

The main responsibility of any drug regulatory authority is to ensure the quality, efficacy, and safety of all marketed products. The first two criteria can be established through data obtained from in vitro testing to ensure compliance with acceptable standards and data obtained from animal studies, preclinical and clinical trials involving humans. It is a well-established fact that pre-marketing clinical trials do not have the statistical power to detect rare adverse drug reactions (ADRs) nor do they have significant follow-up to identify delayed ADRs or effects from long-term exposure. In view of this, pharmacovigilance plays a prominent role in establishing the safety profile of marketed drugs. Originally a modest appendix of drug regulation; it has become a major activity now.
Definitions
“Adverse drug reaction” or an “adverse reaction” means a response to a medicine in the humans or animals, which is noxious and unintended, including lack of efficacy, and which occurs at any dosage and can also result from an overdose, misuse or abuse of a medicine.
“Pharmacovigilance”: As per World Health Organization (WHO), “Pharmacovigilance is the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other drug related problems”. Unfortunately, when this term is mentioned, it is very much a case of “Pharmaco what?” There is still a lack of understanding on this topic like how it functions, what are the benefits of sharing ADR knowledge and its purpose and importance.
On the other hand, the term “post-marketing surveillance (PMS) study” implies a scientifically rigorous study of a product that is approved for registration in a particular country, designed to produce reliable information about drug safety. It is not appropriate to apply the term to clinical trials of registered products or to studies designed primarily for marketing purposes regardless of the scientific validity of the study design.
Post-marketing surveillance studies are generally performed on the initiative of the sponsoring company, but may be suggested or requested by other parties. They should generally be designed to address a specific drug safety question or hypothesis (the latter often identified initially by voluntary reporting).
ADR Monitoring Systems: Structural and Functional Aspects
The pharmacovigilance concept rests on three pillars:
Collecting new information from reliable scientific resources such as marketing authorization holders, healthcare professionals, consumers, international/public bodies, journals, published and updated literature, etc.
Classifying and analyzing the above information.
Circulating its contents as well as any action taken on specific drug to all health sectors [Figure 1].
Figure 1.

ADR monitoring system
Four Elements of ADR Reporting
Any ADR report should have the following four main elements:
Patient
A drug
An adverse reaction
Composer/reporter of the report
Past Experiences
In the past, it was only the developed countries that carried out Research and Development (R&D) activities and new drug launches. The developing countries had a passive “wait and watch” kind of attitude. But now with improving facilities and economics, many of the developed countries are outsourcing their activities, especially those related to clinical trials to these destinations. Hence, now new drugs are almost launched simultaneously here. This changing scenario provides the impetus for more active, vigilant and efficient pharmacovigilance guidelines and control. Pharmacovigilance plays a major role in pharmacotherapeutic decision making, be it individual, regional, national or international. In addition, pharmacovigilance is becoming a scientific discipline in its own right.
Issues and Challenges Faced by Developing Countries
There is a long list of challenges faced by the developing countries. These are as follows:
As most drugs are developed in the West, most of the efficacy data are based on the Caucasians, with little or no information available on the Asians.
There is no data on ADRs that may occur due to the interaction between the established medicines and traditional, herbal medicines and vaccines used locally.
ADR reporting for traditional and herbal medicines, especially the multicomponent and adulterated ones is not done seriously and reports present are negligible.
Attrition from local manufacturers who usually manufacture generics in carrying the burden of pharmacovigilance when innovator products are taken off the market for economic reasons. In addition, most of them do not care to invest in ADR monitoring, as they believe they deal with off-patent products. But ADRs associated with generics although less in frequency, can still arise.
It may require structural and policy changes within the drug regulatory authorities.
Moreover, a lot of sensitization and setting up of new systems will consume resources such as good labs, patient′s ability to pay for tests, infrastructure for proper causality assessment, identification of funding agency and certainty of funding, etc.
Success Factors
The success of any pharmacovigilance system depends upon the following factors:
Public awareness on need to report suspected ADRs.
Government support and well-defined policies with proper financial assistance.
Presence of national coordinator and an advisory committee.
Trained healthcare workers.
Quality control of laboratories.
Free and open communication between public and the policy makers.
Ability to have free flow of information, i.e. inquiries, feedback, etc.
Systems Prevalent in the Selected Countries
All the countries except Kenya and Turkey have a pharmacovigilance center or a unit responsible for carrying out the activities related to ADR monitoring, pharmacovigilance and PMS studies.
These units are mainly concerned with the collection of spontaneous and suspected ADR reports that have to be submitted on expedite basis. All other reports like non-serious ADRs, etc. need not be submitted on an expedite basis but should be properly archived and made available as and when requested by the regulatory authority.
All these reports are then sent to the WHO ADR Monitoring Centre at Uppsala, Sweden, which helps to update the world pool of information on this topic.
The status of ADR monitoring and pharmacovigilance in the selected 10 countries can be highlighted through Table 1.
Table 1.
Status of pharmacovigilance initiatives
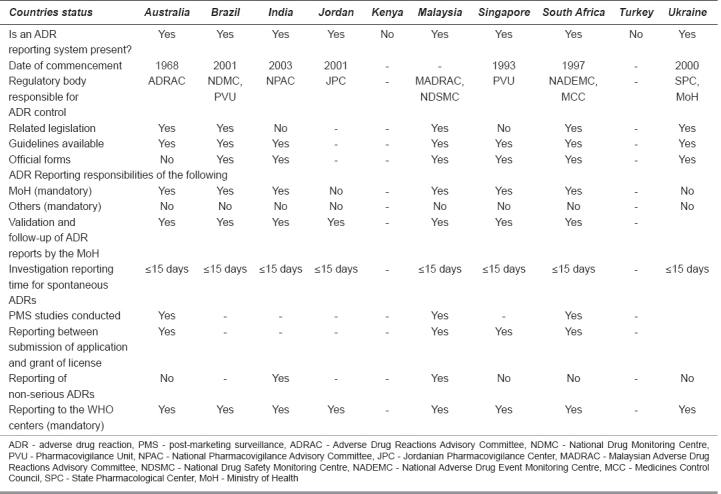
ADR Monitoring and Pharmacovigilance Activities in Selected Countries: General Notes
Australia
Consistent with the legislation, the therapeutic goods administration (TGA)[1] has established a pharmacovigilance system for the collection and evaluation of information relevant to the benefit to risk balance of registered medicinal products. The TGA continually monitors the safety profile of the products available in Australia and takes appropriate action where necessary. This is done through the Adverse Drug Reactions Advisory Committee (ADRAC). The guidelines provided cover the topic of adverse reaction reporting and reporting requirements in special situations.
The sponsor should report suspected adverse reactions to TGA in an expedited manner in accordance with the following:
Serious unexpected and serious expected reactions occurring in Australia must be reported on an expedited basis;
Sponsors are not required to submit on expedited basis, reports of foreign (i.e. not occurring in Australia) serious unexpected or serious expected adverse medicine reaction reports. Sponsors are required instead to advise the TGA within 72 h of any;
Significant safety issue identified by the sponsor as a result of its ongoing review and analysis of all information (including foreign reports of ADRs) that is pertinent to the safety or benefit-risk assessment of the product; or
Action, which has been taken by a foreign regulatory agency, including the basis for such action.
The 72-h clock starts from the time of awareness of any personnel of the sponsor. This is considered to have occurred where the sponsor′s review and analysis have been completed and a conclusion is drawn that a significant safety issue exists, or when the sponsor becomes aware of the actions of an overseas regulatory agency.
In the period between the submission of a registration application and prior to registration, routine single case expedited reporting is not required except according to the separate guidelines where the product is being used in Australia in a clinical trial. However, in the pre-registration period, information that impacts on the benefit/risk evaluation may become available from the applicant or countries where the medicine is already in use on a compassionate basis, or from countries where the medicine is marketed. Australian Drug Evaluation Committee (ADEC) sponsors are required to submit with their pre-ADEC response a tabulation of any serious unexpected ADRs that are not mentioned in the proposed Australian Product Information and have not been submitted previously.
Also cases related to ADRs and pregnancy together with other reports of abnormalities in pregnancy should also be available on request and be included in the next Periodic Safety Update Report (PSUR) together with aggregated data of overall exposure and details of normal/abnormal outcomes. If, in the period between PSURs, a sponsor becomes aware of a signal of a possible teratogenic effect (e.g. a cluster of similar abnormal outcomes), the TGA should be informed immediately.
Brazil
The National System of Pharmacovigilance, managed by the Pharmacovigilance Unit (PVU), is a part of the Health Products Post-Marketing Surveillance Department (HPPMSD). The most recent tragedy that occurred in Brazil was in 2000, and it concerned the use of meglumine antimonite. Over 300 patients had serious lesions caused by the injections. This fact reinforced the initiative of developing drug-monitoring processes in Brazil. Decree 696 created the Brazilian National Drug Monitoring Centre (NDMC) in 2001 by the Ministry of Health (MoH), and takes place in the PVU to obtain legal background to expand its field of action. Brazil was admitted, in 2001, as the 62ndcountry in the WHO International Drug Monitoring Program coordinated by the Uppsala Monitoring Centre, in Sweden, a WHO collaborative center. The PVU is responsible for planning, coordinating and supervising the formulation and implementation of the guidelines and operational technical norms on the safe use and surveillance of drugs.
The main objectives of this unit are:
Spontaneous reporting of ADRs.
Establishment of a network of 100 sentinel hospitals.
Establishment of regional centers.
Creation of a net of sentinel pharmacies.
Along with the Department of Inspection of the National Health Surveillance Agency,[2] the PVU monitors the processes of mandatory and voluntary drug recalls in Brazil through the international communication network.
Rational drug use has been the object of training and multiple courses sponsored by the National Health Surveillance Agency, with the objective of enhancing health professionals' capability to prescribe in a rational basis, to identify drug-related problems, and to notify ADRs to the National System of Pharmacovigilance. The PVU made three National Pharmacovigilance Alerts in 2000, 12 in 2001, and 12 in 2002. It translated 90 International Pharmacovigilance Alert versions and published seven Technical Bulletins in 2001 and 10 in 2002.
India
The National Pharmacovigilance Advisory Committee (NPAC) monitors the performance of various zonal, regional, and peripheral centers and performs the functions of “Review Committee” for this program. The NPAC also recommends possible regulatory measures based on pharmacovigilance data received from various centers. The Zonal Pharmacovigilance Centre (ZPC) and Regional Pharmacovigilance Centre (RPC) have also been established.
The Central Drugs Standard Control Organization (CDSCO)[3] is initiating a countrywide pharmacovigilance program under the aegis of DGHS, MoH and Family Welfare, and Government of India. The National Pharmacovigilance Centre at CDSCO shall coordinate the program. The National Centre will operate under the supervision of the NPAC to recommend procedures and guidelines for regulatory interventions.
The National Pharmacovigilance Program will have the following milestones:
Short-term objectives: To foster a culture of notification.
Medium-term objectives: To engage several healthcare professionals and Non-Government Organizations (NGOs) in the drug monitoring and information dissemination processes.
Long-term objectives: To achieve such operational efficiencies that would make Indian National Pharmacovigilance Program a benchmark for global drug monitoring endeavors.
Periodic Safety Update Reports shall be expected to be submitted every 6 monthly for the first 2 years of marketing in India, and annually for the subsequent 2 years. In addition, training programs and interaction meetings shall be held every 6 months after the initial training [Figure 2].
Figure 2.
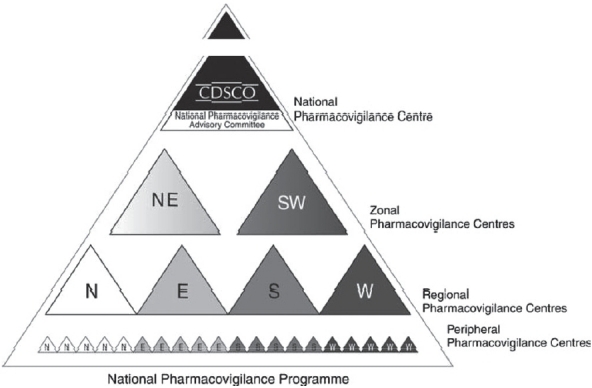
Hierarchical structure of the proposed centers in India
All data generated (including reporting forms) will be stored and preserved for the purpose of archiving for a minimum period of 5 years at the ZPCs. The reporting of seemingly insignificant or common adverse reactions would be important because it may highlight a widespread prescribing problem.
Jordan
The Jordanian Pharmacovigilance Center (JPC) was established in January 2001 in cooperation with Sweden International Development Agency (SIDA) and the Higher Council for Science and Technology.
The main task is to:
enable the technical committee for drug registration at the Ministry of Health/Drug Directorate[4] to take the appropriate decision toward drugs registered and applied for registration under the light of information acquired from JPC;
provide a database for medical sector on drug safety matters, which help enabling physicians to take the proper treatment decision for his patient;
check the existence of pharmacovigilance centers for drugs used in clinical experiments on humans to encourage drug companies to clinically test their drugs in Jordan through research centers, which acts as a promotion for foreign investments; and
enable Jordanian drug manufacturers to export their pharmaceutical products to countries stipulating the existence of pharmacovigilance center in the exporting country.
Kenya
Kenya[5] does not have any such system at present, but a protocol and a pilot project has been prepared to set up a unit on similar lines.
Malaysia[6,7]
The Malaysian Adverse Drug Reactions Advisory Committee (MADRAC), a sub-committee of the Drug Control Authority (DCA), reviews Malaysian reports of suspected drug reactions. The National Drug Safety Monitoring Centre (NDSMC), which is the secretariat to MADRAC, was accepted as the 30thmember of the WHO safety monitoring centers in 1990.
The MADRAC encourages healthcare professionals to report all suspected adverse reactions but it is a compulsory requirement that the marketing authorization holder of a product should inform the DCA of any adverse reactions to the product in accordance to the Malaysian guidelines for ADR monitoring.
Wholesalers and Importers
Licensed wholesalers and importers in Malaysia should maintain records of every transaction involving a registered pharmaceutical product. The records should be kept for at least 5 years from the date of the transaction. Licensed wholesalers are responsible for keeping records with the following information: (1) date of sale, (2) name and address of the purchaser, (3) name and quantity of the product sold, (4) product registration number, and (5) number of the invoice or delivery order.
Likewise, importers in Malaysia are responsible for keeping records with the following: (1) date of importation, (2) name and address of the supplier, (3) name and quantity of the imported product, (4) number of the bill of lading, (5) date of the sale, and (6) name and address of the purchaser.
Important: The registration holder is also obliged to submit a “NULL” report at 6-monthly intervals for the first 2 years should there be no ADR reports submitted to them. Registration holders who have registered a product containing a new chemical entity after 1 January 2002 must routinely submit PSURs on that product 6 monthly for the first 2 years after approval in Malaysia and annually for the subsequent 3 years.
Singapore
The PVU (formerly known as the Adverse Drug Reaction Monitoring Unit, ADRMU) in Singapore was established in 1993. The unit joined the WHO in 1994 as the 40thmember of the WHO International Drug Monitoring Program for international collaboration on drug safety. The unit serves as the national center for the collation and review of ADR reports. The Health Sciences Authority[8] has also appointed a Pharmacovigilance Advisory Committee (PVAC), which comprises experts in the fields of medicine, pharmacy, pharmacology, and forensic sciences. One of their main roles is to assess the impact of major drug safety issues and advise on the appropriate regulatory actions to be taken to enhance drug safety.
The license holder can submit the ADR report to the PVU, using the reporting form prescribed by the unit or the Council for International Organizations of Medical Sciences (CIOMS) I form. The ADR reporting form can be easily downloaded. PSURs may be requested for selected registered medicinal products. PSURs are required to be submitted to the Product Evaluation and Registration Branch (PERB), 6 monthly for the first 2 years after marketing approval, after which they are to be submitted on a yearly basis for the subsequent 3 years. This timeframe may be varied to harmonize periodic safety updates internationally.
A CIOMS-I form is an adverse reaction reporting form developed by the Council for International Organizations of Medical Sciences (CIOMS), intended for notifying the regulatory authorities of countries other than the country where the report originated.
South Africa
Legislation responsible for carrying out these activities is Regulations 34 and 37 of Act 90 of 1997. For the purpose of pharmacovigilance control, the authorities responsible are the Medicines Control Council (MCC)[9,10] and the National Adverse Drug Event Monitoring Centre (NADEMC).
After initial receipt of an adverse reaction report, a notice of acknowledgment is sent to the applicant quoting the number assigned to the case report. Any follow-up correspondence from the applicant, relating to the same case report, should be cross-referenced to the assigned database number or to this unique number assigned to the applicant. This is the only reliable way to minimize the duplication of reports submitted by the applicant. Moreover, the applicant should ensure that an internal pharmacovigilance system exists with in the company.
Turkey
No provision for such a control by any unit but MoH[11] encourages marketing authorization holders and the healthcare professionals to report ADR events.
Ukraine
The present instructions were developed according to the Law of Ukraine “On Medicinal Products”. Surveillance over adverse reactions/effects of medicinal products is monitored by the State Pharmacological Center MoH[12,13] Ukraine (hereinafter - the Center).
Regulatory Action Taken
Regulatory authorities can take a wide range of actions in response to a reported ADR. These range from a change in PI to stricter actions like product recalls. Except for Kenya and Turkey, similar actions taken by the regulatory authorities of other countries are tabulated in Table 2.
Table 2.
Regulatory actions taken by the authorities

Information Dissemination
As has been discussed before, an important task of ADR monitoring and pharmacovigilance planning is to collect new information from reliable scientific resources such as marketing authorization holders, healthcare professionals, consumers, international/public bodies, journals, published and updated literature, etc. followed by classifying and analyzing this information and last but not the least circulating its contents as well as any action taken on specific drug to all health sectors. It is this last provision, which provides important impetus to healthcare professionals and general public.
The same has been tabulated in Table 3.
Table 3.
Information dissemination

Recommendations and Suggestions
Consolidating ADR monitoring and pharmacovigilance practices
Pharmacovigilance is a broad concept, and includes the re-evaluation of marketed drugs, risk management, communicating drug information, promoting rational drug use, and crisis preparedness. It is becoming increasingly important to provide training in all of these activity areas and to carry out intensive monitoring of new drugs to evaluate the risk/benefit.
There should be an involvement of all categories of healthcare professionals in ADR and pharmacovigilance planning to incorporate the sense of ownership.
It should be ensured that the reporting forms are always available. An acknowledgment receipt should always be issued along with a reference number for all further correspondence to avoid any duplication of reports. There should be timely and easy feedback for all the cases reported.
All ADRs should be published in journals, lay press, etc. to keep people as well as healthcare professionals informed. ADR monitoring and reporting issues should be introduced in school and college curricula of academic institutions. Also ADR monitoring should be combined with the public health programs e.g. acquired immune deficiency syndrome (AIDS) program, malaria control, etc.
Interaction with pharmacovigilance centers in other countries and with Uppsala monitoring center, Sweden should be encouraged.
Increasingly, certain medicines are approved based on special conditions such as finalization and reporting of phase IV studies. National regulatory authorities should collaborate on harmonizing the terms of conditional approval, and develop systems to allow sharing of information on medicines in this category.
Obligations to conduct PMS as a public health protection measure should lie with sponsors and donors, as appropriate. International agencies and aid programs should make every effort to comply with these requirements and provide the necessary data.
Regulatory authorities should be encouraged to establish databases of clinical information suitable for epidemiological studies to examine and quantify signals of possible emerging risk. The regulatory authorities should provide, upon request, technical advice and support to member states on the appropriateness of PMS plans submitted by sponsors when a medicine is being introduced to manage a specific public health campaign in that country.
The regulatory authorities should investigate the feasibility and potential utility of creating a database of “recommendations for action” arising from evaluations made by national regulatory authorities of the PSURs to improve the usefulness of such information by making this generally available.
References
- 1.Therapeutic Goods Administration. Available from: http://www.tga.gov.au.
- 2.National Health Surveillance Agency. Available from: Available from: http://www.anvisa.gov.br.
- 3.Central Drugs Standard Control Organization. Available from: http://www.cdsco.gov.in.
- 4.Drug Directorate, Ministry of Health, The Hashemite Kingdom of Jordan. Available from: http://www.drugdirect.gov.jo.
- 5.Pharmacy and Poisons Board Kenya. Available from: http://www.pharmacyboardkenya.org.
- 6.National Pharmaceutical Control Bureau. Available from: http://www.bpfk.gov.my.
- 7.Pharmaceutical Services Division. Available from: http://www.pharmacy.gov.my.
- 8.Health Sciences Authority. Available from: http://www.hsa.gov.sg.
- 9.Medicines Control Council. Available from: http://www.tga.gov.au.
- 10.Advertising Standards Authority of South Africa (ASA) Available from: http://www.asasa.org.za/Default.aspx?mnu_id = 9 .
- 11.Ministry of Health. Available from: http://www.saglik.gov.tr .
- 12.Ministry of Health of Ukraine. Available from: http://http://www.moz.gov.ua .
- 13.State Pharmacological Expert Center. Available from: http://www.pharma-center.kiev.ua .