

Abstract
The inherent instability of heptahelical G protein-coupled receptors (GPCRs) during purification and reconstitution is a primary impediment to biophysical studies and to obtaining high-resolution crystal structures. New approaches to stabilize receptors during purification and to screen reconstitution procedures are needed. Here we report the development of a novel homogeneous time-resolved fluorescence assay (HTRF) to quantify properly folded CC-chemokine receptor 5 (CCR5). The assay permits high-throughput thermal stability measurements of femtomole quantities of CCR5 in detergent and in engineered nanoscale apolipoprotein bound bilayer (NABB) particles. We show that recombinant expressed CCR5 can be incorporated into NABB particles in high yield, resulting in greater thermal stability compared with CCR5 in detergent solution. We also demonstrate that CCR5 binding to the HIV-1 cellular entry inhibitors maraviroc, AD101, CMPD 167, and vicriviroc dramatically increases receptor stability. The HTRF assay technology reported here is applicable to other membrane proteins and could greatly facilitate structural studies of GPCRs.
Keywords: GPCR, lipoprotein particle, CCR5, NABB, thermal stability, maraviroc, europium cryptate, time-resolved fluorescence, energy transfer
G protein-coupled receptors (GPCRs) constitute a large family of heptahelical membrane proteins that recognize a broad array of extracellular ligands and couple to multiple intracellular signaling pathways.(1, 2) Their involvement in the molecular pathophysiology of a number of diseases has made GPCRs a major drug target.(3) Recent high-resolution structures of several GPCRs, including rhodopsin, β2-adrenergic receptor, β1-adrenergic receptor, A2A adenosine receptor, and opsin have provided insight into the molecular mechanism of receptor activation, but further advances are necessary to understand their physiological function and to enhance drug discovery. Stabilizing these complex polytopic membrane proteins in purified and/or defined systems remains the most significant present challenge for biochemists and structural biologists. Successful approaches to generating stable GPCRs in detergent solution include truncations or deletions of disordered regions, insertion of domains such as T4 lysozyme, complex formation with an antibody Fab fragment, ligand binding, and introduction of a combination of stabilizing mutations.(4) These strategies are often difficult or impossible to rationalize and success with one particular receptor does not guarantee generality.
The study of GPCRs in detergents has limits, even when stabilizing conditions are known. Typically, GPCRs, with the possible exception of rhodopsin, are only marginally stable in detergents. Furthermore, the most stabilizing detergents tend to have a low critical micellar concentration (CMC) and relatively large micelle size, which can lead to aggregation phenomena that complicate interpretation of results.(5) In general, G protein activity is sharply limited by detergent concentration and assays of receptor function are extremely difficult, making study of reconstituted ligand-receptor-G protein “signalosomes” in solution impractical.(6) Nanodisc technology is an attractive alternative to detergents because of its defined lipid bilayer.(7) We have recently reported the incorporation of rhodopsin, a prototypical GPCR, into discoidal lipoprotein particles called nanoscale apolipoprotein bound bilayers (NABBs).(8) Though incorporation of β2-adrenergic receptor and CCR5 into HDL particles has also been recently reported, further functional characterization and optimization of reconstitution conditions are necessary.(9, 10) For these reasons, a sensitive, versatile, and high-throughput assay to quantify properly folded GPCRs and to characterize their stability would have significant utility.
We report here a novel homogeneous time-resolved fluorescence (HTRF) immuno-sandwich assay based on the simultaneous binding of anti-receptor monoclonal antibodies (mAbs) labeled with a europium cryptate (EuK) fluorescent donor and a modified allophycocyanin (XL665) acceptor. The HTRF technology has been applied earlier to monitor ligand binding to GPCRs in cell membranes.(11-13) Our HTRF assay enables high-throughput measurements of the thermal stability of recombinant expressed CCR5 in detergents and NABBs using extremely minute quantities of receptor compared with existing approaches.(14, 15) We show that CCR5-NABBs are more stable than detergent-solubilized receptor, suggesting that NABBs are a viable platform for biochemical and biophysical study. We also studied the effects of the small molecule antagonists maraviroc, AD101, CMPD 167, and vicriviroc on CCR5 thermal stability. Using the HTRF assay, we found that the antagonists inhibit 2D7 mAb binding to its conformationally sensitive epitope on the EC2 loop to varying degrees, but none completely. Further, the melting curves demonstrate that the small molecules confer a striking increase in CCR5 thermal stability. This suggests that these high-affinity ligands trap the receptor in a single conformation and prevent denaturation. The antagonist-CCR5 complexes should be promising candidates for structural studies.
Materials and Methods
Cells, Plasmids, and Reagents
HEK-293T cells were obtained from the American Type Culture Collection (Manassas, VA). Transfection reagents were obtained from Invitrogen (Carlsbad, CA). Lipids were obtained from Avanti Polar Lipids (Alabaster, AL) except cholesterol, which was obtained from Sigma-Aldrich (St. Louis, MO). Detergents were obtained from Anatrace, Inc. (Maumee, OH). HTRF reagents were obtained from Cisbio (Bedford, MA). All time-resolved FRET assays were performed on a PerkinElmer EnVision Multilabel Plate Reader. Buffer N: 20 mM Tris-HCl, pH 7.0, 0.1 M (NH4)2SO4, 10% (v/v) glycerol, 0.07% cholesteryl hemisuccinate (CHS), 0.018% 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 0.008% 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS), 0.33% n-dodecyl β-D-maltoside (DM), 0.33% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS). Buffer E1: Buffer N supplemented with 400 μM 1D5-nonapeptide corresponding to the engineered C-terminal receptor epitope. Buffer S: 20 mM Tris-HCl, pH 7.0, 100 mM (NH4)2SO4, and 10% (v/v) glycerol. Buffer C: 100 mM phosphate, 150 mM NaCl, pH 7.2. Buffer P: 100 mM phosphate, 150 mM NaCl, pH 7.5. Buffer E2: Buffer P supplemented with 200 mM imidazole. Buffer G: 100 mM phosphate, 150 mM NaCl, 0.5 mg/mL BSA, pH 7.0.
Preparation of lipids
Lipids were dissolved in chloroform at approximately 100 mg/mL in glass round bottom flasks. The solvent was evaporated under a gentle stream of dry argon while rotating the flask, forming a thin lipid film. The lipids were re-dissolved in dichloromethane and the procedure was repeated. Remaining dichloromethane was removed under vacuum overnight. The lipid films were then hydrated with the appropriate detergent-containing buffers, sonicated, and freeze-thawed in liquid N2 until fully solubilized.
Transient transfection of HEK cells and solubilization of heterologously expressed CCR5
HEK-293T cells were grown in Dulbecco's Modified Eagle's Medium (DMEM; 4.5 g/L glucose, 2 mM glutamine; Gibco) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals) at 37°C in a 5% CO2 atmosphere. HEK-293T cells were transfected with human CCR5 cDNA tagged with the C-terminal 1D4 epitope, TETSQVAPA, in pcDNA3.1(+) using Lipofectamine Plus as previously described for earlier experiments with rhodopsin.(16) Briefly, for one 10-cm plate, 0.75 mL of pre-warmed DMEM was mixed with 3.5 μg plasmid DNA. To this was added 10 μL of PLUS reagent, incubated for 15 min, then transferred to a separate mixture containing 0.5 mL DMEM and 17 μL Lipofectamine Plus. After another 15 min incubation, the volume was made up to a total of 4 mL with DMEM and added to HEK-293T cells at 70-80% confluence. Four hours after transfection, an additional 4 mL of DMEM supplemented with 20% FBS was added. Cells expressing CCR5 were harvested 48 h after transfection in PBS buffer containing the protease inhibitors aprotinin and leupeptin. Cell pellets from 10-cm plates were lysed in 1 mL Buffer N (supplemented with protease inhibitors) per 5 × 106 cells. After thorough resuspension, the solution was probe-tip sonicated with 6 × 1-s pulses and incubated for 2 h at 4°C. The solution was then centrifuged at 20,000 × g for 20 min at 4°C. The supernatant fraction was collected and stored at -80°C until further use.
Immobilization of 1D4 monoclonal antibody
Anti-rhodopsin antibody 1D4 was immobilized on cyanogen bromide activated Sepharose 2B (Sigma-Aldrich), a bead-formed agarose-based gel filtration matrix with 2% agarose content, using established protocols with slight modifications.(17, 18) In a fume hood, 30 mL of packed Sepharose 2B in a total volume of 60 mL 2 M sodium carbonate buffer, pH 11, was treated with 3 g of CNBr in 3 mL of acetonitrile on an ice bath under gentle agitation with a suspended magnetic stirrer. The pH of the reaction was maintained at 10-11 by titration with 1 N NaOH over a period of 30 min. The activated beads were washed under suction four times with 50 mL of ice cold water and twice with 50 mL of cold coupling buffer (150 mM sodium chloride and 10 mM sodium borate buffer, pH 8.2). 25 mL packed beads were immediately incubated with 50 mg 1D4 mAb in 50 mL total volume of coupling buffer with head-over-head mixing at 4°C overnight. Finally, the matrix was washed at 4°C with 100 mL aliquots of phosphate buffered saline, pH 7.4, once with 10 mM glycine for 20 min and four times without glycine. The matrix was stored in the presence of 0.065% sodium azide. The protein binding was quantitative with 2 mg antibody per mL of settled beads, and the resulting binding capacity for rhodopsin was 0.4 mg/mL, as determined spectrophotometrically using solubilized rod outer segment disk membranes.(8)
Purification of heterologously expressed CCR5 and incorporation into NABBs
Solubilized lysate from 2 × 10-cm plates was thawed on ice and added to 50 μL packed beads of 1D4-sepharose resin with 2 mg/mL immobilized 1D4 mAb. The lysate-resin mixture was incubated for 16 h at 4°C. The resin was transferred to a Micro-Spin column (Pierce), centrifuged to remove the supernatant fraction, and washed twice with buffer N. Purified CCR5 was eluted by incubating the resin with 2 × 50 μL buffer E1 for 30 min. The NABB mixture was prepared by adding the combined CCR5 elutions to 3.75 nmol purified zap1 prepared as described,(8) 280 nmol 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and 22.7 nmol cholesterol in a total volume of 200 μL containing 1.5% sodium cholate, 0.33% DM, 15 mM Tris-HCl, pH 7.0, 75 mM (NH4)2SO4, and 7.5% glycerol. When desired, a POPC solution containing 0.5% lissamine rhodadmine B-labeled 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (LRB-DOPE) was used to allow visualization of NABB elution. After vortexing and incubating on ice for 30 min, the mixture was added to 1 mL of Pierce Detergent Removal Resin pre-equilibrated with at least one column volume of Buffer S. Elution was carried out at 4°C under gravity flow by addition of Buffer S to the column and collection of 200 μL fractions. Fractions were analyzed for protein content by monitoring absorbance at 280 nm. Protein-containing fractions were pooled and applied to a Superose 6 10/300 gel filtration column. Absorbance at 280 nm (protein) and, when applicable, 570 nm (LRB-DOPE) was monitored with an in-line detector in the FPLC instrument (Äkta Explorer, GE). Fractions of the peak corresponding to NABBs were combined and, if necessary, concentrated using an Amicon Ultra 30-kDa cutoff centrifugal filter device (Millipore). For the 2D7 immunoprecipitation experiment, CCR5-NABBs were mixed with Dynabeads Protein G (Invitrogen) +/- 2D7 mAb (BD Biosciences) according to manufacturer's protocol. After overnight incubation at 4°C, the supernatant fractions were subjected to 1D4 immunoblot analysis as described below.
Immunoblot analysis
Samples were resolved by SDS-PAGE (NuPage Novex 4-12% Bis-Tris Gel) and transferred onto Immobilon, a polyvinylidene difluoride (PVDF) membrane (Millipore) according to manufacturer's protocol. For CCR5 detection, membranes were probed with primary antibodies against the engineered 1D4 epitope (National Cell Culture Center; 1:5,000) followed by a horseradish peroxidase (HRP) – conjugated anti-mouse secondary antibody (Amersham Biosciences GE; 1:10,000). For zap1 detection, α-His6 tag monoclonal antibody was used (Sigma; 1:5,000) followed by HRP-α-mouse. Signals were visualized by enhanced chemiluminescence treatment (Amersham Bioscences GE) and exposed to HyBlot CL autoradiography film (Denville Scientific, Inc.).
Fluorescein-maraviroc binding to solubilized CCR5
Solubilized CCR5 lysate from approximately one-fifth of a plate was incubated with 100 nM fluorescein-labeled maraviroc analogue (FL-maraviroc; generously provided by Martin Teintze, Montana State University) and 12.5 uL of 1D4-sepharose resin (2 mg of 1D4 mAb per 1 mL of packed resin) for 16 h at 4 °C in the presence and absence of 1 μM unlabeled maraviroc (Toronto Research Chemicals). The beads were washed three times with Buffer N and samples were eluted by incubating the beads with 1% SDS for 1 h at room temperature and shaking. Eluted samples were transferred to a black 96-well round bottom polypropylene microtiter plate (Nunc/Thermo Fisher Scientific). A serial dilution of FL-maraviroc in 1% SDS was also included for a standard curve. The plate was read on a Cytofluor II Fluorescence Multi-well Plate Reader (Perbio Science) with an excitation wavelength of 485 nm and emission wavelength of 530 nm. A linear fit of the standard curve was used to calculate the concentration of FL-maraviroc bound to the CCR5 lysate. The difference between the values +/- excess unlabeled maraviroc was taken to calculate CCR5 concentration with a correction for nonspecific binding of FL-maraviroc.
Biotinylation of IgG
A 20 mM sodium-m-periodate solution was made in 100 mM sodium acetate, pH 5.5 and mixed with an equal volume of 2 mg/mL 1D4 mAb in Buffer C. The mixture was covered in aluminum foil to protect it from light and incubated for 30 min at 4°C. The mixture was then dialyzed against Buffer C for 4 h to remove excess periodate. Biocytin-hydrazide (Pierce) was prepared as a 50 mM stock in DMSO and diluted to a final concentration of 2.9 mM in the 1D4 solution. The coupling reaction was allowed to proceed for 3.5 h at room temperature and unreacted material was removed by dialysis against Buffer C. To improve the stability of the conjugate, 1D4-biotin was reduced with sodium cyanoborohydride in the following manner. A 5 M stock of NaBH3(CN) was prepared in 1 M KOH. To 200 μL of 1D4-biotin was added 1 mL Buffer C and 12 μL 5 M NaBH3(CN). The reaction proceeded overnight at 4°C while protected from light and was then dialyzed against Buffer C. The biotin/1D4 ratio was determined to be 0.7 using the Pierce Biotin Quantitation kit according to manufacturer's protocol. 1D4-biotin was stored at 4°C with 0.02% NaN3.
Solid-phase labeling of IgG with europium cryptate
Ni-NTA magnetic agarose beads (QIAGEN; 200 μL; 5% slurry) were washed with 1 mL Buffer P. 2D7 mAb (100 μg; 1.3 nmol) was bound to beads by incubating for 30 min at room temperature with shaking. In all solid-phase reactions, 0.0037% (w/v) DM was added to facilitate mixing. The beads were spun down and the tube was placed on a magnetic rack to ease removal of the supernatant fraction. The antibody was activated by adding 2 μL of 3.75 mM sulfo-SMCC (Pierce; 7.5 nmol; 5.6 eq) and shaking for 60 min at room temperature. In a separate reaction, 3.68 μL of 0.68 mM europium trisbipyridine di-amino cryptate (EuK; Cisbio; 2.5 nmol), 0.67 μL of 0.5M sodium borate pH 8.2, 1.32 μL of 1M NaF, and 0.67 μL of 7.58 mM SPDP (Pierce; 5.0 nmol; 2 eq) were mixed for 60 min at room temperature with shaking. The EuK-SPDP reaction was stopped by addition of 0.67 μL of 7.58 mM Tris. SPDP was reduced by adding 0.67 μL of 8.27 mM TCEP and mixing for 15 min. The activated EuK was added to activated IgG and mixed overnight at 4°C. The conjugation reaction was stopped with 1 μL of 80 mM N-acetylcysteine. The beads were washed twice with 1-mL Buffer P. IgG was eluted with two 50 μL volumes Buffer E2 for 30 min each. Combined elutions were supplemented with 0.5 mg/mL bovine serum albumin (BSA) and run on a Superdex 200 10/300 GL gel filtration column in Buffer G. Absorbance was monitored at 280 nm and 305 nm for protein and EuK content, respectively. 2D7-EuK was stored at 4°C with 0.01% thiomersal.
HTRF assay to quantify folded CCR5
All HTRF assays were carried out with the appropriate buffer supplemented with 50 mM NaF and 1 mg/mL BSA. Biotinylated 1D4 mAb was mixed with streptavidin-XL665 (SA-XL665; Cisbio) for 15 min on ice at a final concentration of 128 nM each. To this was added an equal volume of 8 nM EuK-labeled 2D7 mAb (2D7-EuK). For each well of the experiment, 20 μL of this mixture was added to a black bottom 384-well microplate (Greiner). Solubilized CCR5 or CCR5 in NABBs was added to a final volume of 40 μL. The final concentrations of labeled assay components were thus 32 nM 1D4-biot, 32 nM SA-XL665, and 2 nM 2D7-EuK. In competition experiments, CCR5 was preincubated with competitors for 30 min on ice prior to addition to fluorescently labeled components in the 384-well plate. To obtain maximum signal, the plate was incubated overnight at 4°C. Dual-channel fluorescence was measured with excitation at 320 nm and emission collection in ten 200-μs windows at 615 nm and 665 nm. Acceptable signal-to-background was achieved with 5000 flashes per well. Emission counts were summed over all windows except the first, and the ratio of F665 nm / F615 nm was used as a normalized measure of sensitized emission. As an assay-to-assay control the signal enhancement ΔF was calculated:
| Eq. 1 |
where the negative control values are obtained from the mixed labeled components in the absence of receptor.
CCR5 thermal stability measurements using HTRF
Detergent-solubilized CCR5 or CCR5-NABBs were diluted to a concentration within the dynamic range of the HTRF assay and 11 μL aliquots were added to PCR tubes. For experiments testing the stabilizing effects of the small molecule antagonists maraviroc, AD101, CMPD 167, and vicriviroc, the receptor was preincubated with 8 μM ligand for 1 h on ice before the next step. The CCR5-containing tubes were transferred to a Multigene Gradient Thermal Cycler (Labnet International, Inc). The thermal cycler was set up to apply a gradient of temperatures across a row of the heating block for 30 min, followed by cooling to 4°C. Each sample was diluted by a factor of two in the appropriate buffer and 20 μL of the heat-treated material was added to each well of a 384-well microplate containing 20 μL the fluorescently labeled components.
Modeling thermal denaturation of CCR5 and CCR5-ligand complexes
Denaturation of unliganded CCR5 in detergent solution and NABBs was modeled as a simple first order process.
| Eq. 2 |
The HTRF signal ΔF is the sum of contributions from folded and unfolded receptor with appropriate scaling factors.
| Eq. 3 |
Because denatured receptor does not bind 2D7-EuK, α2 was set to zero. The signal is then:
| Eq. 4 |
, with
| Eq. 5 |
The curve was fit with parameters α1, A, and Ea; TM was defined as the temperature at which one-half of the receptor is unfolded. The time of incubation is designated as t. The temperature of incubation, T, was specified in Kelvin.
Melting of CCR5-ligand complexes was modeled similarly, but as a two-step sequence with a first order reaction describing the irreversible conversion of the “loose” and “tight” binding states, and a second irreversible denaturation step from the tight state. This model assumes that no unliganded receptor exists in the system, which is likely to be true because of the high affinity of the antagonists tested.
| Eq. 6 |
The HTRF signal in this system is the sum of contributions from these three species.
| Eq. 7 |
Again, the scaling factor for unfolded receptor, α3, was set to zero, making the total signal:
| Eq. 7 |
, with
| Eq. 8 |
and
| Eq. 9 |
The curve was fit with parameters, α1, A1, Ea,1, α2, A2, and Ea,2; TM was defined as the temperature at which one-half of the receptor is unfolded. It is important to note that these models assume that the conversion from the loosely to the tightly bound state and the denaturation are irreversible processes. Moreover, we restrict our interpretation of the model to a single derived parameter, the apparent melting temperature TM.
Results
Solid-phase labeling of IgG with EuK
We first developed a solid-phase strategy to label directly 2D7 mAb with EuK. Magnetic Ni-NTA beads were used to immobilize 2D7 by binding to a histidine-rich region of the Fc stem, permitting efficient wash and elution steps. 2D7-EuK was then prepared in a three-step reaction scheme (Figure 1A). We found that activating 2D7 with SPDP, as previously reported, resulted in significant reduction of IgG disulfide linkages after addition of TCEP (data not shown).(19, 20) To avoid this problem, we inverted the activation steps, reacting 2D7 with sulfo-SMCC and EuK with SPDP. The reduction step was thus carried out in the absence of antibody, resulting in minimal antibody fragmentation as shown by comparing labeled 2D7 to starting material on a Coomassie Blue-stained SDS-PAGE gel (Figure 1B, right).
FIGURE 1.

(A) Scheme depicting strategy to synthesize EuK-labeled 2D7 mAb (9). EuK (1) is activated by addition of a sulfhydryl by reaction with SPDP (2) and reduction by TCEP (top). Separately, 2D7 mAb (6) is immobilized on Ni-NTA resin and reacted with sulfo-SMCC (7) to generate a maleimide derivative (8). A crystal structure of IgG (PDB ID: 1IGY) is shown as a model. The two activated reagents are combined and labeled 2D7 is eluted from the resin with imidazole. (B) Left: Size-exclusion chromatography with monitoring of absorbance at 280 nm (blue; protein) and 305 nm (red; EuK) to determine yield and labeling ratio. Right: Coomassie blue-stained non-reducing SDS polyacrylamide gel electrophoresis showing a single band of the intact 2D7-EuK after labeling. Note the impurities of the initial 2D7 sample have been largely removed by the procedure.
After eluting labeled 2D7 from the magnetic beads, the sample was purified by gel filtration chromatography (Figure 1B, left). Absorbance was monitored at 280 nm (protein) and 305 nm (EuK). Using the integrated absorbance peaks and known molar extinction coefficients,(21) the labeling ratio was estimated to be 1.3 EuK/IgG. The main peak in the chromatogram is monomeric 2D7-EuK. Dimeric 2D7 elutes slightly earlier, and the right shoulder of the peak is likely to represent small quantities of antibody fragments. EuK fluorescence at 615 nm was measured for each column fraction (data not shown), the profile was fit to three Gaussian peaks, and fractions determine to most likely contain monomer were pooled. Recovery of 2D7-EuK was >30 μg from the 100 μg of 2D7 starting material, with monomeric IgG constituting an estimated 90% of the protein content. Including bovine serum albumin (BSA) as a carrier protein during the size exclusion chromatography purification step contributed significantly to the high yield of labeled antibody.
Characterization of HTRF assay with solubilized CCR5
CCR5 was heterologously expressed in transiently transfected HEK293 cells and solubilized in a detergent-containing buffer previously shown to preserve proper CCR5 folding.(22) We estimated the total receptor concentration by incubating CCR5 with a saturating amount of a fluorescein-derivitized maraviroc analogue (FL-maraviroc). CCR5 was immunoprecipitated on 1D4-sepharose beads, and the amount of receptor was calculated by comparing the fluorescent signal in the elution to a FL-maraviroc standard curve (Supporting Figure S1). Using this method, the concentration of CCR5 in cell lysate was estimated to be 13.9 ± 0.2 nM. This concentration represents a lower bound, though we assume that all folded receptor can bind the high-affinity ligand.
The HTRF assay relies on simultaneous binding of 2D7-EuK and 1D4-biotin to CCR5, and 1D4-biotin binding to SA-XL665 (Figure 2A). Preliminary optimization experiments demonstrated that maximal signal was achieved with an excess of 1D4-biotin and SA-XL665 compared to 2D7-EuK. For signal reproducibility, the assay mixtures were incubated overnight at 4°C before reading fluorescence. The assay was characterized using negative controls without the labeled antibodies (Figure 2B). 1D4-biotin/SA-XL665 in the absence of fluorescent donor results in minimal counts at 615 nm and some emission at 665 nm. 2D7-EuK in the absence of the acceptor complex gives high signal at 615 nm and lower emission at 665 nm than XL665. The standard negative control was a mixture of 1D4-biotin, SA-XL665, and 2D7-EuK. Under these conditions, the 615 nm signal was roughly the same as 2D7-EuK alone, and the 665 nm signal was only slightly greater than that from 1D4-biotin/SA-XL665 only. This result suggests minimal sensitized emission of XL665 in the absence of CCR5. CCR5-specific signal manifests as a decrease in 615 nm fluorescence and increase in 665 nm fluorescence as the FRET donor and acceptor are brought into proximity by binding receptor.
FIGURE 2.

(A) HTRF sandwich immunoassay schematic. A hypothetical model of CCR5 based on the crystal structure of rhodopsin (PDB ID: 1U19) is shown. 2D7-EuK recognizes a conformation-sensitive split epitope on the extracellular side of CCR5. Biotinylated 1D4 (1D4-biot) binds an engineered nine-residue C-terminal epitope (red) and is linked to streptavidin-conjugated XL665. FRET is observed between EuK and XL665 when 2D7 binds properly folded CCR5. The Förster radius for this donor-acceptor pair is approximately 95 Å. (B) Assay controls with fluorescence counts at 615 nm (blue) and 665 nm (red) after excitation at 320 nm. CCR5-specific signal is seen as a signal increase at 665 nm and decrease at 615 nm. (C) A serial dilution of CCR5 shows the dynamic range of the assay. The signal saturates at ∼200% enhancement over background. ΔF is defined as:
(D-F) Competition experiments with the 1D5 nonapeptide (D), 1D4 mAb (E), and 2D7 mAb (F) demonstrate signal specificity. The apparent IC50 values are 130 nM, 2.7 nM, and 0.79 nM for the three competitors, respectively.
Measurements with a serial dilution of solubilized lysate containing CCR5 revealed the dynamic range of the assay (Figure 2C). A characteristic linear range precedes signal saturation near 200% over background at high concentrations. The measurement errors are approximately 2% of the background signal as determined by standard error propagation. Using the FL-maraviroc binding experiment for calibration, an estimated 0.02-1.0 nM CCR5 can be effectively quantified. In a volume of 40 μL, this corresponds to 0.8-40 fmol of receptor. Signal saturation near 2 nM, the concentration of 2D7-EuK, suggests that this antibody has a high affinity compared to the concentrations involved. In subsequent experiments, receptor concentrations near the middle of the dynamic range were used to minimize measurement errors.
The specificity of the HTRF signal for CCR5 epitopes was demonstrated in competition experiments. CCR5 was preincubated with a serial dilution of TETSQVAPA nonapeptide (“1D5”), 1D4, and 2D7 (Figure 2D-F). 1D5-nonapeptide corresponds to the engineered C-terminal epitope on the receptor. These competition data were fit to a sigmoidal curve. Signal enhancement ΔF was normalized to the endpoints at 0% inhibition and 100% inhibition. The calculated IC50 values for 1D5, 1D4, and 2D7 were 130 nM, 2.7 nM, and 0.79 nM, respectively. The IC50 values of 1D4 and 2D7 are lower than the concentrations of 1D4-biotin and 2D7-EuK in the assay, suggesting that the labeled antibodies may have slightly reduced binding capacity and/or affinity. Having shown the specificity of the signal, we next focused on applying the assay technology to the problem of CCR5 reconstitution.
Optimizing microincorporation of CCR5 into NABBs using HTRF
We have previously reported the incorporation of rod outer segment bovine rhodopsin into NABB particles.(8) Unlike rhodopsin, CCR5 cannot be purified in large quantities from natural sources, so we employed a microscale approach to optimize a procedure to reconstitute functional expressed CCR5 into NABBs. The general procedure is outlined in Supporting Figure S2. Recombinant expressed CCR5 was immunopurified from solubilized lysate using 1D4-sepharose beads. After several washing steps, receptor was eluted by addition of 1D5-nonapeptide. Roughly one-half of the receptor is lost in this purification step due to incomplete elution from the beads. (Elution is also the theoretical limiting step in rhodopsin purification using 1D4-sepharose beads, unpublished observation, T.P.S..) The NABB assembly mixture was formed by mixing purified zebrafish Apo-A1 (zap1) and lipids at a molar ratio of 1:75, which we showed yields 10-12 nm diameter discs in our earlier rhodopsin-NABBs study.(8) Purified CCR5 elution was added to this mixture, and after incubation on ice, applied to a detergent-removal resin. NABBs were eluted under gravity flow by addition of detergent-free buffer and fractions were collected. Protein-containing fractions were determined by measurement of 280 nm absorbance and pooled.
The combined elutions were run on a gel filtration column for characterization and purification (Figure 3A). Co-elution of protein and lipids was monitored by measuring absorption at 280 nm and 570 nm, which detected rhodamine-DOPE labeled POPC. Immunoblots showed the relative content of CCR5 (1D4 mAb detection) and zap1 (α-His6 mAb detection) in each fraction. The majority of the CCR5-NABBs elute at 15.6 mL in the shoulder of the first peak, which is centered at ∼16.1 mL. The second peak, at ∼17 mL, contains mainly empty NABBs. Note that the ratio of CCR5 to NABBs is approximately 1:100. In our previous study of rhodopsin-NABBs reconstituted at a similar ratio, we also observed that the receptor-containing NABBs elute about 0.5 mL earlier than the majority of empty NABBs. This strategy reduces the likelihood of incorporating more than one receptor per NABB and guarantees a relatively homogeneous CCR5-NABB preparation. Free zap1, which elutes at ∼18.6 mL (Figure 3A, inset), likely constitutes the right peak shoulder that is rather lipid-poor as judged by the 570 nm absorbance. The CCR5-NABBs were mixed with Protein G beads with (left) and without (right) 2D7 and the supernatant fractions were subjected to 1D4 immunoblot analysis. The near-quantitative immunoprecipitation of CCR5-NABBs with the conformationally sensitive 2D7 antibody suggests that most of the receptor is properly folded.
FIGURE 3.
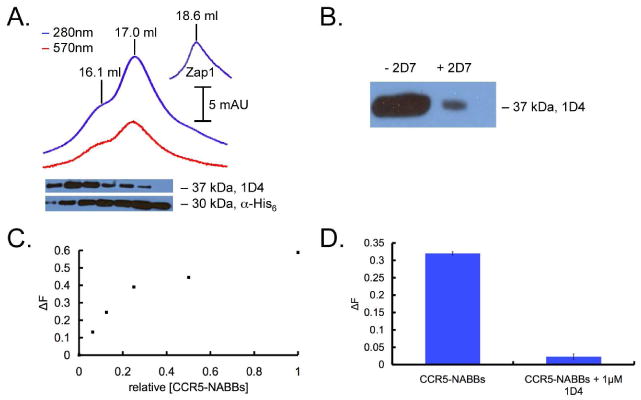
(A) Chromatogram showing co-elution of protein (blue; 280 nm) and fluorescent lipids (red; 570 nm). The 1D4 immunoblot below shows that CCR5-NABBs elute in the peak centered at 15.6 mL, exhibiting larger hydrodynamic radius than the majority of the NABBs at 16.1 and 17.0 mL. The anti-His6 blot detects His-tagged zap1 belt protein. Free zap1 elutes at 18.6 mL (inset). (B) CCR5-NABBs were incubated with Protein G beads and 2D7, and the supernatant fraction was probed with a 1D4 immunoblot. The majority of CCR5 in NABBs is immunoprecipitated by 2D7, indicating properly folded receptor. (C) HTRF signal from a serial dilution of CCR5-NABBs, demonstrating the ability to quantify properly folded CCR5 in NABBs. (D) The HTRF signal is efficiently competed with 1 μM 1D4, showing signal specificity.
The HTRF assay was used in conjunction with gel filtration chromatography and immunoblot analysis to facilitate optimization of preparation conditions. The HTRF assay complements immunoblots because it reports on receptor folding and not just the total amount of receptor protein. Moreover, the HTRF assay is much more quantitative and reproducible. 2D7-EuK and 1D4-biotin bind to CCR5 in NABBs, resulting in a dynamic range for the HTRF assay as with detergent-solubilized receptor (Figure 3C). Competition with 1 μM 1D4 ablates the signal, demonstrating signal specificity as in detergent solution (Figure 3D). Assay conditions were identical to those in detergent experiments, except that phosphate buffered saline (PBS) was used as the starting buffer.
Using the HTRF signal as a guide, we screened a number of conditions for optimal formation of CCR5-NABBs containing correctly folded receptor. Variables included changing the detergents used, the zap1:lipid ratio, the lipid composition, and the detergent removal resin elution buffer. We found that several factors were particularly important for optimal CCR5 recovery. CCR5 elution from 1D4-sepharose beads in Buffer N and inclusion of 0.3% (w/v) n-dodecyl-β-D-maltopyranoside (DM) in the final NABB mixture enhanced stability. Notably, the detergent removal resin adequately extracted DM despite its low CMC. Including cholesterol at a cholesterol/POPC ratio of 7.5:92.5 (mol/mol) also improved CCR5 incorporation. Finally, equilibrating and eluting from the detergent removal resin with Buffer S, 20 mM Tris-HCl pH 7.0, 0.1 M (NH4)2SO4, and 10% (v/v) glycerol further increased recovery. Comparing the HTRF signal from CCR5-NABBs to that of the solubilized CCR5 starting material, the overall yield of correctly folded receptor in the optimized procedure was >15%. This estimate likely represents a lower bound because the NABB might prevent head-to-tail donor-acceptor interactions that theoretically may cause a reduction of donor-acceptor distance and increase in HTRF signal intensity in detergent. Indeed, it appears that the HTRF signal at a saturating concentration of CCR5-NABBs is lower than the maximum signal in detergent solution (Figures 2C, 3C).
High-throughput thermal stability measurements of CCR5
Thermal stability is a crucial indicator of receptor tractability outside of its native membrane environment. We adapted the HTRF assay to measure CCR5 thermal stability under a variety of conditions. In short, a range of temperatures was applied to CCR5 aliquots for 30 min, followed by cooling the samples to 4°C and adding them to the microplate containing labeled 1D4 and 2D7 FRET pair. Plotting the ratio of the fluorescence signal as a function of temperature generates thermal denaturation or melting curves (Figure 4). Receptor denaturation and subsequent loss of 2D7-EuK binding presumably causes this loss of signal. It is important to note that these experiments probe the kinetics of receptor denaturation rather than the equilibrium distribution. The denaturation was modeled as an irreversible first order process, and the data are fit using nonlinear regression analysis. The melting temperature (TM) is defined as the point at which a calculated 50% of the receptor is unfolded (see Materials and Methods section for a description of models and curve fits). Because the process is kinetically controlled, the apparent TM is dependent on the time of heat treatment. For further discussion of this subject, please see the Supporting Information.
FIGURE 4.

The HTRF assay was applied to make high-throughput thermal stability measurements with femtomole quantities of CCR5. A range of temperatures was applied to detergent-solubilized CCR5 or CCR5-NABBs before adding to labeled HTRF components in a 384-well plate. (A) Melting curves of unliganded CCR5 (black) and CCR5 preincubated with the small molecule antagonist maraviroc (red). Maraviroc shifts the TM of detergent-solubilized CCR5 from 47.1°C to 66.0°C (rightward-pointing arrow) and appears to display a two-step binding profile. The first ligand-receptor state reduces the accessibility of the 2D7 epitope on the EC2 loop compared with unliganded receptor (downward-pointing arrow). The second ligand-receptor state results in a higher HTRF signal (upward-pointing arrow). (B) Thermal denaturation of CCR5-ligand complexes. CCR5 was preincubated with maraviroc (red), AD101 (green), vicriviroc (blue), and CMPD 167 (cyan). All of these antagonists appear to have two binding states. The calculated TM for maraviroc, AD101, vicriviroc, and CMPD 167 is 66.0°C, 59.9°C, 59.5°C, and 62.7°C, respectively. The unliganded receptor, which melts at a lower temperature, is shown as a black dashed line for reference. (C) Molecular structures of the CCR5 antagonists tested. (D) Melting curve of CCR5-NABBs. The assembly denatures at 54.5°C. CCR5-NABBs melt over a much broader range than CCR5 in detergent solution, suggesting some sample heterogeneity or a distinct denaturation pathway.
The thermal stability of CCR5 was measured in the presence and absence of high-affinity small molecule receptor ligands. CCR5 solubilized in Buffer N has a TM of 47.1 ± 0.6°C (Figure 4A, black). Preincubation of CCR5 with 8 μM maraviroc, AD101, CMPD 167, or vicriviroc before applying heat conferred a significant increase in thermal stability (Figure 4A, B). Maraviroc stabilizes CCR5 to the greatest extent, shifting the TM to 66.0 ± 0.3°C (Figure 4A, red). The TM for AD101, CMPD 167, and vicriviroc is 59.9 ± 1.0°C, 62.7 ± 2.3°C, and 59.5 ± 1.4°C, respectively (Figure 4B). Another interesting effect of these ligands is reduction of 2D7-EuK binding. Preincubation with all ligands results in a decrease in HTRF signal, but to varying degrees. This indicates that antagonist binding reduces the accessibility of the 2D7 epitope on the EC2 loop, in agreement with earlier work.(23) In addition, each of these melting curves display a rise in signal near 50°C before complete loss due to denaturation. This profile suggests that at least two distinct binding states might be involved in the antagonist-CCR5 interaction, with the “slower” being more amenable to 2D7 binding. These ligand-CCR5 denaturation curves were modeled as a sequence of two irreversible first order processes. It is interesting that the conversion of the two inhibitor-bound conformations occurs around the temperature at which the empty receptor denatures. The molecular structures of the antagonists tested are shown in Figure 4C.
Similar measurements were carried out on CCR5-NABBs (Figure 4D). There are several notable features of the CCR5-NABB melting curve. The TM shifts to 54.5 ± 3.4°C, suggesting that incorporation into the native membrane-like environment of NABBs improves CCR5 thermal stability. Interestingly, the dramatic shift in melting temperature after preincubation with maraviroc is not observed for CCR5-NABBs (data not shown). In addition, the temperature range in which the signal decreases is significantly broadened compared with solubilized CCR5, suggesting some sample heterogeneity or a lower activation energy associated with denaturation.
Discussion
CCR5 is an important Class A (rhodopsin-like) GPCR because of its role as a primary HIV-1 coreceptor.(24, 25) Its natural ligands, including MIP-1α, MIP-1β, and RANTES, are potent inhibitors of HIV infection, and a number of small molecule antagonists that block HIV entry have been reported.(26, 27) Detailed molecular studies of purified CCR5 are important to understand its biological function, role in HIV entry, and mode of binding to pharmacological agents, but its high lability in solution makes it a particularly challenging target. Some progress has been made, as detergent conditions that maintain proper CCR5 folding for several hours have been identified.(22, 28) These reports use binding of the well-characterized conformationally sensitive 2D7 mAb as a reporter for receptor folding and integrity.(29-31)
GPCRs are notoriously difficult to stabilize in a purified system. This challenge amplifies when the receptors cannot be expressed at high levels, as with CCR5. We estimate that 1 μg of CCR5 can be obtained from one 10-cm plate of transfected HEK cells, so an extremely sensitive quantitative assay is desirable. Ideally, this assay should also be high-throughput capable so that a large number of potentially stabilizing conditions can be screened in parallel. We decided on a strategy of HTRF, with fluorescently labeled antibodies forming an immuno-sandwich on distinct epitopes of CCR5. The key reporter is binding of EuK-labeled 2D7 mAb, which binds to the conformationally sensitive split epitope QKEGL-TL on the EC2 loop of the receptor.(32, 33)
HTRF occurs when a EuK fluorescent donor is brought in proximity to a modified allophycocyanin (XL665) acceptor.(34, 35) The dual-wavelength detection of donor (615 nm) and acceptor (665 nm) fluorescence after EuK excitation at 320 nm results in a specific signal with excellent well-to-well reproducibility and internal correction for inner filter effects.(34, 35) The signal is time-resolved due to the exceptionally long flourescence lifetime of EuK, reducing the contribution from autofluorescence. The EuK-XL665 FRET pair also has an unusually large Förster radius, approximately 95 Å,(34, 36) which permits resonance energy transfer from the extracellular to intracellular side of the receptor. The 2D7 mAb was directly labeled with EuK to maximize sensitivity. We developed an efficient solid-phase microlabeling strategy for this conjugation, permitting reaction with much smaller antibody and EuK label quantities than previously reported.(19, 20, 37) This procedure resulted in high yields and is generalizable to any IgG molecule or His-tagged protein. This method is particularly advantageous when the labels or antibodies are expensive and a small reaction scale is desired. Biotinylated 1D4 mAb, which recognizes the engineered C-terminal epitope TETSQVAPA, binds streptavidin-XL665 (SA-XL665) to complete the immuno-sandwich. This assay is capable of detecting ∼1 fmol of correctly folded CCR5 in a single 384-microtiter plate well.
The assay is also more informative than immunoblots, as it reports on the quality of receptor as well as the quantity. Unlike surface plasmon resonance (SPR), conditions can be screened without the need for a common binding buffer and the time-consuming microchip regeneration step. If automated, the HTRF approach described herein combines high-throughput capability and sensitivity at a level exceeding even the best available SPR equipment, which is also more expensive.(38)
The assay capabilities enabled optimization of CCR5 incorporation into NABBs, and the best procedure results in >15% overall yield of folded receptor with the majority of receptor able to bind 2D7. Because the expression, purification, and NABB formation steps are scalable, adequate quantities of CCR5-NABBs for structural and biochemical studies should be attainable, especially using inducible suspension-adapted stable cell lines or Sf9 insect cells. The defined lipid bilayer of NABBs could be a particularly attractive platform for single molecule studies of receptor activation, possibly in combination with microfluidics approaches, because GPCR aggregation phenomena endemic to detergents are of less concern.(39)
Most auspiciously, the HTRF assay permits high-throughput measurements of CCR5 thermal stability with small amounts of material. These experiments are crucial to determine conditions suitable for structural studies. Because CCR5 retained proper folding in the optimal detergent-lipid mixture even after heating at >40 °C for 30 min, solubilized receptor might be more stable than previously thought. Preincubation with small molecule CCR5 antagonists resulted in a marked TM shift to 59-66 °C.
The antagonists maraviroc,(40) AD101,(41) CMPD 167,(42) and vicriviroc(43) block CCR5 interaction with the HIV envelope glycoprotein gp 120 and demonstrate high antiviral potency. Maraviroc, in particular, has proven to be a promising adjuvant anti-HIV drug. Key residues in the transmembrane domains that are required for antagonist binding have been identified, but precisely how gp 120 inhibition occurs remains unknown.(44, 45) The melting curves generated (Figure 4) demonstrate that ligand-receptor complexes under optimized conditions could be sufficiently stable for high-temperature NMR or crystallization studies. The profiles also shed some light on the binding dynamics of these antagonists, which appear to consist of two steps. The first state, with a decreased HTRF signal, could be due to slightly decreased 2D7-EuK affinity or an alteration of the antibody binding orientation that results in a longer donor-acceptor distance. The second state, characterized by a notable rise, seems to more closely resemble CCR5 in the absence of antagonist. Interestingly, the four antagonists tested inhibit 2D7-EuK binding to varying extents. The fact that maraviroc has the smallest effect on 2D7 binding might prove useful for combination therapy with CCR5-based vaccines or therapeutic anti-CCR5 antibodies.
Incorporation of CCR5 into NABBs also confers improved thermal stability to CCR5. Surprisingly, the lipid composition of the bilayer or preincubation of CCR5 with maraviroc did not significantly change the melting profile (data not shown). In contrast, rhodopsin in NABBs exhibits the high thermal stability of the inverse agonist-bound state in membranes.(8)
In summary, we report a rapid and highly efficient solid-phase method to label IgG with EuK, and we used the labeled IgG to establish an HTRF method for sensitive and high-throughput quantification of CCR5. The extension of the methodology to other GPCRs is limited only by the availability of suitable antibodies. Such mAbs have already been produced for CXCR4(46) and the 5-hydroxytryptamine (5HT) receptor.(47) The assay enables optimization and characterization of GPCR reconstitution procedures that would otherwise be extremely burdensome or expensive. Thermal stability measurements of CCR5 and other GPCRs under a variety of conditions could greatly facilitate structural studies. We show, for example, that maraviroc, AD101, CMPD 167, and vicriviroc greatly enhance thermal stability of CCR5 in detergent solution. In addition, these experiments provide some insight into the dynamics of antagonist binding, which could be therapeutically valuable for a highly targeted receptor like CCR5.
Supplementary Material
Acknowledgments
We wish to thank Pallavi Sachdev, Ana Vitória Botelho, Manija A. Kazmi and members of the Sakmar Laboratory. We also acknowledge the generous support and encouragement of several private foundations and individuals.
Funding information: This work was supported by the Tri-Institutional Program in Chemical Biology and NIH Grant EY12049.
Abbreviations
- GPCR
G protein-coupled receptor
- HTRF
homogeneous time-resolved fluorescence
- CCR5
CC-chemokine receptor 5
- NABB
nanoscale apolipoprotein bound bilayer
- CMC
critical micellar concentration
- EuK
europium cryptate
- mAb
monoclonal antibody
- CHS
cholesteryl hemisuccinate
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- DOPS
1,2-dioleoyl-sn-glycero-3-phospho-L-serine
- DM
n-dodecyl β-D-maltoside
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- LRB
lissamine rhodadmine B
- zap1
zebrafish Apo-AI
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- SPR
surface plasmon resonance
Footnotes
Supporting Information Available
The FL-maraviroc standard curve used to calibrate the HTRF assay and a flowchart showing the procedure to incorporate CCR5 into NABBs on a microscale. Additional discussion of the denaturation model, including dependence of apparent TM on the incubation time. This material may be accessed free of charge online at http://pubs.acs.org.
References
- 1.Deupi X, Kobilka B. Mechanisms and Pathways of Heterotrimeric G Protein Signaling. Elsevier Academic Press Inc; San Diego: 2007. Activation of G Protein Coupled Receptors; pp. 137–166. [Google Scholar]
- 2.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 3.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bartfai T, Benovic JL, Bockaert J, Bond RA, Bouvier M, Christopoulos A, Civelli O, Devi LA, George SR, Inui A, Kobilka B, Leurs R, Neubig R, Pin JP, Quirion R, Roques BP, Sakmar TP, Seifert R, Stenkamp RE, Strange PG. The state of GPCR research in 2004. Nat Rev Drug Discov. 2004;3:574–626. doi: 10.1038/nrd1458. [DOI] [PubMed] [Google Scholar]
- 6.Ernst OP, Gramse V, Kolbe M, Hofmann KP, Heck M. Monomeric G protein-coupled receptor rhodopsin in solution activates its G protein transducin at the diffusion limit. Proc Natl Acad Sci U S A. 2007;104:10859–10864. doi: 10.1073/pnas.0701967104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayburt TH, Grinkova YV, Sligar SG. Self-assembly of discoidal phospholipid bilayer nanoparticles with membrane scaffold proteins. Nano Lett. 2002;2:853–856. [Google Scholar]
- 8.Banerjee S, Huber T, Sakmar TP. Rapid incorporation of functional rhodopsin into nanoscale apolipoprotein bound bilayer (NABB) particles. J Mol Biol. 2008;377:1067–1081. doi: 10.1016/j.jmb.2008.01.066. [DOI] [PubMed] [Google Scholar]
- 9.Whorton MR, Bokoch MP, Rasmussen SGF, Huang B, Zare RN, Kobilka B, Sunahara RK. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc Natl Acad Sci U S A. 2007;104:7682–7687. doi: 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshiura C, Kofuku Y, Ueda T, Mase Y, Yokogawa M, Osawa M, Terashima Y, Matsushima K, Shimada I. NMR analyses of the interaction between CCR5 and its ligand using functional reconstitution of CCR5 in lipid bilayers. J Am Chem Soc. 132:6768–6777. doi: 10.1021/ja100830f. [DOI] [PubMed] [Google Scholar]
- 11.Maurel D, Kniazeff J, Mathis G, Trinquet E, Pin JP, Ansanay H. Cell surface detection of membrane protein interaction with homogeneous time-resolved fluorescence resonance energy transfer technology. Anal Biochem. 2004;329:253–262. doi: 10.1016/j.ab.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Albizu L, Balestre MN, Breton C, Pin JP, Manning M, Mouillac B, Barberis C, Durroux T. Probing the existence of g protein-coupled receptor dimers by positive and negative ligand-dependent cooperative binding. Mol Pharmacol. 2006;70:1783–1791. doi: 10.1124/mol.106.025684. [DOI] [PubMed] [Google Scholar]
- 13.Albizu L, Teppaz G, Seyer R, Bazin H, Ansanay H, Manning M, Mouillac B, Durroux T. Toward efficient drug screening by homogeneous assays based on the development of new fluorescent vasopressin and oxytocin receptor ligands. J Med Chem. 2007;50:4976–4985. doi: 10.1021/jm061404q. [DOI] [PubMed] [Google Scholar]
- 14.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the beta 1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci U S A. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alexandrov AI, Mileni M, Chien EYT, Hanson MA, Stevens RC. Microscale fluorescent thermal stability assay for membrane proteins. Structure. 2008;16:351–359. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Yan ECY, Kazmi MA, Ganim Z, Hou JM, Pan DH, Chang BSW, Sakmar TP, Mathies RA. Retinal counterion switch in the photoactivation of the G protein-coupled receptor rhodopsin. Proc Natl Acad Sci U S A. 2003;100:9262–9267. doi: 10.1073/pnas.1531970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oprian DD, Molday RS, Kaufman RJ, Khorana HG. Expression of a synthetic bovine rhodopsin gene in monkey kidney cells. Proc Natl Acad Sci U S A. 1987;84:8874–8878. doi: 10.1073/pnas.84.24.8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goding JW. Monoclonal antibodies: Principles and practice: Production and application of monoclonal antibodies in cell biology biochemistry and immunology. Third. Academic Press Inc.; Academic Press Ltd.; 1996. [Google Scholar]
- 19.Cummings RT, McGovern HM, Zheng S, Park YW, Hermes JD. Use of a phosphotyrosine-antibody pair as a general detection method in homogeneous time-resolved fluorescence: Application to human immunodeficiency viral protease. Anal Biochem. 1999;269:79–93. doi: 10.1006/abio.1999.4021. [DOI] [PubMed] [Google Scholar]
- 20.Kane SA, Fleener CA, Zhang YS, Davis LJ, Musselman AL, Huang PS. Development of a binding assay for p53/HDM2 by using homogeneous time-resolved fluorescence. Anal Biochem. 2000;278:29–38. doi: 10.1006/abio.1999.4413. [DOI] [PubMed] [Google Scholar]
- 21.Lopez-Crapez E, Malinge JM, Gatchitch F, Casano L, Langlois T, Pugniere M, Roquet F, Mathis G, Bazin H. A homogeneous resonance energy transfer-based assay to monitor MutS/DNA interactions. Anal Biochem. 2008;383:301–306. doi: 10.1016/j.ab.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 22.Navratilova I, Sodroski J, Myszka DG. Solubilization, stabilization, and purification of chemokine receptors using biosensor technology. Anal Biochem. 2005;339:271–281. doi: 10.1016/j.ab.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 23.Tsamis F, Gavrilov S, Kajumo F, Seibert C, Kuhmann S, Ketas T, Trkola A, Palani A, Clader JW, Tagat JR, McCombie S, Baroudy B, Moore JP, Sakmar TP, Dragic T. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J Virol. 2003;77:5201–5208. doi: 10.1128/JVI.77.9.5201-5208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen SJ, Crown SE, Handel TM. Chemokine: Receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- 25.Lusso P. HIV and the chemokine system: 10 years later. Embo J. 2006;25:447–456. doi: 10.1038/sj.emboj.7600947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhmann SE, Hartley O. Targeting chemokine receptors in HIV: A status report. Annu Rev Pharmacol Toxicol. 2008;48:425–461. doi: 10.1146/annurev.pharmtox.48.113006.094847. [DOI] [PubMed] [Google Scholar]
- 27.Horuk R. OPINION Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8:23–33. doi: 10.1038/nrd2734. [DOI] [PubMed] [Google Scholar]
- 28.Navratilova I, Dioszegi M, Myszka DG. Analyzing ligand and small molecule binding activity of solubilized GPCRs using biosensor technology. Anal Biochem. 2006;355:132–139. doi: 10.1016/j.ab.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 29.Wu LJ, LaRosa G, Kassam N, Gordon CJ, Heath H, Ruffing N, Chen H, Humblias J, Samson M, Parmentier M, Moore JP, Mackay CR. Interaction of chemokine receptor CCR5 with its ligands: Multiple domains for HIV-1 gp120 binding and a single domain for chemokine binding. J Exp Med. 1997;186:1373–1381. doi: 10.1084/jem.186.8.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farzan M, Mirzabekov T, Kolchinsky P, Wyatt R, Cayabyab M, Gerard NP, Gerard C, Sodroski J, Choe H. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell. 1999;96:667–676. doi: 10.1016/s0092-8674(00)80577-2. [DOI] [PubMed] [Google Scholar]
- 31.Mirzabekov T, Bannert N, Farzan M, Hofmann W, Kolchinsky P, Wu LJ, Wyatt R, Sodroski J. Enhanced expression, native purification, and characterization of CCR5, a principal HIV-1 coreceptor. J Biol Chem. 1999;274:28745–28750. doi: 10.1074/jbc.274.40.28745. [DOI] [PubMed] [Google Scholar]
- 32.Lee B, Sharron M, Blanpain C, Doranz BJ, Vakili J, Setoh P, Berg E, Liu G, Guy HR, Durell SR, Parmentier M, Chang CN, Price K, Tsang M, Doms RW. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in chemokine and coreceptor function. J Biol Chem. 1999;274:9617–9626. doi: 10.1074/jbc.274.14.9617. [DOI] [PubMed] [Google Scholar]
- 33.Khurana S, Kennedy M, King LR, Golding H. Identification of a linear peptide recognized by monoclonal antibody 2D7 capable of generating CCR5-Specific antibodies with human immunodeficiency virus-neutralizing activity. J Virol. 2005;79:6791–6800. doi: 10.1128/JVI.79.11.6791-6800.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mathis G. Probing molecular interactions with homogeneous techniques based on rare earth cryptates and fluorescence energy transfer. Clin Chem. 1995;41:1391–1397. [PubMed] [Google Scholar]
- 35.Trinquet E, Maurin F, Preaudat M, Mathis G. Allophycocyanin 1 as a near-infrared fluorescent tracer: Isolation, characterization, chemical modification, and use in a homogeneous fluorescence resonance energy transfer system. Anal Biochem. 2001;296:232–244. doi: 10.1006/abio.2001.5298. [DOI] [PubMed] [Google Scholar]
- 36.Mathis G. Rare earth cryptates and homogeneous fluoroimmunoassays with human sera. Clin Chem. 1993;39:1953–1959. [PubMed] [Google Scholar]
- 37.Lopez E, Chypre C, Alpha B, Mathis G. Europium(III) trisbipyridine cryptate label for time-resolved fluorescence detection of polymerase chain reaction products fixed on a solid support. Clin Chem. 1993;39:196–201. [PubMed] [Google Scholar]
- 38.Rich RL, Miles AR, Gale BK, Myszka DG. Detergent screening of a G-protein-coupled receptor using serial and array biosensor technologies. Anal Biochem. 2009;386:98–104. doi: 10.1016/j.ab.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jastrzebska B, Maeda T, Zhu L, Fotiadis D, Filipek S, Engel A, Stenkamp RE, Palczewski K. Functional characterization of rhodopsin monomers and dimers in detergents. J Biol Chem. 2004;279:54663–54675. doi: 10.1074/jbc.M408691200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–4732. doi: 10.1128/AAC.49.11.4721-4732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trkola A, Kuhmann SE, Strizki JM, Maxwell E, Ketas T, Morgan T, Pugach P, Xu S, Wojcik L, Tagat J, Palani A, Shapiro S, Clader JW, McCombie S, Reyes GR, Baroudy BM, Moore JP. HIV-1 escape from a small molecule, CCR5-specific entry inhibitor does not involve CXCR4 use. Proc Natl Acad Sci U S A. 2002;99:395–400. doi: 10.1073/pnas.012519099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veazey RS, Klasse PJ, Ketas TJ, Reeves JD, Piatak M, Kunstman K, Kuhmann SE, Marx PA, Lifson JD, Dufour J, Mefford M, Pandrea I, Wolinsky SM, Doms RW, DeMartino JA, Siciliano SJ, Lyons K, Springer MS, Moore JP. Use of a small molecule CCR5 inhibitor in macaques to treat simian immunodeficiency virus infection or prevent simian-human immunodeficiency virus infection. J Exp Med. 2003;198:1551–1562. doi: 10.1084/jem.20031266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strizki JM, Tremblay C, Xu S, Wojcik L, Wagner N, Gonsiorek W, Hipkin RW, Chou CC, Pugliese-Sivo C, Xiao YS, Tagat JR, Cox K, Priestley T, Sorota S, Huang W, Hirsch M, Reyes GR, Baroudy BM. Discovery and characterization of vicriviroc (SCH 417690), a CCR5 antagonist with potent activity against human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2005;49:4911–4919. doi: 10.1128/AAC.49.12.4911-4919.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seibert C, Ying W, Gavrilov S, Tsamis F, Kuhmann SE, Palani A, Tagat JR, Clader JW, McCombie SW, Baroudy BM, Smith SO, Dragic T, Moore JP, Sakmar TP. Interaction of small molecule inhibitors of HIV-1 entry with CCR5. Virology. 2006;349:41–54. doi: 10.1016/j.virol.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Kondru R, Zhang J, Ji C, Mirzadegan T, Rotstein D, Sankuratri S, Dioszegi M. Molecular interactions of CCR5 with major classes of small-molecule anti-HIV CCR5 antagonists. Mol Pharmacol. 2008;73:789–800. doi: 10.1124/mol.107.042101. [DOI] [PubMed] [Google Scholar]
- 46.McKnight A, Wilkinson D, Simmons G, Talbot S, Picard L, Ahuja M, Marsh M, Hoxie JA, Clapham PR. Inhibition of human immunodeficiency virus fusion by a monoclonal antibody to a coreceptor (CXCR4) is both cell type and virus strain dependent. J Virol. 1997;71:1692–1696. doi: 10.1128/jvi.71.2.1692-1696.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mancia F, Brenner-Morton S, Siegel R, Assur Z, Sun Y, Schieren I, Mendelsohn M, Axel R, Hendrickson WA. Production and characterization of monoclonal antibodies sensitive to conformation in the 5HT2c serotonin receptor. Proc Natl Acad Sci U S A. 2007;104:4303–4308. doi: 10.1073/pnas.0700301104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.