

Abstract
The gene encoding the Ras-related GTPase RhoB-specific is immediate-early inducible by genotoxic treatments. Regulation of transcriptional activation of rhoB is still unclear. Here we show that cells lacking either p53 or c-Fos are not different from wild-type cells with respect to the level of rhoB induction upon UV irradiation, indicating that these transcription factors are not crucial for stimulation of rhoB mRNA expression. Extracts from UV-irradiated and non-irradiated cells revealed similar DNA-binding activities to a 0.17 kb rhoB promoter fragment harboring the functional element(s) necessary for stimulation of rhoB by UV light. By means of immunoprecipitation we found that an ATF-2-specific antibody co-precipitates the 32P-labeled 0.17 kb rhoB fragment, whereas an anti-AP1 antibody did not. Since no consensus sequence for binding of ATF-2 is present within the rhoB promoter, ATF-2 is likely to be associated with another factor that binds to the minimal promoter. Deletion analysis and site-directed mutagenesis of the 0.17 kb rhoB fragment revealed a CCAAT box to be an essential requirement for stimulation of rhoB by UV light and methyl methanesulfonate. Moreover, immunoprecipitation experiments showed that the CCAAT-binding factor NF-YA is complexed with ATF-2. Overall, the data strongly indicate that transcriptional activation of the rhoB gene by genotoxic stress is regulated via a CCAAT box and that interaction of CCAAT-binding factor and ATF-2 triggers the stress-inducible expression of rhoB.
INTRODUCTION
Exposure of mammalian cells to DNA-damaging agents elicits a variety of cellular responses, including stimulation of gene expression among others. Growth factor receptors such as the EGF receptor (1), cytokine receptors (2) and PI 3-kinase coupled receptors (3) have been suggested as primary targets for UV-stimulated signaling leading to activation of protein kinase cascades involving ERKs, N-terminal c-Jun kinases/stress-activated protein kinases (JNKs/SAPKs) (4) and p38 MAP kinases (4–6). Phosphorylation of transcription factors by these MAP kinases finally results in transcriptional activation of genes (7). Prototoypes of genes that are transcriptionally activated within minutes after exposure encode the proto-oncoproteins c-Fos and c-Jun (8–11), which form part of the heterodimeric AP1-like transcription factors (e.g. c-Jun/c-Fos or c-Jun/ATF-2). Via trans-activation of the corresponding target genes, these transcription factors control the expression of proteins involved in the regulation of cell cycle progression and cell death (7). For example, as compared to wild-type cells, fibroblast cells derived from c-fos–/– mice are hypersensitive to the cytotoxic and apoptotic effects of a large variety of genotoxic agents (12). This shows that c-Fos protects cells from cell killing induced by DNA-damaging treatments.
Another type of ‘immediate-early’ inducible gene that is also transcriptionally activated by DNA-damaging treatments encodes the Ras-related GTPase RhoB (13,14). Transcriptional activation of rhoB by genotoxic stress is of particular interest because RhoB is a GTP-binding protein and is, therefore, capable of modulating the activity of downstream targets in a very fast manner via GTP binding and GTP hydrolysis. As compared to other members of the Rho protein family, the physiological function of RhoB has been poorly investigated. Data available so far point to a potential regulatory function of RhoB in S phase cells (15) and indicate that RhoB participates in the regulation of EGF receptor trafficking (16). Other interesting features of RhoB are that it is essentially required for Ras-mediated transformation (17), is subject to negative autoregulation (18) and represses NF-κB signaling (19). The identification of RhoB as a gene that is rapidly inducible by DNA-damaging treatments further indicates that RhoB is part of the cellular response to genotoxic stress. In line with this hypothesis, overexpression of RhoB renders cells hypersensitive to the apoptosis-inducing effects of alkylating agents, including antineoplastic drugs (20). Notably, the regulation of rhoB induction by genotoxic stress appears to be different from that of other rapidly inducible genes such as c-fos and c-jun. One argument supporting this hypothesis is that the phorbol ester TPA elicits an increase in expression of c-fos and c-jun mRNA but does not affect rhoB mRNA expression (13,14). Furthermore, MAP kinases (i.e. JNKs and p38 kinase) and AP1-like transcription factors are believed to be most relevant for regulation of c-jun expression upon UV irradiation (21). In contrast, analysis of the rhoB promoter indicated that activation of rhoB by UV irradiation is independent of MAP kinases and so far known UV-stimulated transcription factors and regulatory elements (18). Therefore, a novel mechanism was hypothesized to be involved in the regulation of rhoB induction by UV light. Here we present evidence that a CCAAT box is an essential requirement for the transcriptional activation of rhoB by genotoxic stress.
MATERIALS AND METHODS
Materials
Isolation and characterization of the rhoB promoter as well as the generation of a 0.17 kb rhoB promoter fragment that is still inducible by UV irradiation were described previously (18). Mutational analysis of the 0.17 kb rhoB promoter fragment was performed by use of a standard PCR-based method. Anti-ATF-2 (sc-187x) and anti-c-Jun/AP1 (sc-45x) antibodies used in this study were obtained from Santa Cruz (San Diego, CA). Anti-NF-YA antibody was purchased from Pharmingen (San Diego, CA). c-Fos-deficient cells originated from A. Wagner (Vienna, Austria); p53-deficient cells were generously provided by A. Balmain (Glasgow, UK). Mutational inactivation of CCAAT boxes in the rhoB promoter was done by standard PCR-based methods.
Cell culture and transfection experiments
NIH 3T3 fibroblast cells were routinely grown in Dulbecco’s modified Eagle’s medium containing 10% fetal calf serum. For transient transfection experiments, 5 × 105 cells were seeded per 5 cm dish and transfected 16–24 h later with 5 µg promoter–CAT constructs using the calcium phosphate co-precipitation technique (22). Salmon sperm DNA was added to a final concentration of 10 µg/assay. If not stated otherwise, cells were treated 16–24 h after transfection. After a further incubation period of 24 h, cells were harvested for determination of the level of CAT protein expression using an ELISA-based assay system (CAT-Elisa Kit; Roche Diagnostics GmbH). Determination of protein concentration was done according to the method of Bradford (23).
Northern blot analysis
In order to analyze rhoB expression at the mRNA level, total RNA was prepared according to the method of Chomczynski and Sacchi (24). RNA was separated on a 1% agarose gel and transferred overnight onto Hybond N+ filters. For prehybridization filters were incubated for 2 h at 60°C in a solution containing 7% SDS, 1 mM EDTA, 0.5 M phosphate buffer pH 7.4. Hybridization was done overnight in the same solution additionally containing 1% BSA and 32P-labeled probe. For rhoB-specific hybridization a 0.95 kb EcoRI–XhoI fragment from the 3′-region of the rat rhoB cDNA was used. This rhoB fragment does not cross-hybridize to other rho mRNA species (13). After washing with decreasing concentrations of salt (2–0.5× SSC, 1 mM EDTA, 0.5% SDS) filters were subjected to autoradiography. As an internal standard for the amount of RNA loaded, filters were rehybridized with a glyceraldehyde 3-phosphate dehydrogenase (GAPDH) cDNA probe as described previously (13). For quantitation, densitometric analysis of the autoradiograms was performed. Relative gene expression was calculated by referring the level of rhoB mRNA to the amount of GAPDH mRNA. The relative rhoB mRNA level in treated cells was related to that of control cells, which was set to 1.0.
Gel retardation analysis
32P-labeling of the 0.17 kb rhoB promoter fragment as well as of shorter oligonucleotides was done by use of T4 kinase. Total cell extract was prepared as described (25). Binding reactions were performed by incubation of 5–10 µg protein with ∼5 fmol 32P-labeled oligonucleotide for 30 min at room temperature [binding buffer, 10 mM HEPES pH 7.9, 60 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM DTT, 1 µg/ml BSA, 10% glycerol, 0.5 µg poly(dI·dC)]. For competition experiments the reaction mixture was preincubated with 10–50-fold molar excess of unlabeled oligonucleotide for 10 min at room temperature before 32P-labeled probe was added. After the binding reaction the reaction mixture was separated on 4% polyacrylamide gels at room temperature. After electrophoresis, gels were dried and subjected to autoradiography. The following oligonucleotides were used for DNA binding analyses: NF-YrhoB (5′-GGCTTCCCATTGGGTGGCTAT-3′); AP-1-specific oligonucleotide (5′-AGTGGTGACTCATCACT-3′); two different oligonucleotides for detection of binding of C/EBP proteins, C/EBPa consensus sequence (sc-2525; Santa Cruz) (5′-TGCAGATTGCGCAATCTGCA-3′) and C/EBPb (5′-ATTCAATTGGGCAATCAG-3′) (26); ATF/CREB (5′-AGAGATTGCCTGACGTCAGAGAGCTA-3′).
Southwestern analysis, cross-linking and immunoprecipitation
For southwestern analysis 50 µg protein from total NIH 3T3 cell extact was separated by SDS–PAGE (10% gel). After transfer onto nitrocellulose, nitrocellulose-bound proteins were renatured and hybridized with the 32P-labeled 0.17 kb rhoB promoter fragment as described (27). Proteins binding to the 32P-labeled DNA were detected by autoradiography. For UV cross-linking experiments the 32P-labeled rhoB fragment was incubated with nuclear extract under identical conditions to those described for the gel retardation analysis. Afterwards the reaction mixture was irradiated with a 254 nm hand lamp for 10 min. The lamp was placed on top of an unscrewed tube which was kept on ice during irradiation. After cross-linking the products were separated by SDS–PAGE and cross-linked 32P-labeled protein–DNA complexes were visualized by autoradiography. Immunoprecipitation of UV cross-linked protein was performed basically as described (28).
RESULTS AND DISCUSSION
Expression of the gene encoding the Ras-related GTPase RhoB is rapidly inducible by genotoxic agents such as UV irradiation and alkylating compounds (13,18,20). Data obtained by cloning and analysis of the rhoB promoter indicated that the regulatory mechanism involved in rhoB induction is different from that triggering expression of other genotoxic stress-inducible genes such as c-fos and c-jun (18). As shown in Figure 1, the increase in the level of rhoB mRNA by UV irradiation in mouse fibroblasts deficient in c-Fos or p53 protein is similar to that of wild-type cells. This finding demonstrates that the stimulation of rhoB mRNA expression by UV light is independent of c-Fos and p53, both of which are important transcription factors triggering expression of various genes upon genotoxic stress. Moreover, since c-Fos is part of the heterodimeric transcription factor AP1, the data support our previous conclusion that rhoB induction is independent of AP1-like transcription factors. Yet, the main argument for the involvement of a novel mechanism in the regulation of rhoB expression by DNA-damaging agents is based on the identification of a 0.17 kb rhoB promoter fragment which is still inducible by UV light (18). The sequence of this fragment, which is outlined in Figure 2, lacks consensus sequences for binding of known UV-activated transcription factors. We would like to note that the sequence around position –45 (i.e. 5′-TGAGCTCA-3′) is not identical to the ATF/CREB consensus sequence (TGACGTCA) because of a CG→GC replacement found in the rhoB sequence. Lack of function of this ‘ATF-like’ sequence in the rhoB promoter was confirmed by gel retardation analysis and competition experiments (data not shown). Thus, overall the 0.17 kb rhoB fragment does not contain any known consensus sequences for binding of transcription factors characterized to be rapidly activated by genotoxic treatments (e.g. AP-1, c-Jun/ATF-2, Elk-1 and CHOP). The sequence (85 bp in length) located 5′ of the putative transcription initiation site (+1) only harbors consensus sequences corresponding to CCAAT and TATA boxes (Fig. 2). Based on computational analysis, CCAAT box 2 is thought to be a NF-Y site.
Figure 1.
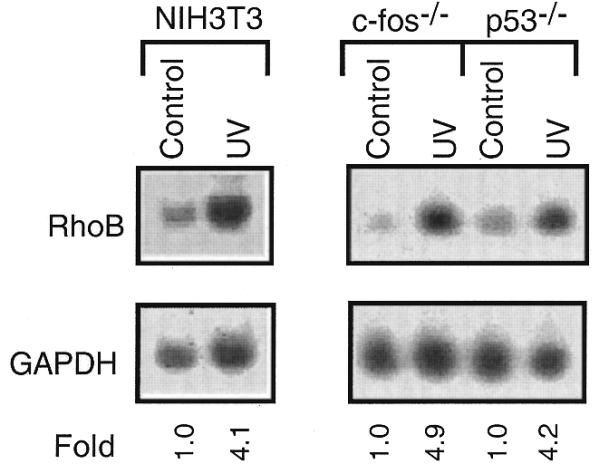
Induction of rhoB mRNA expression by UV irradiation is independent of c-Fos and p53. NIH 3T3 wild-type cells and c-fos–/– and p53–/– fibroblast cells were left untreated (Control) or were irradiated with UV light (40 J/m2). Thirty minutes after treatment, total RNA was isolated and the expression of rhoB mRNA was analyzed by northern blot analysis as described in Materials and Methods. The autoradiogram is shown. The relative amount of rhoB mRNA in untreated cells was set to 1.0.
Figure 2.

Sequence of the 0.17 kb rhoB promoter fragment that is still inducible by UV irradiation. The gene structure and sequence of the 0.17 kb rhoB promoter fragment which is still inducible by UV light are derived from a previous report (18). X, XbaI; P, PstI; TI(+1), transcription initiation site. Putative CCAAT and TATA boxes are underlined and in bold.
Band shift analysis using the 32P-labeled 0.17 kb rhoB promoter fragment revealed a single specific band, which largely disappeared upon competition with 10-fold molar excess of the non-labeled fragment (Fig. 3A). In contrast, addition of up to 50-fold molar excess of an AP-1-specific oligonucleotide exerted no competitive effects (Fig. 3A). This finding agrees with the assumption that AP1-like elements do not interfere with the regulation of rhoB expression by UV irradiation and further excludes the possibility that novel AP1-like elements are involved in rhoB regulation. Extracts prepared from UV-irradiated cells did not show enhanced DNA binding to the 32P-labeled 0.17 kb fragment as compared to extracts from non-irradiated cells (Fig. 3B). Moreover, no additional [32P]DNA–protein complex emerged upon UV irradiation (Fig. 3B). Thus it appears unlikely that transcriptional activation of rhoB is due to additional binding of a novel factor to a particular sequence motif within the 0.17 kb fragment. Both AP1 and ATF-2-specific antibodies failed to induce supershifts (Fig. 3C), suggesting again that these factors are not involved in [32P]DNA–protein complex formation. In order to characterize the proteins which bind constitutively to the 0.17 kb rhoB promoter fragment, DNA cross-linking and southwestern analyses were performed. Upon UV cross-linking of the DNA–protein complex and subsequent SDS–PAGE a 32P-labeled complex of ∼48 kDa was observed (Fig. 4A). Taking into account the DNA within the cross-linked complex, which contributes to an overestimation of the actual size of the protein bound, the molecular weight of the UV cross-linked protein can be estimated to be <48 kDa. As determined by southwestern analysis, the molecular weight of proteins which predominantly bind to the 0.17 kb rhoB fragment is ∼36 kDa (Fig. 4B). This is similar to the molecular weight of TATA-binding factors. Because the 0.17 kb rhoB promoter fragment contains TATA boxes, it is rational to assume that TATA-binding factors were UV cross-linked to the 32P-labeled rhoB fragment.
Figure 3.

Gel retardation analysis using the 0.17 kb rhoB promoter fragment. (A) An aliquot of 5 µg protein from NIH 3T3 cell extracts was analyzed for DNA binding activity to the 32P-labeled 0.17 kb rhoB promoter fragment by gel retardation as described in Materials and Methods. For competition experiments the unlabeled fragment (SComp) was added in 10-fold molar excess. As a control, an AP-1-specific oligonucleotide was added in 10- and 50-fold molar excess. The arrow points to the specific band. (B) Up to 2 h after irradiation (40 J/m2) of NIH 3T3 cells extracts were prepared and analyzed for DNA binding activity using the 32P-labeled 0.17 kb rhoB promoter fragment. (C) Extracts obtained from cells left untreated (Control) and treated with UV light (40 J/m2, 30 min) were used for supershift analysis using 1 µg ATF-2- and c-Jun/AP-1-specific antibodies.
Figure 4.
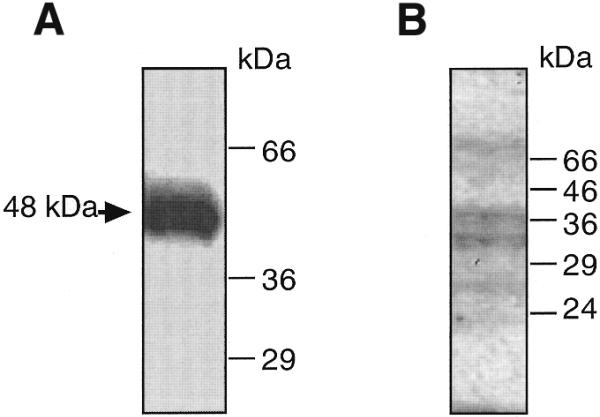
An ∼48 kDa protein is UV cross-linked to the 0.17 kb rhoB promoter fragment. (A) 32P-labeled rhoB fragment was incubated with extracts from UV-irradiated cells under identical conditions to those used in the gel retardation experiments. Afterwards the reaction was irradiated with UVC light as described in Materials and Methods. Products were separated by SDS–PAGE and the cross-linked protein–DNA complex was visualized by autoradiography. (B) An aliquot of 50 µg protein from total cell extract was separated by SDS–PAGE. After blotting to nitrocellulose, southwestern analysis using 32P-labeled rhoB promoter fragment was performed as described in Materials and Methods. The autoradiogram is shown.
The lack of consensus sequences for binding of known UV-activated transcription factors together with the failure to detect UV-induced changes in the DNA-binding activity of extracts from UV-treated cells to the 32P-labeled rhoB promoter fragment raises the question of whether CCAAT- or TATA-binding factors might be involved in the transcriptional activation of rhoB by UV irradiation. Previously, it was reported that the transcriptional activity of the c-fos gene can be enhanced via TATA-binding protein-associated factors (TAFs) (29). Moreover, CCAAT-binding proteins were also shown to increase gene expression upon treatment of cells with serum or calcium (30,31). Most interestingly, the CCAAT-binding factor NF-YA is reported to interact with ATF-2 in regulation of the fibronectin gene (28). Notably, the stimulatory effect of ATF-2 on fibronectin gene expression is supposed to be independent of DNA binding of ATF-2. Rather, it is assumed that ATF-2 exerts its stimulatory effect exclusively via binding to NF-Y, without contacting DNA (28). In this context it is important to note that the ATF-2/NF-Y complex is believed to be unstable under conditions of gel electrophoresis (28). Therefore, the interaction of ATF-2 with NF-Y cannot be detected by gel retardation experiments. Overall, these data prompted us to investigate whether CCAAT-binding factors and ATF-2 might also be relevant for the transcriptional activation of rhoB by genotoxic stress. In a first step a possible involvement of ATF-2 in the regulation of rhoB expression was analyzed by immunoprecipitation experiments (28). After incubation of the 32P-labeled rhoB promoter fragment with nuclear proteins (under identical experimental conditions to those of the gel retardation experiments) and subsequent UV cross-linking, immunoprecipitation with ATF-2-specific antibody was performed and the radioactivity in the precipitate was determined. The experiments showed that the ATF-2-specific antibody used was able to co-precipitate a significantly higher amount of 32P-labeled DNA than either heat-inactivated ATF-2 antibody or an AP1-specific antibody, which was included as a further control (Fig. 5). Overall, these data strongly indicate that ATF-2 is associated with factors that bind to the 0.17 kb rhoB fragment.
Figure 5.

ATF-2 is associated with the 0.17 kb rhoB promoter fragment. The 32P-labeled 0.17 kb rhoB promoter fragment was incubated with cell extracts under the same experimental conditions as used in the gel retardation analyses. Afterwards UV cross-linking and subsequent immunoprecipitation with anti-ATF-2- and c-Jun/AP-1-specific antibodies were performed as described in Materials and Methods. As a further control antibodies were inactivated by heat (5 min, 95°C) prior to use for immunoprecipitation. After washing, the amount of immunoprecipitable radioactivity was determined by scintillation counting. Data shown are means ± SD from at least three independent experiments, each performed in duplicate.
In a next step we analyzed whether or not CCAAT-binding factors might be relevant for rhoB induction by genotoxic stress. To this end we examined the consequences of mutational inactivation of the CCAAT boxes in the rhoB promoter on its inducibility by UV irradiation. As shown in Figure 6, deletion of CCAAT box 1 did not attenuate the induction of rhoB by UV irradiation or methyl methanesulfonate (MMS) treatment, indicating that this CCAAT element is not crucial for the activation of rhoB by genotoxic treatment. In line with this, inactivation of CCAAT box 1 by point mutation also failed to affect transcriptional stimulation of rhoB by UV light (Fig. 6). However, functional disruption of CCAAT box 2 by point mutation blocked the increase in rhoB gene expression induced by UV irradiation as well as by MMS treatment (Fig. 6). Obviously, CCAAT box 2 (i.e. CATTGGG), which is located close to the functional TATA box, is an essential requirement for induction of rhoB by UV light and MMS. Notably, based on computational sequence analysis, this CCAAT site is thought to be a NF-Y (CBF, CP1) binding site. Yet, besides NF-Y, many other factors are also described to bind to CCAAT elements. One important group is the CCAAT enhancer-binding proteins (C/EBP). Therefore, we analyzed whether members of the C/EBP family are able to bind to the functionally relevant CATTGGG sequence (= NF-YrhoB) of the rhoB promoter. To this end, competition experiments were performed. As shown in Figure 7, binding of proteins to the NF-YrhoB oligonucleotide can only be competed by the non-labeled NF-YrhoB sequence, and not by an oligonucleotide harboring a C/EBP consensus sequence (Fig. 7A). Moreover, binding of proteins to two different types of 32P-labeled C/EBP-specific oligonucleotides (i.e. C/EBPa and C/EBPb) was not competed with excess unlabeled NF-YrhoB-specific oligonucleotide (Fig. 7B and C). Based on the data we suggest that proteins of the C/EBP family are very likely not involved in binding to the CATTGGG sequence present in the rhoB promoter. Having in mind a previous report showing an interaction of ATF-2 with the CCAAT-binding factor NF-YA (28), we speculate that NF-YA, together with ATF-2, is involved in the regulation of rhoB by genotoxic stress. To ascertain that association of CCAAT-binding factor NF-Y with ATF-2 really occurs in our cell system, immunoprecipitation experiments were performed (Fig. 8). Upon immunoprecipitation with anti-NF-YA antibody followed by western blot analysis using anti-ATF-2-specific antibody we found that anti-NF-YA antibody is able to co-precipitate proteins cross-reacting with anti-ATF-2 antibody (Fig. 8A). As concluded from cloning and in vitro translation experiments, ATF-2 protein is ∼46 kDa in size, however, splice variants and post-translational modifications might lead to different migration on SDS gels (32). The amount of co-precipitated ATF-2 protein was not enhanced if extract from UV-irradiated cells was used (Fig. 8A). This indicates that NF-YA is constitutively associated with AFT-2. Complexation of NF-YA and ATF-2 was further confirmed by immunoprecipitation using an anti-ATF-2 antibody and subsequent analysis of co-precipitated NF-YA protein by western blot analysis (Fig. 8B). Thus, overall, the previously reported data (28) were confirmed by our own results.
Figure 6.

The CCAAT box in the rhoB promoter mediates its inducibility by genotoxic stress. The 0.17 kb rhoB promoter fragment was used for PCR-based mutational disruption of the CCAAT boxes. For mutational changes of CCAAT box 1 and CCAAT box 2 (see Fig. 2) the standard PCR technique was used. Twenty-four hours after transient transfection of wild-type and mutated constructs, cells were UV irradiated (40 J/m2) or treated with MMS (0.75 mM). A further 24 h later cells were harvested for determination of the amount of CAT protein. Δ, deletion; *, mutational inactivation (by introduction of a point mutation). Data shown are means ± SD from at least two independent experiments, each performed in duplicate.
Figure 7.
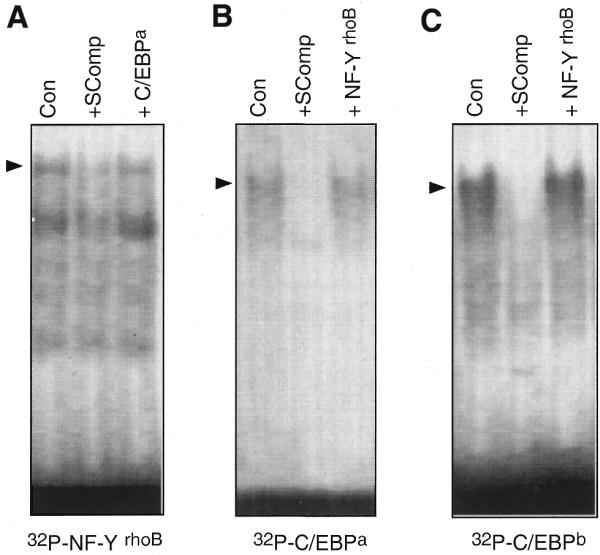
Members of the C/EBP family are not involved in the regulation of rhoB. (A) For gel retardation analysis the 32P-labeled oligonucleotide derived from the sequence harboring the CCAAT box essentially required for activation of the rhoB promoter by genotoxic stress was used (NF-YrhoB, 5′-GGCTTCCCATTGGGTGGCTAT-3′). Competition experiments were performed using 20-fold molar excess of either non-labeled NF-YrhoB oligonucleotide (specific competition) or an oligonucleotide containing the consensus sequence for binding of C/EBP (C/EBPa, 5′-TGCAGATTGCGCAATCTGCA-3′) (sc-2525; Santa Cruz). SComp, specific competition. (B) For band shift analysis the 32P-labeled C/EBP-specific oligonucleotide (C/EBPa) described in (A) was used. Competition experiments were done as described using either unlabeled C/EBPa (SComp) or NF-YrhoB-specific oligonucleotide. SComp, specific competition. (C) A second consensus sequence described to bind C/EBP proteins (26) was used for band shift analysis (C/EBPb, 5′-ATTCAATTGGGCAATCAG-3′). Specific competition (SComp) and competition with NF-YrhoB-specific oligonucleotide was done as described before.
Figure 8.
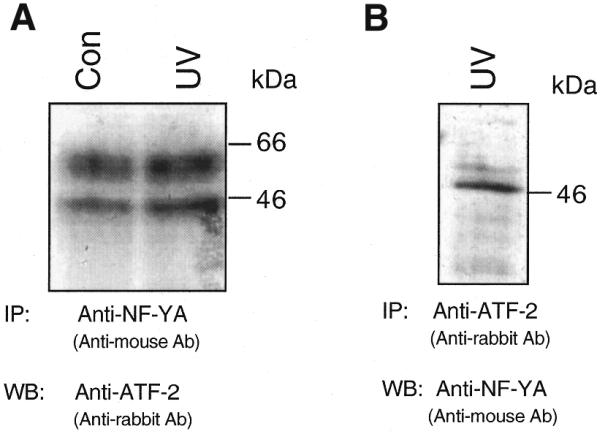
CCAAT-binding factor NF-YA is complexed with ATF-2. (A) Extracts from untreated (Con) and UV-treated (60 J/m2, 30 min) cells were used for immunoprecipitation (IP) with anti-NF-YA-specific antibody. After precipitation, western blot analysis (WB) using antibody directed against ATF-2 protein was performed. The autoradiogram is shown. (B) ATF-2 protein was immunoprecipitated from UV-treated cells. Subsequently, immunoprecipitate was applied to western blot analysis using anti-NF-YA-specific antibody. The autoradiogram is shown.
To summarize, the data available strongly indicate that CCAAT-binding factors, putatively NF-YA, act in concert with the transcription factor ATF-2 in stimulating rhoB gene expression upon genotoxic stress. A possible function of ATF-2 might be to increase the frequency of transcription initiation after a particular type of stimulus. Notably, ATF-2 is a substrate for JNKs/SAPKs and p38 MAP kinase and is believed to play a central role in the stimulation of c-jun expression by UV irradiation (21). With respect to rhoB regulation, it is therefore tempting to speculate that, in analogy to the regulation of c-jun (with c-Jun/ATF-2 being of particular relevance), genotoxic stress leads to activation of ATF-2/NF-Y, which finally leads to an increase in rhoB gene expression. Bearing in mind that MAP kinases (i.e. JNK and p38 kinase) are important for the stimulation of c-jun expression by UV light (21), this hypothesis at the same time implies that MAP kinases are involved in rhoB regulation. Our initial hypothesis that MAP kinase-related pathways do not participate in the regulation of rhoB was based on the observation that pharmacological inhibition of MAP kinases does not prevent activation of rhoB expression by UV irradiation (18). However, recently we showed that even induction of c-jun by UV light is not blocked by pharmacological inhibiton of JNK1 activity (3). It appears that JNK1 is not an essential requirement for expression of c-jun, possibly because under conditions of JNK1 inhibition other JNK isoenzymes come into play and compensate for loss of JNK1 activity. Therefore, with respect to a possible involvement of MAP kinase-related pathways in rhoB regulation, conclusions drawn from experiments using pharmacological MAP kinase inhibitors might be misleading. Nevertheless, the essential requirement of a CCAAT box for transcriptional activation of rhoB by DNA-damaging treatments represents a novel mechanism of regulation of an early responsive gene in cells exposed to genotoxic agents.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by the Deutsche Forschungsgemeinschaft (Fr 1241/1-3).
References
- 1.Sachsenmaier C., Radler-Pohl,A., Zinck,R., Nordheim,A., Herrlich,P. and Rahmsdorf,H.J. (1994) Involvement of growth factor receptors in the mammalian UVC response. Cell, 78, 963–972. [DOI] [PubMed] [Google Scholar]
- 2.Rosette C. and Karin,M. (1996) Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science, 274, 1194–1197. [DOI] [PubMed] [Google Scholar]
- 3.Fritz G. and Kaina,B. (1999) Activation of c-Jun N terminal kinase 1 by UV irradiation is inhibited by wortmannin without affecting c-jun expression. Mol. Cell. Biol., 19, 1768–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Derijard B., Hibi,M., Wu,I.H., Barrett,T., Deng,T., Karin,M. and Davis,R.J. (1994) JNK1: a protein kinase stimulated by UV-light and Ha-ras that binds and phosphorylates the c-Jun activation domain. Cell, 76, 1025–1037. [DOI] [PubMed] [Google Scholar]
- 5.Price M.A., Cruzalegui,F.H. and Treisman,R. (1996) The p38 and ERK MAP kinase pathways cooperate to activate ternary complex factors and c-fos transcription in response to UV light. EMBO J., 23, 6552–6563. [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X.Z. and Ron,D. (1996) Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science, 272, 1347–1349. [DOI] [PubMed] [Google Scholar]
- 7.Canman C.E. and Kastan,M.B. (1996) Three paths to stress relief. Nature, 384, 213–214. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh R., Amstad,P. and Cerutti,O. (1993) UVB-induced DNA breaks interfere with transcriptional induction of c-fos. Mol. Cell. Biol., 13, 6992–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg M.E. and Ziff,E.B. (1984) Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature, 311, 433–437. [DOI] [PubMed] [Google Scholar]
- 10.Hollander C. and Fornace,A.J. (1989) Induction of fos RNA by DNA damaging agents. Cancer Res., 49, 1687–1692. [PubMed] [Google Scholar]
- 11.Sherman M.L., Datta,R., Hallahan,D.E., Weichselbaum,R.R. and Kufe,D.W. (1990) Ionizing radiation regulates expression of the c-jun protooncogene. Proc. Natl Acad. Sci. USA, 87, 5663–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaina B., Haas,S. and Kappes,H. (1997) A general role for c-Fos in cellular protection against DNA-damaging carcinogens and cytostatic drugs. Cancer Res., 57, 2721–2731. [PubMed] [Google Scholar]
- 13.Fritz G., Kaina,B. and Aktories,K. (1995) The Ras-related small GTP-binding protein RhoB is immediate-early inducible by DNA-damaging treatments. J. Biol. Chem., 270, 25172–25177. [DOI] [PubMed] [Google Scholar]
- 14.Jähner D. and Hunter,T. (1991) The ras-related gene rhoB is an immediate early gene inducible by v-Fps, epidermal growth factor and platelet-derived growth factor in rat fibroblasts. Mol. Cell. Biol., 11, 3682–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zalcman G., Closson,V., Linares-Cruz,G., Lerebours,F., Honore,N., Tavitian,A. and Olofsson,B. (1995) Regulation of Ras-related RhoB protein expression during cell cycle. Oncogene, 10, 1935–1945. [PubMed] [Google Scholar]
- 16.Gampel A., Parker,P.J. and Mellor,H. (1999) Regulation of epidermal growth factor receptor traffic by the small GTPase RhoB. Curr. Biol., 9, 955–958. [DOI] [PubMed] [Google Scholar]
- 17.Prendergast G.C., Khosravi-Far,R., Solski,P.A., Kurzawa,H., Lebowitz,P.F. and Der,C.J. (1995) Critical role of Rho in cell transformation by oncogenic Ras. Oncogene, 10, 2289–2296. [PubMed] [Google Scholar]
- 18.Fritz G. and Kaina,B. (1997) rhoB encoding a UV-inducible Ras-related small GTP-binding protein is regulated by GTPases of the Rho family and independent of JNK, ERK and p38 MAP kinase. J. Biol. Chem., 272, 30637–30644. [DOI] [PubMed] [Google Scholar]
- 19.Fritz G. and Kaina,B. (2001) Ras-related GTPase RhoB represses NF-kB signaling. J. Biol. Chem., in press. [DOI] [PubMed] [Google Scholar]
- 20.Fritz G. and Kaina,B. (2000) Ras-related GTPase RhoB forces alkylation-induced apoptotic cell death. Biochem. Biophys. Res. Commun., 268, 784–789. [DOI] [PubMed] [Google Scholar]
- 21.Van Dam H., Wilhelm,D., Herr,I., Steffen,A., Herrlich,P. and Angel,P. (1995) ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J., 14, 1798–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graham F.L. and Van der Erb,A.J. (1973) A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology, 52, 456–467. [DOI] [PubMed] [Google Scholar]
- 23.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 24.Chomczynski P. and Sacchi,N. (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem., 162, 156–159. [DOI] [PubMed] [Google Scholar]
- 25.Van Dam M., Müllberg,J., Schooltink,H., Stoyan,T., Brakenhoff,J.P.J., Graeve,L., Heinrich,P.C. and Rose-John,S. (1993) Structure-function analysis of interleukin-6 utilizing human/murine chimeric molecules. J. Biol. Chem., 268, 15285–15290. [PubMed] [Google Scholar]
- 26.Bakker O. and Parker,M.G. (1991) CAAT/enhancer binding protein is able to bind to ATF/CRE elements. Nucleic Acids Res., 19, 1213–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huebscher U. (1987) Double replica southwestern. Nucleic Acids Res., 15, 5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alonso C.R., Pesce,C.G. and Kornblihtt,A.R. (1996) The CCAAT binding proteins CP1 and NF-1 cooperate with ATF-2 in the transcription of the fibronectin gene. J. Biol. Chem., 271, 22271–22279. [DOI] [PubMed] [Google Scholar]
- 29.Rothofsky M.L. and Lin,S.L. (1997) CROC-1 encodes a protein which mediates transcriptional activation of the human FOS promoter. Gene, 195, 141–149. [DOI] [PubMed] [Google Scholar]
- 30.Framson P. and Bornstein,P. (1993) A serum response element and a binding site for NF-Y mediate the serum response of the human thrombospondin 1 gene. J. Biol. Chem., 268, 4989–4996. [PubMed] [Google Scholar]
- 31.Roy B., Li,W.W. and Lee,A.S. (1996) Calcium-sensitive transcriptional activation of the proximal CCAAT regulatory element of the grp78/BiP promoter by the human nuclear factor CBF/NF-Y. J. Biol. Chem., 271, 28995–29002. [DOI] [PubMed] [Google Scholar]
- 32.Ivashkiv L.B., Liou,H.C., Kara,C.J., Lamph,W.W., Verma,I.M. and Glimcher,L.H. (1990) mXBP/CRE-BP2 and c-Jun form a complex which binds to the cyclic AMP, but not to the 12-O-tetradecanoylphorbol-13-actetate, response element. Mol. Cell. Biol., 10, 1609–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]