

Abstract
Objective
The aim of this study was to better understand how MLL fusion proteins deregulate the expression of genes critical for leukemia.
Materials and Methods
The transforming domain of one of the most common MLL fusion partners, AF9, was immuno-purified after expression in myeloblastic M1 cells and associating proteins were identified by mass spectrometric analysis. Chromatin immunoprecipitation followed by quantitative PCR was used to determine how binding of associating proteins compare across Hoxa9 and Meis1 in cell lines with and without MLL fusion proteins and how binding is altered during gene down-regulation and differentiation.
Results
Consistent with earlier purifications of ENL and AF4 from 293 cells, the 90 amino acid C- terminal domain of AF9 associates with many other MLL translocation partners including Enl, Af4, Laf4, Af5q31, Ell, and Af10. This complex termed EAP for Elongation Assisting Proteins, also contains the RNA polymerase II C terminal domain kinase Cdk9/Cyclin T1/T2 (pTEFb) and the histone H3 lysine 79 methyltransferase Dot1L. Myeloid cells transformed by MLL fusions show higher levels and a broader distribution of EAP components at genes critical for leukemia. Inhibition of EAP components pTEFb and Dot1l show that both contribute significantly to activation of Hoxa9 and Meis1 expression. EAP is dynamically associated with the Hoxa9 and Meis1 loci in hematopoietic cells and rapidly dissociates during induction of differentiation. In the presence of MLL fusion proteins, its dissociation is prevented.
Conclusion
The findings suggest that MLL fusion proteins deregulate genes critical for leukemia by excessive recruitment and impaired dissociation of EAP from target loci.
Keywords: MLL, Af9, Enl, Hox, leukemia
Introduction
Rearrangements involving the Mixed Lineage Leukemia (MLL) gene are associated with acute lymphoid and myeloid leukemias. Balanced translocations involving MLL result in the production of fusion proteins in which the amino terminus of MLL is fused in-frame to one of over 50 different translocation partners. The most common fusion partners are the nuclear proteins ENL, AF4 and AF9, all of which are transcriptional activators. Upregulation of A cluster HOX genes and the HOX cofactor MEIS1 is pivotal for MLL fusion protein-mediated leukemogenesis [1].
The mechanism by which the translocation partners deregulate transcription is beginning to be defined. One of the first insights was provided by Bitoun et al. who identified proteins in human embryonic kidney cells that associate with the MLL translocation partner AF4, including ENL, AF5q31, CDK9 and CYCLIN T1 [2]. AF4, in association with ENL and AF9, was found to stimulate activity of the RNA polymerase II (RNA pol II)-CTD kinase pTEFb and the histone methyltransferase DOT1L. Subsequently, we reported a complex of proteins termed EAP that interacts with the MLL translocation partner ENL in 293 cells [3]. EAP is similar to the complex reported by Bitoun et al. but includes additional interacting proteins such as AF10, PC3, RING1b and others. Importantly, the amino acids in ENL that directly interact with DOT1L were found to be essential for MLL-ENL to transform bone marrow progenitors. This, together with earlier data [4], suggested that DOT1L methyltransferase activity is crucial for Hox gene deregulation and transformation seen in leukemias with MLL rearrangements.
pTEFb, which interacts with AF4 family members [5–7] has also been shown to be critical to MLL fusion protein-mediated transformation [7]. Yokoyama et al highlight the importance of pTEFb, which is shown to be part of a core complex termed AEP that is physically distinct from the Dot1L complex. They show that fusing the pTEFb-interacting domain of AF4 family members to MLL is necessary and sufficient for leukemic transformation, while DOT1L is not sufficient. However, the authors do not rule out the importance of DOT1L for transformation. Thus, both CTD kinase and histone methyl transferase activities of MLL fusion protein complexes appear to be critical to their ability to aberrantly activate the expression of genes critical for leukemia.
In this study we focused on further defining the mechanisms of transactivation by one of the most common MLL fusion proteins, MLL-AF9, and explored pTEFb as a possible therapeutic target. We first identified the proteins that interact with the C-terminal 90 amino acids of AF9, the minimal domain required for transformation when fused to the N-terminal fragment of MLL. These studies showed that, in myeloblastic cells, a complex similar to EAP interacts with the transforming domain of AF9 and the MLL-AF9 fusion protein. ChIP studies for EAP subunits showed that EAP is localized to MLL target loci in cells with and lacking MLL rearrangements. Notably, in cells transformed by MLL fusion proteins, increased amounts of EAP are localized to the Hox loci and the complex is more broadly distributed. Furthermore, while EAP recruited independently of MLL fusion proteins dissociates from target loci in response to differentiation cues, MLL fusion protein-associated EAP remains at target genes, maintaining Hox and Meis expression. These findings suggest that a key activity of MLL fusion proteins is to prevent the normal dissociation of the EAP complex from loci critical for leukemogenesis.
Materials and Methods
Cloning and transduction of AF9C-term into M1 cells
Bases 1691–1963 encoding residues 478–568 of human AF9 (Accession NP_004520) were PCR-amplified from an MLL-AF9 expression vector and cloned into the Xho1 and Not1 sites of a modified murine stem cell virus (MSCV) vector. This vector contains two N-terminal FLAG tags, three nuclear translocation signals, and two C-terminal MYC tags and expresses GFP from an internal ribosome entry site (MIGR1-2XFLAG-3XNLS-2XMYC). MIGR1-2XFLAG-AF9-3XNLS-2XMYC and the control MIGR1-2XFLAG-3XNLS-2XMYC were packaged into viruses using the 293-derived PLAT-E cell line [8]. One ml of viral supernatant and 5 μg/ml polybrene was added to three million M1 cells (from ATCC) in a six-well tissue culture dish. Cells were spin-infected for 90 minutes at 2500 RPM in an Eppendorf 5810 R centrifuge. The next day the media was changed. Cells were GFP sorted at the University of Michigan Flow Cytometry Core one week after transduction.
Cell lines
M1 (ATCC), M1-control, M1-AF9, MonoMac6 (DSMZ), RS4;11 (ATCC), KOPN-8 (DSMZ), REH (ATCC), Raji (ATCC), 697 (DSMZ), NALM-1 (DSMZ), HAL-01 (DSMZ), and NALM-6 (DSMZ) cells were maintained in RPMI media containing 10% fetal bovine serum. MV4:11 (ATCC) cells were maintained in IMDM with 15% FBS and 10 ng/mL IL-3. 293 cells (ATCC) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco). E2A-HLF, MLL-AF9, and MLL-ENL transformed bone marrow cells were established by retroviral transduction. In brief, 6–8 week old C57/BL6 mice were injected with 150 mg/kg 5-fluorouracil. Bone marrow was harvested after four days and placed in a pre-stimulation IMDM media containing 100 ng/ml SCF, 10 ng/ml IL3, and 10 ng/ml IL6. The next day, cells were transduced with pMSCV-E2A-HLF (a generous gift from Dr. Michael Cleary), pMSCV-MLL-AF9, or pMSCV-MLL-ENL that was packaged into retroviruses using PLAT-E cells as described above for the MIGR1 constructs. Forty-eight hours post-transduction, cells were placed in selection media containing 1 mg/ml G418 (Invitrogen). Fusion protein-transformed cells were maintained in IMDM media containing 15% FBS and 10 ng/ml IL-3.
Immunoprecipitation and Western blot for EAP
Nuclei were isolated from 200 ml to 1000 ml of M1 suspension culture by douncing cells in hypotonic buffer (10 mM KCl, 10 mM Tris-Cl, pH 7.9) and spinning at 4000 rpm for 15 minutes, 4° C. Nuclei were then lysed in BC-300 lysis buffer: 0.1% NP40, 10% glycerol, 300–500 mM KCl, 100 uM PMSF, 1 protease inhibitor cocktail tablet (Roche), 1 mM EDTA, 50 mM Tris-CL pH 7.9 for 30 minutes at 4ºC. Some immunoprecipitations were done in the presence of 100 μg/ml ethidium bromide or after treatment with 2500 units of benzonase (in presence of 2 mM MgCl2). Cell lysate was spun at maximum speed in an Eppendorf 5424 microcentrifuge for 15 minutes and supernatant was pre-cleared with protein-G-agarose (Roche) beads for 1 hour at 4ºC. After pre-clearing, the lysate was incubated with 100 μl anti-FLAG M2 agarose beads (Sigma) for 1 hour rocking at 4ºC. Beads were subjected to six five-minute washes with lysis buffer (excluding protease inhibitors). For mass spectrometry analysis, the purified proteins were eluted with 2X FLAG peptide two times for fifteen minutes each at 4ºC, then resolved by SDS-PAGE. For Western blotting, the beads were heated at 90ºC for 5 minutes in SDS Tris-Glycine sample buffer (Invitrogen). The supernatant was loaded onto 4–20% Tris-Glycine gels (Invitrogen), and proteins were transferred to a nitrocellulose membrane for 1 hour. Membranes were blocked in 5% non-fat dry milk (Bio-Rad) and then incubated with the appropriate primary antibody overnight (antibodies listed in Supplemental Information). Goat anti-rabbit or goat anti-mouse secondary antibodies (Santa Cruz) were used at 1:2000 for 30 minutes. Proteins were detected using SuperSignal West Pico Chemiluminescent Substrate (Pierce).
Protein identification of EAP by LC-tandem MS
This procedure is described in detail in Supplemental Information. In brief, samples were resolved by SDS-PAGE and visualized with MS-compatible silver stain (PROTSIL-2, Sigma). Gel resolved proteins were digested with sequencing grade, modified trypsin according to standard protocol. Data-dependent MS/MS data on the resulting peptides was collected using an ion-trap instrument (LTQ XL, ThermoFisher). Proteins were identified by searching the data against mouse IPI data (v 3.32) appended with decoy (reverse) sequences using X!Tandem/TPP software suite [9].
Transient Transfections
105 human embryonic kidney 293 cells were seeded in 10 cm dishes overnight. Cells were transfected with 10 μg empty vector, 5 μg empty vector and 5 μg pcDNA-HA-Dot1l, 5 μg pcDNA-HA-Dot1l and 5 μg pFLAG-HA-MLL1116–1397, or 5 μg pcDNA-HA-Dot1l and 5 μg pFLAG-MLL-AF9and harvested after 48 hours for whole cell lysis in BC-300 buffer and immunoprecipitation overnight with anti-FLAG M2 affinity resin (Sigma). After five five-minute washes with lysis buffer, beads were boiled in SDS Tris-Glycine loading buffer, loaded onto 6% Tris-Glycine gels, and transferred to nitrocellulose membranes. After 1 hour blocking in 5% non-fat milk, blots were incubated with anti-HA (Covance), anti-FLAG (Sigma), or anti-CDK9 (Santa Cruz) overnight, with secondary antibody for 30 minutes, and subjected to enhanced chemiluminescence with SuperSignal West Pico Chemiluminescent Substrate.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed according to Upstate’s protocol using their recommended lysis, dilution, and wash buffers. In brief, cells were fixed in 1% formaldehyde for 15 minutes at room temperature. Cells were pelleted, PBS-rinsed, and snap frozen. After thawing, cells were lysed and sonicated at high intensity 30 second on-off cycles for 15 minutes in the Diagenode Bioruptor XL. These conditions generated an average DNA fragment size of 500 bp as determined by DNA electrophoresis. Protein-DNA complexes were collected using EAP-conjugated Protein G Dynabeads that were prepared as described above for standard immunoprecipitations (antibodies listed in Supplemental Information). After washing the beads, DNA was eluted by incubation with SDS buffer (1% SDS, 100 mM sodium bicarbonate) for 30 minutes at 42ºC. DNA-protein crosslinks were reversed in 200 μM NaCl overnight at 65ºC. DNA was purified using the QIAquick PCR Purification Kit (Qiagen) and subjected to qPCR as described below. Primer and probe sequences are provided in Supplemental Information (Supplemental Table 2). The student’s t-test was performed to show statistical significance of some comparisons.
Knockdown of DOT1L
MV4:11 cells at a concentration of 10 million cells/ml were transfected with a 500 nM mixture of 2 siRNA oligonucleotides targeting DOT1L (Dharmacon) by electroporation using a Bio-Rad GenePulser MXcell. RNA was extracted and reverse-transcribed (described above) for qPCR 48 hours post-transfection. Gene expression was normalized to 5S ribosomal RNA.
Flavopiridol experiments
MLL-AF9, MLL-ENL, or E2A-HLF cells were seeded at 105 cells/ml in growth media. The following day, cells were treated with 0 nM-200 nM flavopiridol, which was generously provided by Sanofi-Aventis and the National Cancer Institute, NIH. Gene expression was analyzed after 24 hours of treatment as described below. Viability was assessed by cell counting with Trypan blue staining at 48 hours. Viability curves were generated using Prism 5.0 software.
Screening of human ALL cell lines was performed with assistance from the University of Michigan Center for Chemical Genomics (CCG). Cells were seeded at a density of 1250 cells/well in a 384-well plate overnight in RPMI-1640 medium containing 10% FBS. 200 nl of flavopiridol (stock concentrations of 0.04mM–5mM) was added using a Beckman Biomek FX with a HDR (Beckman Coulter, Fullerton,CA) to give a final concentrations of 0.2 –25μM. 10 μM of staurosporine (BIOMOL) was used as a positive control for complete cell death and DMSO was used as a negative control. The plates were incubated in a 37° C, 5% CO2 incubator for 48 hours. Viability was assessed using CellTiter-Glo Luminescent Cell Viability Assay (Promega). 25μl of CellTiter-Glo reagent was added to each well using a Multidrop (Thermo Scientific) and luminescence was read (gain 3600, 0.76 sec/well read) with a PHERAstar plate reader (BMG Labtech). pIC50 curves were generated using gretl software.
Il-6/LPS-induced differentiation of myeloblastic cells
M1-AF9, MLL-AF9, MLL-ENL, or E2A-HLF cells were seeded at 105 cells/ml in growth media. The next day, cells were treated with 100 ng/ml IL6, 5 μg/ml LPS, or an equal volume of PBS as a control. At different time points following treatment, cells were collected for morphologic staining with the HEMA-3 stain kit (Fisher Scientific). Additional aliquots were prepared for gene expression analysis for Hoxa9, Meis1, GAPDH, and beta-actin (described below) and chromatin immunoprecipitation for EAP components (described above).
qPCR
qPCR for ChIP’ed samples was performed with Taqman primers and Taqman Universal Master Mix (Applied Biosystems) using Applied Biosystems 7500 Real-Time PCR system. Data is expressed as percent of total input using the formula % Total Input = 2^–[Ct (IP)/Ct (Input)] [10].
For expression analysis, RNA was extracted from cell lines with Trizol reagent (Invitrogen) according to the manufacturer’s protocol. Reverse-transcription was performed using Superscript II (Invitrogen) according to the manufacturer’s protocol. FAM-labeled Hoxa9, Meis1, Af9, Enl, Cyclin T2, Dot1L, c-Jun, GAPDH, β-Actin, and U2 snRNA Taqman primers (Applied Biosystems) were used in expression analysis of murine cell lines. The qPCR reactions were carried out with Taqman Universal Master Mix (Applied Biosystems), and gene expression was normalized to GAPDH for all experiments except the flavopiridol and DOT1L knockdown experiments. In the flavopiridol experiments, gene expression was normalized to U2 snRNA, which is not dependent on pTEFb CTD kinase activity [11]. For DOT1L knockdown experiments, primers for qPCR were designed with MacVector sequence analysis program. Single product formation was confirmed by running PCR products on 1% agarose gel. SYBR Universal Master Mix (Applied Biosystems) was used for qPCR. Data were normalized to 5S ribosomal RNA. Primer sequences are provided in Supplemental Information (Supplemental Table 3). Analysis was performed using the comparative Ct method [12].
Results
The transforming domain of AF9 associates with other MLL fusion partners and key transcriptional regulators in hematopoietic cells
A cDNA encoding the C-terminal 90 amino acids of AF9 was modified by addition of two N-terminal FLAG-epitope tags, three nuclear translocations signals, and two C-terminal MYC tags and was retrovirally transduced into myeloblastic M1 cells to establish stably expressing cell lines. Cell extracts were immunopurified with FLAG affinity resin and resolved by polyacrylamide gel electrophoresis (PAGE) followed by silver staining. A protein expressing only the tags and nuclear translocation signals served as a negative control (Fig. 1A). Proteins specifically interacting with AF9 identified by mass spectrometry included MLL fusion partners Af4, Laf4, Af5q31, Af10, Ell, and Enl, in addition to Cdk9, Cyclin T1/T2 (pTEFb), Dot1l, Pc3 and Ring1b. The number of peptides identified for each protein is summarized in Supplemental Table 1. Associations with Af5q31, Laf4, Cdk9, and Pc3 were verified by Western blot (Fig. 1A, 1B). BCoR, AF5q31, Laf4, Cdk9, Cyclin T1/T2, Dot1L, and AF10 were identified by mass spectrometric-based analysis after the immunoprecipitations were carried out in 500 mM KCl and 100 μg/mL ethidium bromide, suggesting that they are strong and are not mediated through DNA interactions. Benzonase treatment was tested on DOT1L, AF5q31, and Cdk9 associations to confirm the ethidium bromide results (Fig. 1C). Mass spectrometric analysis identified Ring1b and Pc3 after AF9 immunoprecipitation in up to 500 mM KCl, but not when ethidium bromide was present. Enl, ELL, and AF4 were identified by mass spectrometry after immunoprecipitation in 300 mM salt, and whether these interactions are DNA-dependent was not tested.
Fig. 1.

The transforming domain of AF9 associates with other MLL fusion partners and key transcriptional regulators (EAP) A) Silver stain of proteins co-purifying with AF9 and the control fragment from M1-AF9 and M1-control cell lysate, respectively. Both lanes were subjected to tandem mass spectrometry. A subset of proteins identified specifically associating with AF9 are listed to the right. B) Western blot confirmation of interactions identified by MS. The asterisk indicates the band corresponding to LAF4. Antibodies used for detection are indicated below blots. Input represents 1% of total lysate. C) Western blot for EAP in M1-AF9 and M1-control cells after FLAG IP with Benzonase treatment. D) Western blot after immunoprecipitations performed on 293 cells transfected with control, a FLAG-HA-tagged N-terminal MLL fragment encoding residues 1116–1397 of MLL (MLL1116–1397) with HA-Dot1L, or FLAG-MLL-AF9 with HA-tagged Dot1L expression vectors shows that that CDK9 and Dot1 specifically interact with MLL-AF9. Antibodies used for detection are indicated below the blots. HA detects HA-FLAG-MLL1116–1397 (second panel) and HA-Dot1L (third panel). The asterisk indicates the band corresponding to F-MLL-AF9. Approximate molecular weights are indicated to the right of the bands. Input represents 5% of total lysate.
To test whether the associations are preserved in the context of the MLL fusion protein, we performed immunoprecipitations in 293 transfectants (Fig. 1D). These experiments showed that, like MLL-ENL and MLL-AF4 [6, 7], MLL-AF9 retains the ability to associate with key EAP subunits, including pTEFb and Dot1L.
MLL fusion proteins increase the amount of pTEFb and Dot1 at target loci
A key question for understanding mechanisms of transformation by MLL fusion proteins is how the amount and distribution of EAP subunits at target loci differs between cells transformed by MLL fusion proteins versus those transformed by other mechanisms. To address this, we established leukemia cell lines by expression of MLL-ENL, MLL-AF9, or E2A-HLF in murine hematopoietic progenitor cells and analyzed EAP component expression levels and binding at Hoxa9. E2A-HLF transformed cells are not dependent on Hox expression for growth [13]. These cells express Hoxa9 at 5–10-fold lower levels than MLL fusion-transformed cell lines and have barely detectable expression of Meis1 (Fig. 2A). The cells express comparable levels of the EAP components Enl, Cyclin T2, and Dot1L while Af9 expression is lower in the MLL fusion protein transformed cells compared to the E2A-HLF transformed cells (Fig. 2A).
Fig. 2.
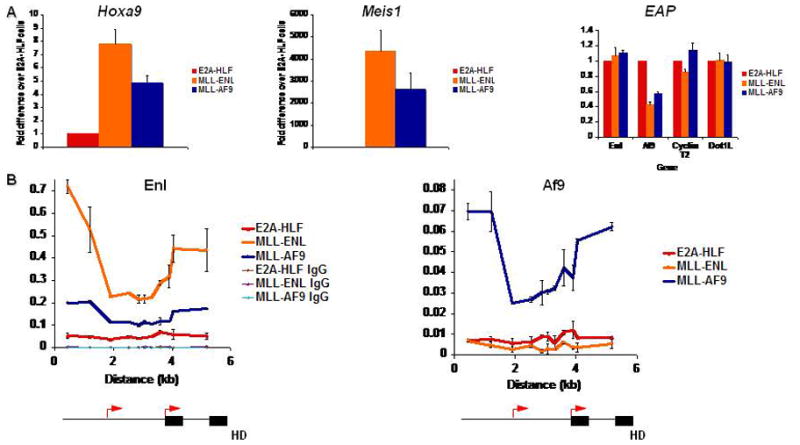
MLL fusion protein binding to target loci A) qPCR for Hoxa9, Meis1, and EAP expression in E2A-HLF, MLL-ENL, and MLL-AF9-transformed myeloblastic cell lines. Expression is normalized to GAPDH. B) ChIP-qPCR for MLL fusion partners across Hoxa9 in leukemic cell lines with or without MLL fusion proteins. Schematics of the Hoxa9 locus are depicted below the graphs. Black arrows indicate transcriptional start sites. Black boxes indicate exons.
ChIP-qPCR experiments on these cell lines using AF9 and ENL antibodies that recognize the endogenous proteins as well as the fusion proteins show a pattern of MLL-AF9 and MLL-ENL binding similar to that we previously reported for a conditional form of MLL-ENL [14] or for MLL-AF4 [15] (Fig. 2B), with peaks in both the promoter and coding region of Hoxa9 that contrast with the distribution of MLL, which is concentrated at the promoter in cells lacking MLL rearrangements [14]. While MLL-AF9 appears to recruit wild-type Enl, wild-type Af9 levels are minimal at target loci in cells transformed with MLL-ENL. This is possibly due to the lower levels of Af9 expression in the MLL fusion-transformed cell lines compared to the E2A-HLF-transformed cells.
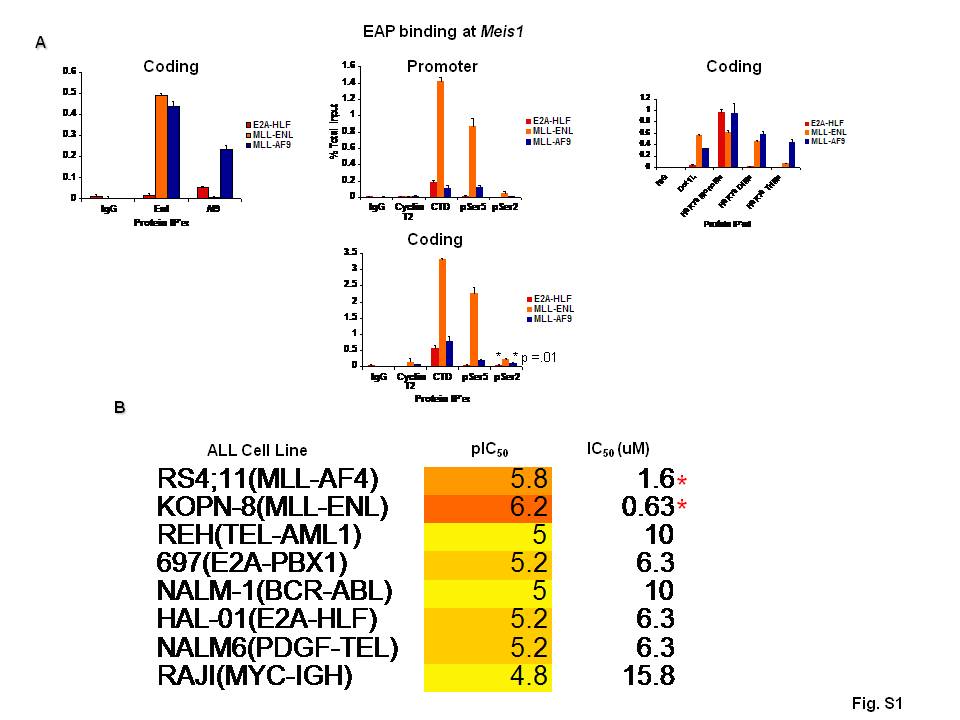
We then examined the distribution of pTEFb, which phosphorylates Serine 2 of the RNA polymerase II (Pol II) CTD as well as the DRB sensitivity inducing factor DSIF, thereby releasing RNA pol II from elongation inhibition [16]. One possibility is that MLL fusion proteins deregulate Hox expression through recruitment of excessive pTEFb to target loci thereby resulting in unregulated transcriptional elongation. In keeping with this, ChIP for the pTEFb component Cyclin T2 showed that more Cyclin T2 is localized to the coding regions of Hoxa9 and Meis1 in cells with MLL fusion proteins than in those lacking MLL rearrangements (Fig. 3A, S1A). In cells transformed by E2A-HLF, ChIP for Pol II showed that it is present at Hoxa9 and Meis1 and shows Serine 5 CTD phosphorylation, a modification associated with Pol II transcriptional initiation, similar to levels in MLL-AF9-transformed cells. However, cells expressing MLL fusion proteins show 2 to 10-fold more Serine 2 phosphorylated Pol II at the Hoxa9 and Meis1 loci (Fig. 3A, S1A).
Fig. 3.

MLL fusion proteins colocalize with elongating Pol II and are associated with elevated levels of H3K79 di- and tri-methylation A) ChIP-qPCR for Pol II-CTD and Ser2 and 5 phosphorylated CTD across Hoxa9 in leukemic cells lines transformed by MLL fusion proteins or E2A-HLF. Graph represents three independent experiments. The p value is shown for difference at the most 3’ site. Schematics of the Hoxa9 locus are depicted below the graphs in A and C. Red arrows indicate transcriptional start sites. Black boxes indicate exons. B) Cell viability and qPCR for gene expression after 24 hours of flavopiridol treatment. Data are normalized to U2 snRNA. C) ChIP-qPCR for Dot1L and H3K79 methylation across Hoxa9. D) qPCR for Hoxa9 and Meis1 in MV4:11 cells (express MLL-AF4) after knockdown with non-targeting control siRNA or siRNA targeting DOT1L.
These findings along with other published work showing the importance of the MLL fusion protein’s interaction with pTEFb through nuclear run-on, RNA tethering, and leukemogenesis assays [3, 6, 7] suggest that the kinase activity of pTEFb may be an effective therapeutic target in leukemias with MLL rearrangements. To assess whether cells transformed by MLL fusion proteins are differentially sensitive to CDK9 inhibition, we treated murine AML cell lines transformed by E2A-HLF, MLL-AF9, and MLL-ENL as well as a range of human ALL cell lines harboring different chromosomal translocations (RS4;11, KOPN-8, REH, 697, NALM-1, HAL-01, NALM6, RAJI) with the cyclin-dependent kinase inhibitor, flavopiridol, which has a Ki of 3 nM with CDK9 and weaker inhibition of other CDKs [17], and measured effects on cell viability and gene expression. Viability assays showed that MLL rearranged cell lines are modestly more sensitive to flavopiridol than those lacking MLL rearrangements (Fig. 3B, S1B). Flavopiridol treatment results in decreased Hoxa9 and Meis1 expression, but other constitutively expressed genes such as GAPDH and β-actin are also affected, a finding that is consistent with the general role of pTEFb in transcriptional regulation [18, 19]. It is also possible that the inhibition of other cyclin-dependent kinases may be leading to the toxicity and gene expression decreases observed in these assays.
ChIP studies performed for Dot1l showed that the enzyme is present at elevated levels at MLL fusion target loci in a pattern similar to the MLL fusion proteins (Fig. 3C, 1C). While mono-methylation of H3K79 is higher in the E2A-HLF cells, di- and tri-methylation of H3K79 is higher in cells transformed by MLL fusion proteins, showing a broad distribution across the locus. These findings are consistent with recent ChIP experiments performed on human ALL cell lines [15] and primary ALL blasts [20], which show that precursor B ALLs with MLL rearrangements show large blocks of histone H3 lysine 79 di-methylation at the HOXA9 locus compared to leukemic cells lacking MLL rearrangements. Elevated levels of Dot1L and its associated H3K79 di- and tri-methyl marks were also seen in the coding region of the Meis1 locus (Fig. S1A). Furthermore, knockdown of Dot1L in cells with MLL rearrangements show decreased Hoxa9 and Meis1 expression, demonstrating the importance of Dot1L to MLL fusion protein-mediated gene activation (Fig. 3D) [20].
EAP association with target loci is dynamic and is altered by recruitment via MLL fusion proteins
Previous studies in 293 cells, as well as the work reported here, indicate that the EAP complex regulates Hoxa9 and Meis1 transcription both in cells with and lacking MLL rearrangements [3]. Since the composition of the complex appears to be similar, the question arises how regulation of the complex is altered by MLL fusion proteins so that this does not allow for down-regulation of these targets. One attractive possibility is that through fusion with MLL, EAP association with target loci becomes dependent not only on interactions mediated by EAP subunits, but also on the amino terminal MLL sequences. The presence of a number of DNA binding motifs including the AT hooks, CXXC domain, as well as interactions with menin, could result in tighter association with target loci, making dissociation of the complex resistant to normal physiologic differentiation cues.
To explore this further, we first developed a model system in which we could study the association of EAP with target loci during the induction of myeloid differentiation. Myeloblastic M1 cells and our stably-transduced M1-AF9 cell line differentiate into macrophages in response to IL-6 [21, data not shown], with dramatic down-regulation of Hoxa9 evident after 24 hours of treatment (data not shown). Meis1 decreases are evident as early as 4 hours after treatment (Fig. S2A). This is accompanied by rapid dissociation of EAP subunits including Af9, Cyclin T2, and Dot1 from the Hoxa9 locus, which is also evident as early as 4 hours (Fig. S2B). Notably, with the exception of endogenous Af9, decreases in EAP expression are not detected until 8 hrs post-treatment (Fig. S2A), indicating that the dissociation precedes EAP down-regulation in differentiating cells. The MYC-AF9 is stably expressed by an MSCV promoter, and its expression is presumably independent of IL-6-mediated gene regulation. Af9 and Enl were the earliest to dissociate from the Meis1 locus (Fig. S2C), and it remains to be seen whether their dissociation precedes Meis1 down-regulation. Thus, there may be differences in regulation between Hoxa9 and Meis1 in this experimental model.
We then utilized the E2A-HLF, MLL-ENL, and MLL-AF9 transformed cell lines to test if EAP dissociation is altered by MLL fusion proteins. E2A-HLF transformed murine bone marrow cell lines are highly sensitive to induction of differentiation with lipopolysaccharide (LPS) (Fig. 4A, S3A), with dramatic downregulation of Hoxa9 (Meis1 is not detectable in these cells) observed after 24 hours of LPS treatment (Fig. 4B). In contrast, cells transformed by MLL-AF9 or MLL-ENL do not show appreciable Hoxa9 or Meis1 downregulation or differentiation (Fig. 4A, 4B, S3B). To show that LPS receptor signaling is intact in the MLL fusion transformed cell lines, we tested the expression of c-Jun, a target gene of LPS signaling [22]. Both MLL transformed cell lines showed upregulation of c-Jun, although the increase in MLL-AF9 transformed cells is relatively modest (Fig. S3B).
Fig. 4.

MLL fusion protein transformed cells are resistant to LPS-induced Hox downregulation and differentiation A) Wright-Giemsa staining on PBS or LPS-treated leukemia cell lines after 24 hours. B) Hoxa9 expression in PBS or LPS-treated cells lines after 24 hours C) ChIP-qPCR for EAP components across Hoxa9 in leukemia cell lines after 24 hours of LPS treatment. Schematic of Hoxa9 locus is below graphs.
One explanation for the observed difference in Hox expression is that the MLL fusion protein alters association of EAP with target loci. In support of this, ChIP shows that in E2A-HLF transformed cells, EAP dissociates from the Hoxa9 and Meis1 loci in response to LPS (Fig. 4C, S3C). In contrast, cells transformed by either MLL-AF9 or MLL-ENL show persistent association of EAP with the loci after LPS treatment. We also detected decreases in total Pol II binding at Hoxa9 and Meis1 in terminally differentiated E2A-HLF cells, while Pol II levels at these loci in MLL fusion-transformed cells were unchanged or elevated (Fig. 4C).
Discussion
Deregulation of the A cluster HOX genes and MEIS1 is emerging as a very common mechanism of transformation in acute leukemia [1]. It is also becoming apparent that controlling transcriptional elongation plays a central role in regulating transcription at these loci. The Drosophila Hox genes Ultrabithorax and Abdominal-B, for example, have Pol II associated with their proximal promoters, even though they are not expressed [23]. Similarly, Pol II is also present at silenced genes in mammalian cells [24]. Furthermore, mutations in elongation factors elongin A or Cdk9 result in homeotic defects associated with Pol II stalling [25].
Just as transcriptional elongation is regulated in the developing embryo, deregulation of transcriptional elongation is emerging as an important mechanism in growth regulation and oncogenesis. For example, high levels of CDK9 expression are associated with a variety of solid tumors including neuroblastoma, primitive neuroectodermal tumors, prostate cancer and a wide range of lymphomas [26]. The mechanisms regulating CDK9 association with promoters are incompletely defined. In some cases, recruitment appears to mediated by the bromodomain containing-protein BRD4, while in other cases, specific transcription factors such as MYC recruit pTEFb directly [27]. From work presented here and in previous experiments [2, 3], it now is clear that another recruitment mechanism, particularly important for Hox gene regulation, involves interactions with the EAP complex.
The evidence that deregulated recruitment of DOT1L plays a role in oncogenesis is also increasing rapidly. One of the first connections was that DOT1L interacts with the MLL translocation partner AF10 and that its methyltransferase activity is important for MLL fusion protein mediated transformation [4]. Subsequent studies and the work reported here implicate DOT1L in transformation by other MLL fusion proteins (MLL-AF4, MLL-ENL, MLL-AF9) as well as by the CALM-AF10 translocation in AML and SET-NUP214 in T-ALL [3, 20, 28]. Our initial studies with Dot1L conditional knockout mice show that transformation by MLL-AF9 is absolutely dependent on Dot1L, while E2A-HLF is not (Jo et al. in preparation), suggesting that loss of Dot1L function is not generally toxic to myeloid cells.
By association with the EAP complex, both CDK9 and DOT1L would be coordinately recruited to target promoters such as the HOX loci. While many mechanisms are possible, one possibility is that EAP is recruited via the YEATs domains in AF9 or ENL, which has been found to bind histones H1 and H3 [29]. The central role of AF9 and ENL in transcriptional regulation of the HOX loci is highlighted by the histone H3 lysine 79 demethylation and loss of HOX transcription with AF9 and ENL knockdown [3] and by the finding that Af9 knockout mice show homeotic transformations and posterior shifts in Hox gene expression [30].
Importantly, our experiments show that EAP association with target loci is dynamic and under active regulation, as the complex rapidly dissociates from the Hox and Meis loci within 4 hours after treatment with differentiation-inducing agents. Defining how these cell signaling pathways impinge upon EAP’s association mechanism, possibly through post-translational modifications, will be an important topic for future investigation. It is noteworthy in this regard that both Lilliputian, the Drosophila homolog of AF4/FMR2, and Dot1 appear to be downstream effectors of the RAS signaling pathway [31].
A number of possible models can be imagined for how the EAP complex results in persistent high-level Hox transcription in cells with MLL fusion proteins. The finding of increased levels of EAP subunits in the presence of MLL fusion proteins and the lack of silencing of the Hox and Meis loci in these cells in response to induction of differentiation suggests the model shown in Fig. 5. In this model, fusion of EAP subunits to N-terminal MLL sequences, as occurs in mixed lineage leukemia, alters association of the EAP complex so that levels of the complex are increased and dissociation is impaired. This results in persistent Hox and Meis gene expression in spite of differentiation signaling cues. It is likely that other mechanisms of EAP deregulation may be observed in leukemias with high level Hox gene expression including over expression of individual subunits, mutation of regulatory phosphorylation sites, or constitutive activation of upstream signaling pathways that impinge upon EAP activity.
Fig. 5.
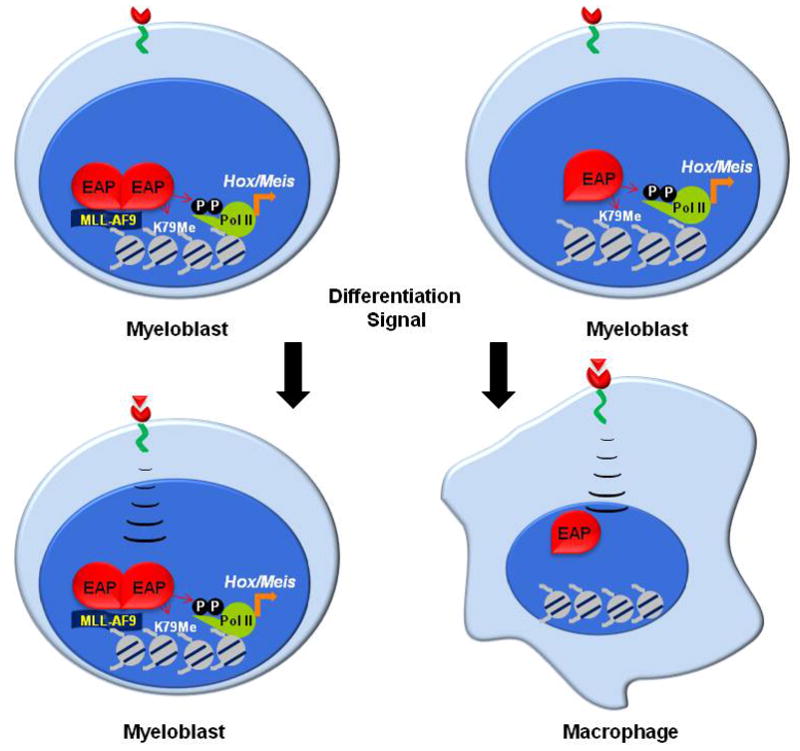
Model for EAP regulation of Hox genes in hematopoietic cells with and lacking MLL rearrangements. In cells lacking MLL fusion proteins (left), the EAP complex is dynamically associated with the Hox locus and readily dissociates on induction of differentiation. In cells transformed by an MLL fusion protein (right), the fusion protein inhibits Hox downregulation and differentiation by preventing EAP dissociation from genes critical for leukemia.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Gregory Dressler for critical review of the manuscript and Martha Larson at the University of Michigan Center for Chemical Genomics for assistance with screening human leukemia cell lines for drug sensitivity. This work was supported by NIH grant RO1 CA116570 and a Specialized Center for Research (SCOR) Grant from the Lymphoma and Leukemia Society of America. Stephanie Jo is supported by NIH Ruth H. Kirchenstein National Pre-doctoral Research Fellowship F30 HL095280. The flavopiridol was generously provided by Sanofi–Aventis and the National Cancer Institute, NIH.
Footnotes
Conflict of Interest
No financial interest/relationships with financial interest relating to the topic of this article have been declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sitwala KV, Dandekar MN, Hess JL. HOX Proteins and Leukemia. Int J Clin Exp Pathol. 2008;1:461–474. [PMC free article] [PubMed] [Google Scholar]
- 2.Bitoun E, Oliver PL, Davies KE. The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum Mol Genet. 2007;16:92–106. doi: 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- 3.Mueller D, Bach C, Zeisig D, et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood. 2007;110:4445–4454. doi: 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Estable MC, Naghavi MH, Kato H, et al. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J Biomed Sci. 2002;9:234–245. doi: 10.1007/BF02256070. [DOI] [PubMed] [Google Scholar]
- 6.Mueller D, Garcia-Cuellar MP, Bach C, Buhl S, Maethner E, Slany RK. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009;7:e1000249. doi: 10.1371/journal.pbio.1000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yokoyama A, Lin M, Naresh A, Kitabayashi I, Cleary ML. A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell. 17:198–212. doi: 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000;7:1063–1066. doi: 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 9.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 10.Milne TA, Zhao K, Hess JL. Chromatin immunoprecipitation (ChIP) for analysis of histone modifications and chromatin-associated proteins. Methods Mol Biol. 2009;538:409–423. doi: 10.1007/978-1-59745-418-6_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medlin J, Scurry A, Taylor A, Zhang F, Peterlin BM, Murphy S. P-TEFb is not an essential elongation factor for the intronless human U2 snRNA and histone H2b genes. EMBO J. 2005;24:4154–4165. doi: 10.1038/sj.emboj.7600876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 13.Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003;17:2298–2307. doi: 10.1101/gad.1111603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milne TA, Martin ME, Brock HW, Slany RK, Hess JL. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005;65:11367–11374. doi: 10.1158/0008-5472.CAN-05-1041. [DOI] [PubMed] [Google Scholar]
- 15.Guenther MG, Lawton LN, Rozovskaia T, et al. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008;22:3403–3408. doi: 10.1101/gad.1741408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, Handa H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell. 2006;21:227–237. doi: 10.1016/j.molcel.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 17.Chao SH, Fujinaga K, Marion JE, et al. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem. 2000;275:28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- 18.Lam LT, Pickeral OK, Peng AC, et al. Genomic-scale measurement of mRNA turnover and the mechanisms of action of the anti-cancer drug flavopiridol. Genome Biol. 2001;2:RESEARCH0041. doi: 10.1186/gb-2001-2-10-research0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu X, Burgan WE, Cerra MA, et al. Transcriptional signature of flavopiridol-induced tumor cell death. Mol Cancer Ther. 2004;3:861–872. [PubMed] [Google Scholar]
- 20.Krivtsov AV, Feng Z, Lemieux ME, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–368. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu CP, Lee F. IL-6 is a differentiation factor for M1 and WEHI-3B myeloid leukemic cells. J Immunol. 1989;142:1909–1915. [PubMed] [Google Scholar]
- 22.Kurata S, Matsumoto M, Tsuji Y, Nakajima H. Lipopolysaccharide activates transcription of the heme oxygenase gene in mouse M1 cells through oxidative activation of nuclear factor kappa B. Eur J Biochem. 1996;239:566–571. doi: 10.1111/j.1432-1033.1996.0566u.x. [DOI] [PubMed] [Google Scholar]
- 23.Zeitlinger J, Stark A, Kellis M, et al. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet. 2007;39:1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Margaritis T, Holstege FC. Poised RNA polymerase II gives pause for thought. Cell. 2008;133:581–584. doi: 10.1016/j.cell.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 25.Chopra VS, Hong JW, Levine M. Regulation of Hox gene activity by transcriptional elongation in Drosophila. Curr Biol. 2009;19:688–693. doi: 10.1016/j.cub.2009.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romano G, Giordano A. Role of the cyclin-dependent kinase 9-related pathway in mammalian gene expression and human diseases. Cell Cycle. 2008;7:3664–3668. doi: 10.4161/cc.7.23.7122. [DOI] [PubMed] [Google Scholar]
- 27.Bres V, Yoh SM, Jones KA. The multi-tasking P-TEFb complex. Curr Opin Cell Biol. 2008;20:334–340. doi: 10.1016/j.ceb.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Vlierberghe P, van Grotel M, Tchinda J, et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood. 2008;111:4668–4680. doi: 10.1182/blood-2007-09-111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeisig DT, Bittner CB, Zeisig BB, Garcia-Cuellar MP, Hess JL, Slany RK. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene. 2005;24:5525–5532. doi: 10.1038/sj.onc.1208699. [DOI] [PubMed] [Google Scholar]
- 30.Collins EC, Appert A, Ariza-McNaughton L, Pannell R, Yamada Y, Rabbitts TH. Mouse Af9 is a controller of embryo patterning, like Mll, whose human homologue fuses with Af9 after chromosomal translocation in leukemia. Mol Cell Biol. 2002;22:7313–7324. doi: 10.1128/MCB.22.20.7313-7324.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittwer F, van der Straten A, Keleman K, Dickson BJ, Hafen E. Lilliputian: an AF4/FMR2-related protein that controls cell identity and cell growth. Development. 2001;128:791–800. doi: 10.1242/dev.128.5.791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.