 mL), and extracted with CH2Cl2 (5 × mL). The combined organic layers were dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexanes:EtOAc 4:1) to give hydrazide 51 (4.8 mg, 12%) as a colorless solid: 1H NMR (500 MHz, CDCl3) δ 7.36–7.26 (m, 5H), 6.04 (bs, 1H), 5.41 (s, 1H), 4.51 (s, 2H), 3.71 (s, 3H), 3.48 (t, J = 6.2 Hz, 2H), 3.15 (bs, 1H), 2.18–1.84 (m, 8H), 1.67–1.41 (m, 9H), 1.16 (m, 1H), 0.95 (d, J = 6.6 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 158.1, 138.9, 128.6, 127.8, 127.7, 126.7, 119.1, 73.1, 70.6, 52.5, 37.81, 34.6, 33.9, 32.2, 30.4, 29.9, 29.6, 24.4, 22.9, 20.7, 14.4 ppm;67 IR (thin film) 3324, 1726, 1455,, 1365, 1261, 1101 cm−1; HRMS (ESI) m/z 423.2618 (423.2624 calcd for C24H36N2O3Na+ [MNa]+).
N′-(6-(4-(Benzyloxy)butyl)-3-methyl-1,2,3,4,4a,5,8,8a-octahydronaphthalen-1-yl)-4-methylbenzenesulfonohydrazide (52)
A solution of tosylhydrazide (0.24 g, 1.3 mmol) in MeOH (15 mL), was added over 30 h by syringe pump to a solution of 50 (0.400 g, 1.23 mmol) and KOH (1.65 g, 29.5 mmol) in MeOH (15 mL) at rt. The reaction was quenched with saturated aqueous NH4Cl (30 mL), and extracted with CH2Cl2 (3×20 mL). The combined organic extracts were dried with MgSO4 and concentrated. The residue was purified by column chromatography (hexanes:EtOAc 9:1→3:1) to provide the corresponding hydrazone (0.070 g) as a colorless solid. A mixture of MeOH and concentrated HCl (9:1) was added dropwise at rt over 1 h to a stirred mixture of hydrazone (0.70 g, 0.14 mmol), NaBH3CN (0.010 g, 0.15 mmol) and a trace (~ 0.2 mg) of methyl orange in MeOH (3 mL). The rate of addition was controlled so that the color of the reaction mixture remained orange-red (pH = 3–4) over 1 h. The reaction was quenched with aqueous 6 N KOH (10 mL). The aqueous layer was extracted with Et2O (5 × 10 mL). The combined organic layers were washed with water (30 mL) and brine (30 mL), dried with MgSO4, and concentrated in vacuo. Purification by column chromatography (hexanes:EtOAc 9:1→3:1) provided 52 as a colorless solid (45 mg, 7%, 2 steps): 1H NMR (500 MHz, CDCl3) δ 7.83–7.78 (m, 3H), 7.36–7.32 (m, 4H), 7.30–7.28 (m, 3H), 5.78 (s, 1H), 5.23 (s, 1H), 4.51 (s, 2H), 3.49–3.44 (m, 2H), 3.32 (m, 1H), 2.44 (s, 3H), 2.01–1.73 (m, 6H), 1.60–1.38 (m, 8H), 1.03–0.86 (m, 5H), 0.82 (d, J = 6.8 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 144.1, 138.9, 135.7, 129.8, 128.6, 128.4, 127.9, 127.8, 127.7, 118.6, 73.1, 70.6, 41.7, 37.8, 37.6, 37.5, 34.4, 33.5, 30.4, 29.9, 29.6, 29.5, 25.5, 24.4, 21.8 ppm; IR (thin film) 1454, 1362, 1207, 1103, 1034, 1011, 814 cm−1.
N′-((1S,3R,4aS,8aR)-6-(4-(Benzyloxy)butyl)-3-methyl-1,2,3,4,4a,5,8,8a-octahydronaphthalen-1-yl)benzohydrazide (53)
A solution of benzoic hydrazide (0.38 g, 2.8 mmol) and 50 (0.90 g, 2.8 mmol) was stirred at rt for 3 days in MeOH (20 mL), and NaCNBH3 (0.190 g, 3.03 mmol) and bromocresol green indicator (~1 mg) were added to the solution. A MeOH/HCl solution then was added dropwise over 30 min at a rate to maintain a pH ~3–5. (A green/blue color was maintained for 30 minutes and the reaction was complete when a yellow color persisted.) The reaction mixture was quenched with 6 N KOH (2 mL), and stirred for 1 h. This mixture was diluted with saturated aqueous NH4Cl, and extracted with CH2Cl2 (5 × 20 mL). The combined organic layers were dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexanes:EtOAc 4:1) to give 53 (0.98 g, 80%) as a colorless solid: mp 98–101 °C; [α]D24 −21 (c = 1.1, CHCl3); 1H NMR (500 MHz, DMSO–d6, 323 K, peak broadening caused by dynamic NMR processes) δ 9.90 (bs, 1H), 7.81 (d, J = 6.5 Hz, 2H), 7.51 (t, J = 7.3 Hz, 1H), 7.44 (t, J = 7.7 Hz, 2H), 7.33–7.24 (m, 5H), 5.35 (bs, 1H), 4.88 (bs, 1H), 4.42 (s, 3H), 3.40 (t, J = 6.3 Hz, 2H), 3.11 (bs, 1H), 2.25–1.93 (m, 5H), 1.90–1.87 (m, 3H), 1.64 (m, 1H), 1.54–1.46 (m, 3H), 1.42–1.38 (m, 2H), 1.24 (m, 1H), 1.03 (m, 1H), 0.97 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO–d6, 340 K) δ 149.5, 138.5, 130.6, 127.8, 127.7, 126.9, 126.8, 126.7, 118.6, 71.5, 69.3, 36.6, 34.1, 33.4, 28.5, 28.3, 23.5, 19.8 ppm; IR (thin film) 3291, 2915, 2862, 1630, 1457, 1315, 1103 cm−1;68 HRMS (ESI) m/z 469.2823 (469.2831 calcd for C29H38N2O2Na+ [MNa]+).
Intramolecular Azomethine Imine Cycloaddition to Form Tetracyclic Pyrazolidine 54
Benzoyl hydrazide 53 (250 mg, 0.56 mmol) was added to a sealable tube containing paraformaldehyde (84 mg, 2.8 mmol), 4 Å molecular sieves powder (1.7 g, activated by flame drying under vacuum), diisopropylethylamine (93 μL, 0.56 mmol) and toluene (12 mL). The resulting suspension was placed in an oil bath pre-heated to 115 °C and stirred at 115 °C until the starting material was completely consumed (~20 h) as monitored by electrospray MS analysis or TLC (hexanes:EtOAc 4:1). The reaction mixture was allowed to cool to rt, and the molecular sieves were removed by filtration through a plug of neutral alumina, eluting with Et2O (50 mL). The solution was concentrated in vacuo, and the residue was purified by column chromatography (hexanes:EtOAc 4:1) to give the tetracyclic product 54 (218 mg, 85%) as a colorless solid: mp 123–126 °C; [α]D23 −16 (c = 1.6, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.52–7.50 (m, 2H), 7.38–7.25 (m, 8H), 4.47 (s, 2H), 3.47 (ddd, J = 21.7, 9.3, 6.2 Hz, 2H), 3.19 (m, 1H), 2.90 (t, J = 4.3 Hz, 1H), 2.73 (d, J = 10.8 Hz, 1H), 2.57 (ddd, J = 13.2, 13.2, 3.9 Hz, 1H), 2.40 (t, J = 4.8 Hz, 1H), 2.24 (d, J = 14.8 Hz, 1H), 2.07 (m, 1H), 1.89 (dd, J = 14.7, 11.1 Hz, 1H), 1.82–1.67 (m, 4H), 1.67–1.48 (m, 6H), 1.35–1.26 (m, 1H), 1.16 (ddd, J = 12.4, 1.9, 1.9 Hz, 1H), 1.04 (ddd, J = 12.9, 12.9, 4.9 Hz, 1H), 0.81 (ddd, J = 13.5, 13.3, 5.7 Hz, 1H) 0.73 (d, J = 6.3 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 167.9, 138.9, 138.2, 129.7, 128.5, 128.1, 127.9, 127.6, 127.6, 73.0, 70.3, 69.9, 62.8, 62.4, 42.2, 40.2, 38.1, 36.3, 34.5, 34.2, 33.7, 33.4, 30.1, 23.5, 22.5, 21.1 ppm; IR (thin film) 1624 cm−1; HRMS (ESI) m/z 459.3009 (459.3011 calcd for C30H39N2O2+ [MH]+).
Tricyclic Amino Amide (59)
A solution of SmI2 (9 mL, ~1 M in THF)51,59 was added at a rapid rate to a stirring solution of tetracyclic pyrazolidine 54 (120 mg, 0.26 mmol) in MeOH (1.0 mL), which had been sparged with argon for 30 min. The deep blue reaction mixture was stirred at rt under argon for 30 min, and then quenched with 37% aqueous formaldehyde (2 mL). Bromocresol green indicator (~1 mg) and NaCNBH3 (50 mg, 0.78 mmol) were added, and a solution of MeOH/HCl was then added dropwise over 30 min at a rate to maintain a pH ~3–5. (A green/blue color was maintained for 30 minutes and the reaction complete when a yellow color persisted.) The reaction mixture was then quenched with aqueous 6 N KOH (2 mL) and stirred for 1 h. The mixture was diluted with saturated aqueous NH4Cl, and extracted with CH2Cl2 (5 × 10 mL). The combined organic layers were dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography (hexanes:acetone 7:3) to give tricyclic amino amide 59 (100 mg, 80%) as a yellow oil: [α]D24 +10 (c = 0.8, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.71 (d, J = 7.4 Hz, 2H), 7.47 (t, J = 7.2 Hz, 1H), 7.40 (t, J = 7.5 Hz, 2H), 7.36–7.19 (m, 5H), 5.74 (s, 1H), 4.44 (s, 2H), 3.43 (dddd, J = 21.8, 9.0, 6.5, 6.5 Hz, 2H), 2.93 (d, J = 11.8, 1H), 2.60 (bs, 1H), 2.42 (t, J = 12.2, 1H), 2.26 (ddd, J = 13.0, 13.0, 4.2 Hz, 1H), 2.14 (dd, J = 11.8, 2.5 Hz, 1H), 2.03 (s, 3H), 1.97–1.87 (m, 4H), 1.81–1.79 (m, 2H), 1.71 (dd, J = 11.8, 5.7 Hz, 1H), 1.59 (ddd, J = 14.5, 14.5, 6.8 Hz, 2H), 1.52–1.37 (m, 4H), 1.29–1.24 (m, 1H), 1.18 (ddd, J = 12.4, 12.8, 4.9 Hz, 1H), 0.89 (m, 1H), 0.84 (d, J = 6.4, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 166.8, 138.9, 136.1, 131.3, 128.7, 128.5, 127.8, 127.6, 126.9, 73.0, 70.6, 63.7, 60.4, 59.3, 43.4, 40.6, 39.0, 37.7, 36.2, 35.4, 34.2, 32.9, 31.1, 30.2, 22.8, 21.2, 20.8 ppm; IR (thin film) 2921, 2852, 2771, 1644, 1528, 1488, 1455, 1102 cm−1; HRMS (ESI) m/z 475.3329 (475.3325 calcd for C31H43N2O2+ [MH]+).
Diamino Alcohol (S5)
A round bottom flask containing diamine 59 (140 mg, 0.29 mmol), Pearlman’s catalyst (20 wt.% Pd(OH)2/C, 41 mg, 0.030 mmol), concentrated HCl (1 drop) and MeOH (10 mL) was fitted with a septa and a balloon of hydrogen gas. The reaction vessel was evacuated and backfilled with hydrogen 5 times. The reaction mixture was stirred at rt for 1 h, and filtered through a syringe filter (25 mm, 0.45 μm pore size), and the filtrate concentrated in vacuo. The residue was purified by column chromatography (MeOH:CHCl3:NH4OH (5:95:1)) to give diamino alcohol S5 (110 mg, 97%) as a colorless solid: mp 60–63 °C; [α]D24 + 14 (c = 1.15, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 7.3 Hz, 2H), 7.48 (t, J = 7.3 Hz, 1H), 7.42 (t, J = 7.8 Hz, 2H), 5.82 (s, 1H), 3.67–3.59 (m, 2H), 2.94 (d, J = 11.8 Hz, 1H), 2.57 (bs, 1H), 2.43 (t, J = 12.4 Hz, 1H), 2.33 (ddd, J = 12.8, 12.8, 4.2 Hz, 1H), 2.15 (dd, J = 11.8, 2.9 Hz, 1H), 2.05 (s, 3H), 1.99–1.82, (m, 7H), 1.72 (dd, J = 11.9, 5.8 Hz, 1H), 1.63–1.47 (m, 5H), 1.40 (m, 1H), 1.30 (m, 1H), 1.19 (ddd, J = 12.7, 12.4 4.9 Hz, 1H), 0.91 (m, 1H), 0.86 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 167.1, 135.9, 131.4, 128.8, 126.9, 63.7, 62.6, 60.5, 59.3, 43.4, 40.6, 39.0, 37.8, 36.4, 35.3, 33.8, 32.9, 32.8, 31.1, 22.8, 21.2, 20.0 ppm; IR (thin film) 3343, 2944, 2921, 2865, 2771, 1644, 1530, 1457 cm−1; HRMS (ESI) m/z 385.2838 (385.2885 calcd for C24H37N2O2+ [MH]+).
Diamino Alcohol (60)
A THF solution of AlH352 (3.6 mL, 1.8 mmol, ~0.5 M) was added to a solution of amide S5 (70 mg, 0.18 mmol) in THF (4 mL). The reaction mixture was stirred at rt for 3 h, and then quenched with Rochelle salt. The resulting mixture was stirred overnight and then extracted with CH2Cl2 (10 × 10 mL). The combined organic extracts were dried over MgSO4 filtered and concentrated in vacuo. Purification of the residue by column chromatography (MeOH:CHCl3:NH4OH 0:99:1→10:90:1) afforded diamino alcohol 60 (50 mg, 74%) as a colorless oil: [α]D23 + 4.3 (c = 0.7, MeOH); 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 7.3 Hz, 2H), 7.31 (t, J = 7.3 Hz, 1H), 7.23 (t, J = 7.3 Hz, 1H), 3.67 (t, J = 6.6 Hz, 2H), 3.62 (d, J = 3.6 Hz, 1H), 3.60 (m, 1H), 3.07 (d, J = 11.6 Hz, 1H), 2.19 (t, J = 12.8 Hz, 1H), 2.13 (dd, J = 11.6, 2.9 Hz, 1H), 2.08 (s, 3H), 2.01–1.84 (m, 4H), 1.83–1.62 (m, 5H), 1.55–1.34 (m, 10H), 1.16 (ddd, J = 12.6, 12.4, 4.9 Hz, 1H), 0.87 (m, 1H), 0.83 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CDCl3) δ 141.9, 128.6, 128.5, 126.9, 63.8, 63.4, 58.6, 57.4, 45.4, 43.6, 41.0, 39.5, 39.2, 36.6, 36.1, 33.9, 33.8, 33.5, 32.0, 22.9, 21.1, 19.4 ppm; IR (thin film) 3342, 2921, 2865, 2769, 1455, 1061 cm−1; HRMS (ESI) m/z 371.3062 (371.3062 calcd for C24H39N2O2+ [MH]+).
N-Benzyl Nankakurine A (61)
Methanesulfonyl chloride (25 μL, 0.32 mmol) was added dropwise to a solution of diamino alcohol 60 (110 mg, 0.29 mmol) and NEt3 (54 μL, 0.38 mmol) in CH2Cl2 (5.0 mL) at −40 °C. The reaction was stirred at −40 °C for 30 min and then the reaction was quenched at −40 °C with MeOH (0.6 mL) and NEt3 (0.2 mL). The resulting solution was allowed to warm to rt and stirred for 3 h. The solvent was removed in vacuo and the residue was purified by column chromatography (MeOH:CHCl3:NH4OH 0:99:1→10:90:1) to afford diamine 61 (100 mg, 96%) as a colorless oil: [α]D24 + 4.6 (c = 1.0, CHCl3); 1H NMR (500 MHz, CD3OD) δ 7.35 (d, J = 7.4 Hz, 2H), 7.24 (t, J = 7.4 Hz, 2H), 7.14 (t, J = 7.4 Hz, 1H), 3.88 (s, 2H), 3.35 (ddd, J = 10.9, 2.3, 2.3 Hz, 1H), 2.80 (m, 1H), 2.72 (t, J = 12.5 Hz, 1H), 2.49 (d, J = 13.8 Hz, 1H), 2.34 (bs, 1H), 2.14 (s, 3H), 2.10–1.93 (m, 5H), 1.85 (m, 1H), 1.75 (m, 1H), 1.65–1.33 (m, 19H), 1.14 (ddd, J = 12.7, 12.7, 4.9 Hz, 1H), 0.88 (m, 1H), 0.82 (d, J = 6.6 Hz, 3H) ppm; 13C NMR (125 MHz, CD3OD) δ 144.6, 129.3, 129.0, 127.3, 64.9, 59.3, 59.1, 50.8, 44.8, 43.5, 42.5, 42.1, 40.5, 37.6, 34.8, 33.3, 32.9, 31.1, 23.2, 22.2, 21.4, 21.2 ppm; IR (thin film) 2944, 2921, 2863, 2769, 1621, 1453, 1104 cm−1; HRMS (ESI) m/z 353.2953 (353.2957 calcd for C24H37N2+ [MH]+).
(+)-Nankakurine A (2)
A round bottom flask containing N-benzylnankakurine A (61) (90 mg, 0.26 mmol), palladium on carbon catalyst (10 wt % Pd/C, 27 mg, 0.03 mmol), concentrated HCl (1 drop) and MeOH (4.0 mL) was fitted with a septa and a balloon of hydrogen gas. The reaction vessel was evacuated and backfilled with hydrogen 5 times. The reaction mixture was stirred at rt for 1 h, and then filtered through a syringe filter (25 mm, 0.45 μm pore size), and the filtrate concentrated in vacuo. The residue was purified by column chromatography (MeOH:CHCl3:NH4OH 5:95:1) to give (+)-nankakurine A (66 mg, 99%) as a colorless solid: [α]D24 + 13 (c = 0.4, MeOH); IR (thin film) 3397, 1671 cm−1; HRMS (ESI) m/z 263.2484 (263.2487 calcd for C17H31N2+ [MH]+).
A portion of this sample was dissolved in 0.60 mL of CD3OD and 150 μL of NaOCD3 (~0.1 M in CD3OD) was added to assure that only the free base was present. Aliquots of a solution of TFA in CD3OD (15–25 μL portions of a 0.15 M solution) were successively added to the sample, and 1H NMR spectra were obtained after each addition (Figure S2). Spectra matching the reported spectrum of (+)-nankakurine A2 were obtained upon addition of several portions of TFA•CD3OD (Figure 3 and S3). NMR spectra of the free base: 1H NMR (500 MHz, CD3OD/CD3ONa) δ 3.00 (ddd, J = 11.9, 5.1, 2.7 Hz, 1H), 2.80 (t, J = 4.5 Hz, 2H), 2.28 (t, J = 12.7 Hz, 1H), 2.13 (dd, J = 12.7, 2.8 Hz, 1H), 2.11 (s, 3H), 2.05–2.02 (m, 2H), 2.01–1.90 (m, 1H), 1.89–1.76 (m, 3H), 1.69–1.67 (m, 3H), 1.66–1.44 (m, 7H), 1.20 (ddd, J = 12.7, 12.7, 5.1 Hz, 1H), 0.89 (m, 1H), 0.84 (d, J = 6.6 Hz, 3H) ppm; 13C NMR (125 MHz, CD3OD) δ 65.2, 58.7, 56.0, 43.6, 42.0, 41.4, 40.1, 37.6, 34.8, 34.6, 32.7, 26.5, 23.2, 22.2, 21.1 ppm.2,66
(+)-Nankakurine B (3)
To a solution of synthetic (+)-nankakurine A (24 mg, 0.9 mmol) in MeOH (3 mL) was added NaCNBH3 (17 mg, 0.28 mmol). To the resulting solution was added a MeOH/HCl solution dropwise over 30 min at a rate to maintain a pH of ~3–5 (no indicator was used because it was difficult to separate from nankakurine B). The reaction mixture was quenched with aqueous 6 N KOH and stirred for 1 h, saturated aqueous NH4Cl was added, and the mixture was extracted with CH2Cl2 (5 × 20 mL). The combined organic layers were dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by column chromatography (MeOH:CHCl3:NH4OH (10:90:1)) to give (+)-nankakurine B (20 mg, 80%) as a colorless solid: [α]D24 + 12 (c = 1.5, MeOH); IR (thin film) 3449, 1680 cm−1; HRMS (ESI) m/z 276.2635 (276.2644 calcd for C18H33N2+ [MH]+)
A portion of this sample was dissolved in 0.60 mL of CD3OD and 150 μL of NaOCD3 (~0.1 M in CD3OD) was added. 1H and 13C NMR spectra of the free base were obtained, and aliquots of a solution of TFA in CD3OD (15–25 μL portions of a 0.15 M solution) were successively added to the sample (Figure 1 and S4). Spectra matching the reported spectrum of (+)-nankakurine B4 were obtained after the addition of TFA•CD3OD (Figure 1 and S5). NMR spectra of the free base: 1H NMR (500 MHz, CD3OD/CD3ONa) δ 3.09 (ddd, J = 11.4, 5.1, 2.7 Hz, 1H), 3.0 (bs, 1H), 2.80 (bs, 1H), 2.49 (t, J = 12.6 Hz, 1H), 2.41 (s, 3H), 2.10 (s, 3H), 2.08 (dd, J = 11.9, 3.0 Hz, 1H), 2.05–1.93 (m, 3H), 1.92–1.77 (m, 4H), 1.76–1.54 (m, 5H), 1.53–1.38 (m, 4H), 1.22 (ddd, J = 12.7, 12.7, 4.6 Hz, 1H), 0.90 (m, 1H), 0.85 (d, J = 6.5 Hz, 3H) ppm; 13C NMR (125 MHz, CD3OD) δ 64.9, 60.4, 58.6, 43.4, 42.2, 40.3, 36.9, 35.2, 34.5, 32.7, 25.9, 23.2, 22.3, 20.6, 19.1 ppm.69
Neurite Growth Assay
H19-7 cells (ATCC) were derived from hippocampi from E17 rat embryos and immortalized by retroviral transduction of SV40 large T antigen. The H19-7 cells were plated at low density (20 000 cell/well) in 48-well cell culture plates in N2-supplemented DMEM. After 24 h in culture, cells were treated with 0.3–10 μM concentrations of either nankakurine A (2) or nankakurine B (3) or with 1 μM of a reference compound in culture medium. After treatment for 24 h, cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with triton X100 and incubated with an antibody against neuronal class b-III tubulin (Tuj1, Covance) overnight at 4 °C. Cells were incubated with a secondary Alexa Fluor® 546 goat anti-mouse antibody for 30 min at room temperature. Both length of neurites stained by Tuj1 and cell counting were quantified using a fluorescence microscope (Zeiss) coupled to an automated image analysis system (ExploraNova). The ratio between these two endpoints (total length of neurites per cell in μm) allowed us to evaluate the activity of nankakurines on neurite outgrowth of H19-7 cells. Data are expressed as percent of neurite length in treated-cells compared to non-treated cells (control).
Supplementary Material
Acknowledgments
This research was supported by the NIH Neurological Disorders & Stroke Institute (NS-12389), and by unrestricted grants from Amgen and Merck. J.R.A. and R.A.A. were funded by the University of California President’s Postdoctoral Fellowship and the NIH (5F32GM085989), respectively. NMR and mass spectra were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation grants. We are grateful to Dr. Hiroshi Morita, Department of Pharmacognosy, Faculty of Pharmaceutical Sciences, Hoshi University, Japan, for copies of 1H and 13C NMR spectra of natural nankakurine B; Dr. Dominique Lesuisse, Aging Department, Sanofi-aventis R&D, for arranging for neurotrophic testing of synthetic nankakurines A and B; and Dr. Joe Ziller, X-Ray Crystallography Facility Director, UC Irvine, for X-ray analyses.
Footnotes
Supporting Information Available: Copies of 1H and 13C NMR spectra of new compounds; 1H NMR comparison of synthetic 1, 2 and 3 with natural 2 and 3; CIF file for compound 54. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of the Lycopodium alkaloids, see: Kobayashi J, Morita H. In: The Alkaloids. Cordell GA, editor. Vol. 61. Academic Press; New York: 2005. p. 1.Ma X, Gang DR. Nat Prod Rep. 2004;21:752–772. doi: 10.1039/b409720n.Ayer WA, Trifonov LS. In: The Alkaloids. Cordell GA, Brossi A, editors. Vol. 45. Academic Press; New York: 1994. p. 233.Ayer WA. Nat Prod Rep. 1991;8:455–463. doi: 10.1039/np9910800455.MacLean DB. In: The Alkaloids. Brossi A, editor. Vol. 26. Academic Press; New York: 1985. p. 241.MacLean DB. In: The Alkaloids. Manske RHF, editor. Vol. 14. Academic Press; New York: 1973. p. 348.MacLean DB. In: The Alkaloids. Manske RHF, editor. Vol. 10. Academic Press; New York: 1968. p. 305.
- 2.Hirasawa Y, Morita H, Kobayashi J. Org Lett. 2004;6:3389–3391. doi: 10.1021/ol048621a. [DOI] [PubMed] [Google Scholar]
- 3.Ayer WA, Ball LF, Browne LM, Tori M, Delbaere LTJ, Silverberg A. Can J Chem. 1984;62:298–302. [Google Scholar]
- 4.Hirasawa Y, Kobayashi J, Obara Y, Nakahata N, Kawahara N, Goda Y, Morita H. Heterocycles. 2006;68:2357–2364. [Google Scholar]
- 5.(a) Dawbarn D, Allen SJ. Neuropath Appl Neurobiol. 2003;29:211–230. doi: 10.1046/j.1365-2990.2003.00487.x. [DOI] [PubMed] [Google Scholar]; (b) Dauer W. Science. 2007;411:60–62. doi: 10.1126/stke.4112007pe60. [DOI] [PubMed] [Google Scholar]
- 6.Kirik D, Georgievska B, Björklund A. Nat Neurosci. 2004;7:105–110. doi: 10.1038/nn1175. [DOI] [PubMed] [Google Scholar]
- 7.(a) Hefti F. Annu Rev Pharmacol Toxicol. 1997;37:239–267. doi: 10.1146/annurev.pharmtox.37.1.239. [DOI] [PubMed] [Google Scholar]; (b) Luu B, González de Aguilar J-L, Girlanda-Junges C. Molecules. 2000;5:1439–1460. [Google Scholar]; (c) Waters SP, Tian Y, Li Y-M, Danishefsky SJ. J Am Chem Soc. 2005;127:13514–13515. doi: 10.1021/ja055220x. and references therein. [DOI] [PubMed] [Google Scholar]
- 8.The scarcity of the natural nankakurines (2 and 3 were isolated in 0.0003% and 0.0002% yields, respectively) prevented evaluation of the neurotrophic properties of the less abundant nankakurine B (2).4
- 9.Hitchcock SA, Pennington LD. J Med Chem. 2006;49:7559–7583. doi: 10.1021/jm060642i. [DOI] [PubMed] [Google Scholar]
- 10.Portions of this work were described in an earlier communication: Nilsson BL, Overman LE, Read de Alaniz J, Rohde JM. J Am Chem Soc. 2008;130:11297–11299. doi: 10.1021/ja804624u.
- 11.After our studies in this area were concluded, an efficient construction of (±)-2 and (±)-3 from (±)-luciduline (5) was described: Cheng, X., Waters, S. P. Org. Lett. 2010, 12, 205–207.
- 12.(a) Heathcock CH, Ruggeri RB, McClure KF. J Org Chem. 1992;57:2585–2594. [Google Scholar]; (b) Lögers M, Overman LE, Welmaker GS. J Am Chem Soc. 1995;117:9139–9150. [Google Scholar]
- 13.(a) Oppolzer W, Petrzilka M. J Am Chem Soc. 1976;98:6722–6724. [Google Scholar]; (b) Oppolzer W, Petrzilka M. Helv Chim Acta. 1978;61:2755–2762. [Google Scholar]
- 14.(a) Chong B-D, Ji Y-I, Oh S-S, Yang J-D, Baik W, Koo S. J Org Chem. 1997;62:9323–9325. [Google Scholar]; (b) Nicolaou KC, Montagnon T, Baran PS. Angew Chem, Int Ed. 2002;41:1386–1389. doi: 10.1002/1521-3773(20020415)41:8<1386::aid-anie1386>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 15.Mondal M, Puranik VG, Argade NP. J Org Chem. 2007;72:2068–2076. doi: 10.1021/jo0624344. [DOI] [PubMed] [Google Scholar]
- 16.(a) Sardina FJ, Johnston AD, Mourino A, Okamura WH. J Org Chem. 1982;47:1576–1578. [Google Scholar]; (b) Masaguer CF, Raviña E, Fontenla JA, Brea J, Tristán H, Loza MI. Eur J Med Chem. 2000;35:83–95. doi: 10.1016/s0223-5234(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 17.(a) Mewshaw RE. Tetrahedron Lett. 1989;30:3753–3756. [Google Scholar]; (b) Clark RD, Heathcock CH. J Org Chem. 1976;41:636–643. doi: 10.1021/jo00870a023. [DOI] [PubMed] [Google Scholar]
- 18.Cossy J, Bellosta V, Ranaivosata J-L, Gille B. Tetrahedron. 2001;57:5173–5182. [Google Scholar]
- 19.Tsunoda T, Suzuki M, Noyori R. Tetrahedron Lett. 1980;21:1357–1358. [Google Scholar]
- 20.Illesinghe J, Campi EM, Jackson WR, Robinson AJ. Aust J Chem. 2004;57:531–536. [Google Scholar]
- 21.In our early studies, sulfonamide 18 was prepared from 16 by reaction with (cyanomethylene)-tributylphosphorane and p-toluenesulfonamide; however, this reaction provided low yields (18–80%) in some runs.10
- 22.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 23.For selected examples of enyne metathesis, see: Hansen EC, Lee D. J Am Chem Soc. 2004;126:15074–15080. doi: 10.1021/ja045422d.Smulik JA, Diver ST. Org Lett. 2000;2:2271–2274. doi: 10.1021/ol006035l.
- 24.Gassman PG, Singleton DA, Wilwerding JJ, Chavan SP. J Am Chem Soc. 1987;109:2182–2184. [Google Scholar]
- 25.Anderson JC, Blake AJ, Graham JP, Wilson C. Org Biomol Chem. 2003;1:2877–2885. doi: 10.1039/b305116a. [DOI] [PubMed] [Google Scholar]
- 26.Kim KS, Song YH, Lee BH, Han CS. J Org Chem. 1986;51:404–407. [Google Scholar]
- 27.These mild conditions were important for conserving the configurational integrity of the cis-octahydronaphthalene ketone product.
- 28.We previously reported the use of an EtAlCl2-catalyzed Diels–Alder reaction to prepare decalone 20 in modest yield as a mixture of epimers (74%, cis:trans = 1:1–1:3).10 This mixture of compounds was converted under kinetic control to a mixture of cis- and trans-oximes 21 under basic conditions (>90% yield, 3:1–5:1 cis:trans). Pure cis-21 could be selectively precipitated in 40–60% yield. For details, see references 10 and 13.
- 29.Ipaktchi J. Chem Ber. 1984;117:856–858. [Google Scholar]
- 30.Overman LE, Jacobsen EJ, Doedens RJ. J Org Chem. 1983;48:3393–3400. [Google Scholar]
- 31.(a) Schlummer B, Hartwig JF. Org Lett. 2002;4:1471–1474. doi: 10.1021/ol025659j. [DOI] [PubMed] [Google Scholar]; (b) Haskins CM, Knight DW. Chem Commun. 2002:2724–2725. doi: 10.1039/b207755h. [DOI] [PubMed] [Google Scholar]
- 32.For recent reviews of metal-catalyzed hydroamination reactions, see: Müller TE, Beller M. Chem Rev. 1998;98:675–704. doi: 10.1021/cr960433d.Hultzsch KC. Adv Synth Catal. 2005;347:367–391.
- 33.Most late transition metals effect hydroamination by initial activation of the olefin, followed by attack of the amine nucleophile onto the alkene from the face trans to the coordinated metal.32 For the metal-catalyzed hydroamination of 24 or 25, the most sterically accessible face of the olefin for metal coordination resides on the convex face of the tricycle, which would require cyclization of the amine to occur from the concave face, and would deliver the incorrect stereochemistry of the piperidine spirocycle. The steric effect of the tricycle could inhibit this cyclization. Metals that promote hydroamination by amine activation represent the most attractive catalysts to prepare epi-nankakurine A (1).
- 34.(a) Anderson LL, Arnold J, Bergman RG. J Am Chem Soc. 2005;127:14542–14543. doi: 10.1021/ja053700i. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hong S, Marks TJ. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r. [DOI] [PubMed] [Google Scholar]
- 35.Komeyama K, Morimoto T, Takaki K. Angew Chem Int Ed. 2006;45:2938–2941. doi: 10.1002/anie.200503789. [DOI] [PubMed] [Google Scholar]
- 36.See, inter alia: Ziegler FE, Spitzner EB. J Am Chem Soc. 1973;95:7146–7149. doi: 10.1021/ja00802a040.Stevens RV, Lee AWM. J Am Chem Soc. 1979;101:7032–7035.Overman LE, Lesuisse D, Hashimoto M. J Am Chem Soc. 1983;105:5373–5379.
- 37.Anet FAL, Yavari I. J Am Chem Soc. 1977;99:2794–2796. [Google Scholar]
- 38.In stereoelectronically more favorable contexts, regioselectivity of deprotonation of this type in aza-Prins cyclizations can be ascribed to an ene mechanism. For an early example, see: Kametani Tetrahedron Lett. 1986;27:1167–1170.
- 39.(a) Katritzky AR, Yao G, Lan X, Zhao X. J Org Chem. 1993;58:2086–2093. [Google Scholar]; (b) Kobayashi S, Ishitani H, Komiyama S, Oniciu DC, Katritzky AR. Tetrahedron Lett. 1996;37:3731–3734. [Google Scholar]; (c) Katritzky AR, Pernak J, Fan W-Q. Synthesis. 1991:868–870. [Google Scholar]; (d) Katritzky AR, Fang Y, Silina A. J Org Chem. 1999;64:7622–7624. [Google Scholar]
- 40.The yield was similar in CHCl3 alone. Although AcOH promoted the desired reaction in 12% yield, reactions employing other acids such as camphorsulfonic acid, methanesulfonic acid, and formic acid did not afford any of the tetracyclic product 27. Moreover the desired spirotetracyclic product was not detected when TFA was employed neat or as a solution in THF, MeOH, DMF, PhMe, or H2O. The use of Lewis acids, such as BF3•OEt, TiCl4, SnCl4, AlCl3, FeCl3, CeCl3 or TMSOTf at −65 °C to room temperature in CH2Cl2 did not provide any detectable 27. The use of formalin or 1,3,5-trioxane instead of formaldehyde under various conditions also did not lead to 27.
- 41.We do not attribute the low yield of 27, in an appreciable extent, to its instability under acidic conditions. Although 27 fragments slowly to a mixture of tricyclic alkenes 40 in the presence of 10 equiv of TFA in CDCl3, its half-life under these conditions was ~ 5 h.
- 42.After eventually completing the synthesis of authentic nankakurine A (2), we became convinced that the spectra in the original report were obtained from a natural sample that contained a mixture of the free base and its conjugate acid (vide infra). As a result, 1H NMR spectra of various mixtures of synthetic (±)-1 and its trifuoroacetate salt were measured. None of these spectra matched the 1H NMR data reported for natural nankakurine A (see data in the Supporting Information).2
- 43.(a) The TURBOMOLE V6.2 quantum chemistry program was utilized for higher-level DFT calculations: TURBOMOLE V6.2. Turbomole GmbH; Karlsruhe, Germany: 2009. http://www.turbomole.com. [Google Scholar]; (b) Treutler O, Ahlrichs R. J Chem Phys. 1995;102:346–354. [Google Scholar]
- 44.For a recent review, see: Schantl JG. Azomethine Imines. In Science of Synthesis. Vol. 27. Georg Thieme Verlag; Stuttgart: 2004. p. 731.For select examples, see: Oppolzer W. Tetrahedron Lett. 1970;11:3091–3094.Oppolzer W. Tetrahedron Lett. 1972;13:1707–1710.Oppolzer W. Angew Chem, Int Ed Engl. 1977;16:10–23.Jacobi PA, Martinelli MJ, Polanc S. J Am Chem Soc. 1984;106:5594–5598.Katz JD, Overman LE. Tetrahedron. 2004;60:9559–9568.Gergely J, Morgan JB, Overman LE. J Org Chem. 2006;71:9144–9152. doi: 10.1021/jo061537j.
- 45.Takami K, Mikami S, Yorimitsu H, Shinokubo H, Oshima K. J Org Chem. 2003;68:6627–6631. doi: 10.1021/jo0344790. [DOI] [PubMed] [Google Scholar]
- 46.Fleming I, Maiti P, Ramarao C. Org Biomol Chem. 2003;1:3989–4004. doi: 10.1039/b305880h. and references therein. [DOI] [PubMed] [Google Scholar]
- 47.The Diels–Alder reaction of enone (+)-9 and diene 48, and subsequent deprotection to prepare 50 was performed prior to the finding that the Diels–Alder reaction of (±)-15 with 19, followed by deprotection provided a higher yield of the cis-octahydronaphthalene ketone. It would be expected that the Diels–Alder reaction of (+)-15 with 48 would increase the yield of 50 obtained after deprotection.
- 48.Hydrazines 51 and 52 were prepared only once; no attempt was made to optimize their syntheses.
- 49.The structure of 54 was confirmed by single-crystal X-ray analysis: CCDC 690534. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 50.For enhancement of yields using NEt3 in bimolecular cycloadditions of formaldehyde-derived azomethine imines see: Kanemasa S, Tomoshige N, Wada E, Tsuge O. Bull Chem Soc Jpn. 1989;62:3944–3949.
- 51.Freshly prepared SmI2 was required to obtain reproducible yields for this reaction.
- 52.Brown HC, Yoon NM. J Am Chem Soc. 1966;88:1464–1472. [Google Scholar]
- 53.To facilitate monitoring reactions and purifying intermediates, the N-benzyl protecting group was retained until the last step.
- 54.Reported optical rotations for the natural products are: (a) nankakurine A: [α]21D +16 (c 0.4, MeOH),2 (b) nankakurine B: [α]19D +12 (c 1.0, MeOH).4
- 55.Eves EM, Tucker MS, Roback J, Downen M, Rosner MR. Proc Natl Acad Sci USA. 1992;89:4373–4377. doi: 10.1073/pnas.89.10.4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Representative images can be found in the Supporting Information.
- 57.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 58.Armarego WLF, Chai CLL. Purification of Laboratory Chemicals. 5. Butterworth Heinemann; Amsterdam: 2003. [Google Scholar]
- 59.Samarium metal (5.64 g, 37.5 mmol, 40 mesh) was transferred into a Schlenk flask in a glove box. The Schlenk flask was sealed, removed from the glove box, and placed under a steady stream of nitrogen. A solution of iodine (6.35 g, 25.0 mmol) in THF (250 mL, freshly distilled from a ketyl still) was transferred by cannula to the Schlenk flask. Upon completion of the cannula transfer, the Schlenk tube was sealed under nitrogen and heated to 50 °C for 24 h. The reaction mixture was cooled to rt and the dark blue stock solution of SmI2 (~0.08 M) was used without further purification.
- 60.(a) Mondal M, Puranik VG, Argade NP. J Org Chem. 2007;72:2068–2076. doi: 10.1021/jo0624344. [DOI] [PubMed] [Google Scholar]; (b) Tietze LE, Spiegl DA, Stecker F, Major J, Raith C, Grosse C. Chem Eur J. 2008;14:8956–8963. doi: 10.1002/chem.200800967. [DOI] [PubMed] [Google Scholar]
- 61.Clark RSJ, Holmes AB, Matassa VG. J Chem Soc, Perkin Trans 1. 1990:1389–1400. [Google Scholar]
- 62.Musser AK, Fuchs PL. J Org Chem. 1982;47:3121–3131. [Google Scholar]
- 63.Watson IDG, Yudin AK. J Am Chem Soc. 2005;127:17516–17429. doi: 10.1021/ja055288c. [DOI] [PubMed] [Google Scholar]
- 64.Rozhiewicz DI, Janczewski D, Verboom W, Ravoo BJ, Reinhoudt DN. Angew Chem, Int Ed. 2006;45:5292–5296. doi: 10.1002/anie.200601090. [DOI] [PubMed] [Google Scholar]
- 65.Sulfonamide 18 was reported previously with no characterization data, see: Luo FT, Wang RT. Tetrahedron Lett. 1992;33:6835–6838.Ochifuji N, Mori M. Tetrahedron Lett. 1995;36:9501–9504.
- 66.One 13C NMR signal was not observed.
- 67.Two 13C NMR signals were not observed.
- 68.Several 13C NMR signals were not observed, because of peak broadening caused by dynamic NMR processes.
- 69.Three 13C NMR signals were not observed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.