

Abstract
Macromolecular Gd(III) complexes are advantageous over small molecular Gd(III) complexes in contrast enhanced magnetic resonance imaging (MRI) because of their prolonged blood circulation and preferential tumor accumulation. However, macromolecular contrast agents have not been approved for clinical applications because of the safety concerns related to their slow body excretion. Polydisulfide Gd(III) complexes have been designed and developed as biodegradable macromolecular MRI contrast agents to alleviate the concerns by facilitating the clearance of Gd(III) complexes from the body. These agents initially behave as macromolecular agents and result in superior contrast enhancement in the vasculature and tumor tissues. They can then be readily degraded in vivo into small molecular chelates that can rapidly excrete from the body via renal filtration after the MRI examinations. Various polydisulfide Gd(III) complexes have been prepared as biodegradable macromolecular MRI contrast agents. These agents have resulted in strong contrast enhancement in the vasculature and tumor tissue in animal models with minimal long-term tissue accumulation comparable to small molecular contrast agents. Polydisulfide Gd(III) complexes are promising for further clinical development as safe and effective biodegradable macromolecular MRI contrast agents for cardiovascular and cancer imaging. The review summarizes the chemistry and properties of polydisulfide Gd(III) complexes.
Keywords: polydisulfide, biodegradable macromolecular contrast agents, MRI, gadolinium
Introduction
Magnetic resonance imaging (MRI) is a non-ionization imaging modality based on the longitudinal or transverse relaxation rates (1/T1 or 1/T2) of water protons [1]. MRI provides anatomical images of soft tissues with high spatial resolution [1, 2]. In many cases, contrast agents are used to enhance the image contrast between normal and diseased tissues for accurate diagnosis and to study physiological properties of diseased tissues. MRI contrast agents are mainly paramagnetic metal chelates, including Gd(III), Fe(III) and Mn(II) complexes, and superparamagnetic nanoparticles. These paramagnetic agents increase relaxation rates of the surrounding water protons, resulting in enhanced signal contrast [1]. The MRI contrast agents approved for clinical applications are mostly Gd(III)-based low molecular weight chelates with high stability, including Gd-DTPA, Gd-DOTA and their derivatives [2, 3].
Macromolecular Gd(III) complexes have been designed and developed to further improve in vivo properties of small molecular MRI contrast agents. Macromolecular MRI contrast agents are prepared by conjugating Gd(III) chelates of DTPA, DOTA or their derivatives to biocompatible synthetic polymers [4–8], natural macromolecules [9–11], and colloids [12], or by copolymerizing low molecular weight contrast agents [5, 13, 14]. Macromolecular contrast agents have shown prolonged plasma circulation and can preferentially accumulate in solid tumor due to the hyperpermeability of tumor vasculature [15, 16]. As a result, they provide superior contrast enhancement in the blood pool and in solid tumor tissue in preclinical studies [17].
Although macromolecular Gd(III) complexes have demonstrated superior efficacy in cardiovascular and cancer MR imaging in animal models, clinical development of macromolecular MRI contrast agents is limited because of safety concerns related to their slow clearance from the body. Free Gd(III) ions are highly toxic with an LD50 as low as 0.5 mmol/kg in rats [3]. The clearance rate decreases with the increase of molecular weight of the contrast agents [18]. The long-term tissue accumulation of macromolecular Gd(III) chelates may result in release of toxic Gd(III) ions [19]. For example, Gd(III) chelate conjugates of carboxymethylhydroxyethylstarch and second generation polypropyleneimine dendrimer conjugates had 47% and 45% retention in rats after 7 and 14 days, respectively [20, 21]. Currently, no macromolecular MRI contrast agent is approved for clinical applications.
To alleviate the safety concerns about macromolecular contrast agents, we have recently designed and developed novel extracellular biodegradable macromolecular Gd(III) complexes based on polydisulfides to facilitate Gd(III) elimination from the body [22]. These polymeric complexes initially act as macromolecular agents for MR contrast enhancement. The disulfide bonds in the polymer chains are then gradually reduced by the endogenous thiols to give small molecular Gd(III) complexes that can readily be eliminated via renal filtration. Here, we summarize the design, synthesis, physicochemical and biological properties and in vivo contrast enhanced MRI of the polydisulfide biodegradable macromolecular contrast agents.
Design of polydisulfide macromolecular MRI contrast agents
MRI contrast agents are mostly extracellular agents and have low cellular uptake. The biodegradable macromolecular contrast agents should be readily degraded into small Gd(III) chelates in plasma. Biodegradable macromolecular Gd(III) complexes have been designed by incorporating disulfide bonds into the backbone of Gd(III) chelates containing polymers. Disulfide-thiol exchange reaction is a reversible reaction and commonly present in biological systems. The endogenous thiols in the plasma can reduce the disulfide bonds in the backbone of the polydisulfides to form small Gd(III) chelates. The concentration of endogenous thiols, including cysteine, glutathione (GSH), cysteinylglycine (Cys-Gly) and homocysteine, is approximately 15 μM in human blood plasma and about 10 mM in the cytoplasm [23, 24]. Since the concentration of free thiols in human blood is relatively low (ca. 15 μM), the degradation reaction of the macromolecules will be a slow and gradual process, and it can provide an acceptable time window for effective contrast enhanced MRI.
Figure 1 shows the general structure of polydisulfide Gd(III) complexes. Functional groups can be introduced around the disulfide bonds to modify the steric hindrance around the disulfide bonds and to control the rate of disulfide-thiol exchange reaction. The degradation rate of polydisulfides can be adjusted by the structure of substituents around the disulfide bonds in order to control the physicochemical properties, pharmacokinetics and in vivo contrast enhancement of the biodegradable macromolecular MRI contrast agents. Polydisulfide chelates can be also modified by biocompatible polyethylene glycol to reduce non-specific tissue interaction of the contrast agents.
Figure 1.

Structure of polydisulfide Gd(III) complexes.
Synthesis of polydisulfide Gd(III) complexes
Polydisulfide Gd(III) complexes are synthesized by the condensation copolymerization of DTPA dianhydride with a disulfide containing diamine monomer, followed by complexation with Gd(III) ions. The synthetic procedure of polydisulfide Gd(III) complexes is described in Figure 2 with Gd-DTPA cystamine copolymers (GDCC) as an example.
Figure 2.

Synthesis of Gd-DTPA cystamine copolymers (GDCC).
The copolymerization of DTPA dianhydride with cystamine can be in either DMSO with a tertiary amine as a catalyst or in water with Na2CO3 or NaOH as a catalyst [22, 25]. Substituted polydisulfide Gd(III) chelates are prepared by the copolymerization of DTPA dianhydride with cystine or its derivatives, followed by the complexation with GdCl3 or Gd(OAc)3 [25–28]. Cystine is an animo acid and a disulfide containing diamine monomer. The carboxylic groups of cystine can be readily functionalized to form different substitutes, e.g. diesters and bisamides. DMSO and tertiary amine are effective for the monomers soluble in organic solvents, and water and Na2CO3 or NaOH for water-soluble monomers. The polydisulfide ligands are then complexed with GdCl3 or Gd(OAc)3 at pH 5–6 to give biodegradable macromolecular contrast agents. Figure 3 shows the synthesis of Gd-DTPA cystine bispropyl amide copolymers [29].
Figure 3.

Synthesis scheme of Gd-DTPA cystine bispropyl amide copolymers.
PEGylated polydisulfide Gd(III) chelates are synthesized by grafting monomethoxy polyethylene glycol (PEG) on Gd-DTPA cystine copolymers (GDCP) in the presence of coupling agents [25–28]. The grafting reaction is carried out in aqueous solution with N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) as the coupling agents, Figure 4. The grafting degree can be readily manipulated by altering the molar ratio of MPEG-NH2 to GDCP. The pegylation increases apparent molecular weights of the copolymers at a low grafting degree depending on the size of PEG. Further increase of grafting degree with PEG of same size does not result in further increase of apparent molecular weight or hydrodynamic volume of the copolymers.
Figure 4.

Synthesis of PEGylated GDCP.
The molecular weight of polydisulfides from the condensation copolymerization can be as high as 100,000 Daltons. The complexation of the polydisulfide ligands with Gd(III) ions result in significant reduction of the hydrodynamic volume of the agents as compared to that of the ligands because of the decrease of the overall charge of the polymers. Table 1 lists the physicochemical parameters of the synthesized polydisulfide Gd(III) complexes.
Table 1.
Physicochemical parameters of some polydisulfide Gd(III) complexes.
| Polymeric Contrast Agent | Molecular Weight (KDa) | Relaxivity | PEGylation | |||
|---|---|---|---|---|---|---|
| Mw | Mn | Mw/Mn | mM−1sec−1 (3.0T) | PEG Size (Da) | PEG/Gd | |
| Gd(DTPA-BMA) | 0.574 | 0.574 | N/A | 4.6 | ||
| GDCC | 23 | 21 | 1.07 | 5.7 | ||
| 43 | 38 | 1.13 | 5.8 | |||
| GDCP | 22 | 20 | 1.10 | 6.8 | ||
| 45 | 42 | 1.08 | 5.9 | |||
| GDCEP | 10.2 | 9.4 | 1.1 | 5.9 | ||
| GDGP | 20 | 19 | 1.04 | 12.0 | ||
| 43 | 38 | 1.13 | 9.5 | |||
| GCAC | 28.3 | 18.2 | 1.55 | 4.4 | ||
| GCPC | 30.3 | 24.2 | 1.25 | 5.3 | ||
| GCIC | 55.3 | 36.7 | 1.51 | 5.6 | ||
| PEG2000-GDCP | 37.7 | 28.5 | 1.3 | 8.7 | 2000 | 1.2 |
| PEG1000-GDCP | 37.8 | 31.1 | 1.2 | 7.8 | 1000 | 1.3 |
| PEG550-GDCP | 33.7 | 30.8 | 1.1 | 7.8 | 550 | 1.3 |
Relaxivity of polydisulfide Gd(III) complexes
MRI contrast agents increase both T1 and T2 relaxation rates of surrounding water protons, resulting in brighter image enhancement in T1 weighted imaging and darker enhancement in T2 weighted imaging. The Gd(III) chelates are commonly used as T1 contrast agents and result in brighter image enhancement. Gd(III) ions have a coordination number of 9. Gd(III) based MRI contrast agents have used eight of the coordination sites to achieve the highest possible stability of the chelates. The remaining coordination site is available for the coordination of one water molecule, which can rapidly exchange with the bulky water molecules. Gd(III) chelates have strong effect on the relaxation rate of water protons in the coordination sphere of Gd(III) ions, weak effect on water molecules immediately surrounding the chelates, and little direct effect on the bulk water molecules. The rapid exchange of the complexed water molecule with bulk water is critical for efficient relaxation. The change of relaxation rate of water protons is linearly proportional to the concentration of Gd(III) chelates. Relaxivity is the measurement of the efficiency of a contrast agent to change the relaxation rate. Both T1 and T2 relaxivities can be calculated from the equation (1/Ti)obs = (1/Ti)d + ri[Gd] (i = 1, 2), where (1/Ti)obs refers to the measured relaxation rate, (1/Ti)d the relaxation rate without a contrast agent and ri the T1 or T2 relaxivity.
Polydisulfide Gd(III) complexes have higher T1 relaxivity than small molecular Gd(III) chelates and the relaxivity varies based on the structure of the complexes (Table 1). However, the disulfide bonds in the polymer backbones can freely rotate and most polydisulfide Gd(III) chelates have relatively low relaxivities as compared to other reported macromolecular Gd(III) complexes [8]. Gd-DTPA cystamine copolymers (GDCC), Gd-DTPA cystine copolymers (GDCP) and Gd-DTPA copolymers of cystine derivatives have similar T1 relaxivity, which do not vary substantially with the size of these agents. Gd-DTPA glutathione (oxidized form) copolymers (GDGP) have higher T1 relaxivity than other polydisulfide Gd(III) complexes because two large substituents of oxidized glutathione may inhibit free rotation of the disulfide bonds in GDGP.
Degradability of polydisulfides
Polydisulfide Gd(III) complexes are readily reduced into oligmeric or smaller Gd(III) chelates by free endogenous thiols, including cysteine, glutathione (GSH), cysteinylglycine (Cys-Gly) and homocysteine, via disulfide-thiol exchange reaction. Figure 5 shows the degradation reaction of GDCC by cysteine. The stability of the polydisulfides against reduction with the endogenous thiols depends on their structures [22]. Generally, the polydisulfide ligands are more stable than the corresponding Gd(III) complexes in the presence of cysteine. This is because negative charges around the disulfide bonds may impede the access of the negatively charged cysteine to the disulfide bonds in the polymers at neutral pH. The complexation of the polydisulfide ligands with Gd(III) ions decreases the negative charges on the polymer chains and reduces charge repulsion of the negatively charged endogenous free thiols, which facilitate the disulfide-thiol exchange reaction. Similarly, neutral polydisulfide Gd(III) chelates, including GDCC and Gd-DTPA copolymers of cystine diesters or bisamides, degrade more rapidly than the negatively charged GDCP. Gd-DTPA glutathione copolymers are more stable than GDCP because of the presence of larger negatively charged substituents around the disulfide bonds. [26, 27, 30]
Figure 5.

Degradation of Gd-DTPA cystamine copolymers (GDCC) in the presence of cysteine.
The PEGylation of GDCP slows down its degradation rate and the effect varies with PEG length. The PEG modified GDCP is stable at a low cysteine concentration (ca. 10 μM) and degraded at an increased cysteine concentration. PEG-GDCP with a short PEG chain is more susceptible to degradation in the presence of cysteine than that with a long chain [26, 27]. The structure of endogenous thiols has little effective on the degradation of neutral agent GDCC, but has a significant impact on the degradation rate of negatively charged GDCP. Degradation rate of GDCP decreases with the increasing size of the endogenous thiols in the order of cysteine, homocysteine > cystinylglycine > glutathione.
It is also interesting to note that Gd-DTPA cystine diester or bisamide copolymers have much lower stability in aqueous solution than GDCC and GDCP. GDCEP and Gd-DTPA cystine bisamide or substituted bisamide copolymer readily degrade into small chelates in aqueous solution during the storage, possibly due to hydrolysis of the substituted cystines. In comparison, GDCC is highly stable in aqueous solution during long-term storage for up to 14 months at room temperature. [31]
The degradability of polydisulfide Gd(III) chelates via disulfide-thiol exchange reaction is controlled by the chemical structure around the disulfide bonds. Generally, degradation rate decreases with the increase of steric hindrance around the disulfide bonds. The presence of negatively charged substituents at neutral pH results in further decrease of the degradation rate. Structural modification of polydisulfide Gd(III) chelates results in biodegradable macromolecular MRI contrast agents with different degradability towards the disulfide-thiol exchange reaction, which may have different pharmacokinetic properties and in vivo contrast enhancement.
Dynamic stability of polydisulfide Gd(III) complexes
Gd(III) chelates of linear ligands, including DTPA and its derivatives, have relatively high thermodynamic stability. However, the linear Gd(III) chelates may dissociate through transmetallation with endogenous metal ions in plasma, such as Zn2+, Cu2+ and Ca2+, to release toxic Gd(III) ions. The dynamic stability of Gd(III) chelates in GDCC has been investigated in rat plasma in comparison with clinical MRI contrast agents, Gd(DTPA-BMA) (Omniscan), Gd-BOPTA (MultiHance) and Gd(HP-DO3A) (ProHance). Zn2+ ions in plasma cause significant transmetallation with linear MRI contrast agents, Figure 6. All of the tested contrast agents are stable against Cu2+ and Ca2+. The linear neutral contrast agent Gd(DTPA-BMA) shows the least stability against transmetallation with Zn2+. Macrocyclic contrast agent Gd(HP-DO3A) shows the highest dynamic stability against Zn2+ ions with no detectable transmetallation. The biodegradable macromolecular contrast agent GDCC has comparable stability as Gd-BOPTA with only one tenth of the transmetallation of Gd(DTPA-BMA) in rat plasma. GDCC also shows similar in vivo stability as Gd-BOPTA against transmetallation with Zn2+ in rats, and much higher stability than Gd(DTPA-BMA). [31]
Figure 6.

Transmetallation of Gd chelating agents with Zn2+ in rat plasma.
Pharmacokinetics of polydisulfide Gd(III) complexes
Polydisulfide Gd(III) complexes as biodegradable macromolecular MRI contrast agents should initially behave as macromolecular agents for effective contrast enhanced MR imaging and then gradually break down into small Gd(III) chelates, which can rapidly excrete from the body. Long-term tissue retention of toxic Gd(III) ions from the contrast agents should be as minimal as the clinically used contrast agents. Pharmacokinetics quantitatively measures the circulation, biodistribution and excretion of the biodegradable macromolecular contrast agents in the body. Table 2 lists the blood distribution (α) and elimination (β) half-lives of GDCC-18 (MW = 18 kDa), GDCC-60 (60 kDa), GDCP, GDCEP and GDHC in rats [32, 33]. The weight-average molecular weight of GDCP, GDCEP and GDHC are 35 kDa. Gd(DTPA-BMA) and Gd-DTPA 1, 6-hexanediamine copolymers (GDHC) are a low molecular weight control agent and a nondegradable macromolecular agent, respectively. The blood circulation half-lives of the biodegradable macromolecular agents are shorter than the non-degradable macromolecular agent and longer than Gd(DTPA-BMA). For GDCC, high molecular weight (60 kDa) results in longer blood half-life than low molecular weight. For GDCP and GDCEP of similar molecular weights, negatively charged GDCP with a slow in vitro degradation rate by cysteine has longer blood half-lives than the neutral agent GDCEP. In comparison, the non-degradable macromolecular contrast agent GDHC has much longer blood half-lives than the biodegradable contrast agents. Since the total thiol concentration in rat plasma is relatively low (ca. 83 μM), the degradation of the polydisulfides is a gradual process. The biodegradable macromolecules may initially reduce into relatively large molecules, which remain in the blood at a high concentration in the early stage of degradation. The large molecules eventually degrade into smaller complexes that are readily cleared via renal filtration.
Table 2.
Pharmacokinetic parameters of Gd(DTPA-BMA), GDCC-18, GDCC-60, GDCEP, GDCP, and GDHC after intravenous injection at a dose of 0.1 mmol-Gd/kg in rats.
| Gd(DTPA-BMA) | GDCC-18 | GDCC-60 | GDCEP | GDCP | GDHC | |
|---|---|---|---|---|---|---|
| t1/2, α (min) | 0.48 ± 0.16 | 1.08 ± 0.24 | 1.74 ± 0.25 | 1.60 ± 0.26 | 3.15 ± 1.26 | 5.92 ± 2.02 |
| t1/2, β (min) | 21.2 ± 5.5 | 26.5 ± 5.9 | 53.7 ± 15.9 | 29.3 ± 15.9 | 33.1 ± 19.3 | 223 ± 64 |
In vivo degradation of the polydisulfide Gd(III) complexes and renal excretion of degradation products are verified by the identification of low molecular weight Gd(III) chelates in the mass spectra of rat urine samples after the intravenous injection of the contrast agents [22]. The in vivo degradation of the polydisulfide Gd(III) complexes are more complicated than the disulfide-thiol exchange reaction. Further metabolism may occur to the Gd(III) complexes after the cleavage of the disulfide bonds. Rapid excretion of the degradation products results in much lower long-term tissue retention of the polydisulfide chelates than the nondegradable GDHC (Figure 7) [32, 33]. The overall tissue accumulation of GDCC and GDCEP is comparable to that of Gd(DTPA-BMA) in rats. The negatively charged GDCP shows higher accumulation in the liver and spleen than the neutral agents GDCC and GDCEP, possibly due to non-specific interaction of the negatively charged GDCP with the liver and spleen.
Figure 7.
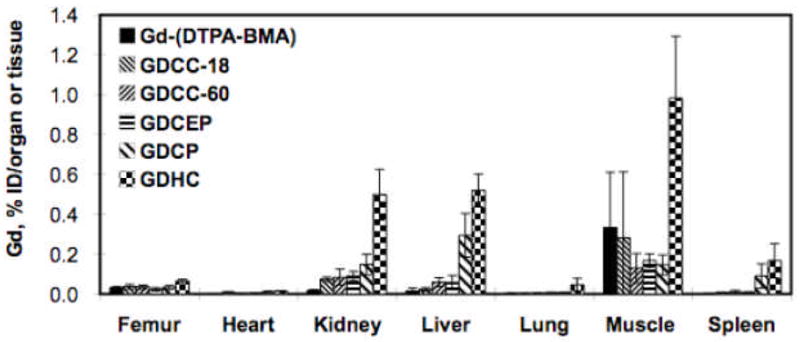
Biodistribution of Gd(III) in rats 10 days after intravenous injection of Gd(DTPA-BMA), GDCC-18, GDCC-60, GDCEP, GDCP and GDHC at a dose of 0.1 mmol-Gd/kg.
PEGylation of GDCP modifies the plasma pharmacokinetics and reduces tissue retention of the agents [28]. Figure 8 shows the blood plasma Gd(III) concentration time profiles for PEG1000H-GDCP (PEG, MW = 1000 Da, PEG/Gd = 0.96), PEG1000M-GDCP (PEG/Gd = 0.65), and PEG1000L-GDCP (PEG/Gd = 0.39). The PEGylated agents with different modification degrees have similar pharmacokinetic properties. The PEGylated GDCP have similar plasma concentrations as GDCP in the first 30 minutes after injection and then are more rapidly eliminated from the blood. As a result, significantly reduced tissue accumulation, especially in the liver and spleen, is observed for the PEGylated GDCP as compared to the unmodified precursor, Figure 9.
Figure 8.

Blood clearance of Gd(III) complexes in rats after intravenous injection of GDCP and PEG-GDCP. Each contrast agent was injected at a dose of 0.1 mmol-Gd/kg. Values are shown as the mean ± SD, N = 6. The inset shows plasma Gd(III) concentration for the first 30 min on a linear-linear scale; error bars are not shown for clarity.
Figure 9.

Biodistribution of Gd(III) in rats 10 days after the intravenous injection of GDCP and PEG1000X-GDCP. Each contrast agent was injected at a dose of 0.1 mmol-Gd/kg. Values are shown as the mean ± SD, N = 6. *p < 0.05 for GDCP compared to all three PEG1000X-GDCP contrast agents according to Bonferroni’s Multiple Comparison Test.
Chemical structure of the polydisulfide Gd(III) complexes has a significant impact on their plasma pharmacokinetics and tissue retention. Neutral polydisulfide Gd(III) complexes degrade more rapidly than negatively charged polydisulfide Gd(III) complexes and result in more rapid clearance from blood circulation and lower tissue retention. PEGylation of the negatively charged GDCP significantly reduces the retention in the liver and spleen. The pharmacokinetics of the biodegradable macromolecular contrast agents can be readily altered by structural modification of polydisulfide to minimize tissue retention of the contrast agents and to achieve effective in vivo contrast enhancement.
Contrast enhanced MRI with polydisulfide Gd(III) complexes
The prolonged blood circulation of polydisulfide Gd(III) complexes result in strong contrast enhancement in the cardiovascular systems as shown in animal models [30]. Figure 10 shows the representative 3D maximum intensity projection (MIP) contrast enhanced MR images of mice before and at various time points after injection of GDCC, GDCP, and GDGP with different molecular weights. All agents result in strong contrast enhancement in mouse heart, vasculature and kidney at 2 minutes post injection and the signal intensity gradually decreased thereafter depending on their structures and sizes. The duration of the contrast enhancement in the heart and vasculature increases with the increasing molecular weight for the same agent. For the agents with similar molecular weights, the duration of cardiovascular enhancement increases with the increasing steric hindrance around the disulfide bonds in the order of GDCC < GDCP < GDGP. GDGP with high relaxivity and slow degradation rate result in strong contrast enhancement in the blood pool within the first 30 minutes post-injection even at the low molecular weight (21 kDa). Gradual enhancement in the urinary bladder indicates urinary clearance of Gd complexes after the degradation for all polydisulfide contrast agents.
Figure 10.
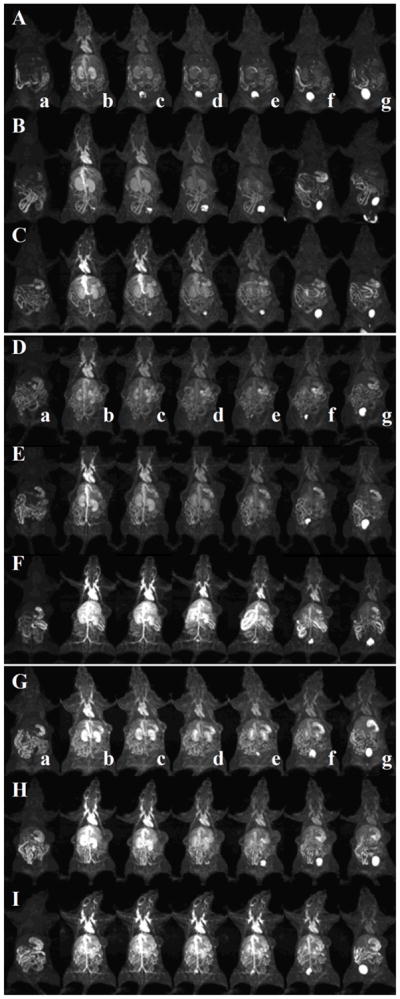
Three dimensional maximum intensity projection MR images of mice before injection (a) and 2 (b), 5 (c), 10 (d), 15 (e), 30 (f) and 60 (g) minutes after injection of GDCC (23 kDa, A; 43 kDa, B; 113 kDa, C), GDCP (23 kDa, D; 43 kDa, E; 109 kDa, F) and GDGP (21 kDa, G; 43 kDa, H; 108 kDa, I) at a dose of 0.1 mmol-Gd/kg via a tail vein.
Figure 11 shows the 3D-MIP rat images acquired with a high resolution TOF-MRA pulse sequence after intravenous injection of the PEGylated contrast agents PEG1000H-GDCP, PEG1000M-GDCP and PEG1000L-GDCP at a low dose (0.03 mmol-Gd/kg) [28]. Strong contrast enhancement in the vasculature, including descending abdominal aorta and small vessels in the liver and shoulder and neck regions, is observed with the PEGylated GDCP and GDCP. Contrast enhancement can also be seen in the renal calyx and renal pelvis for the agents. Drainage by the ureters from the renal pelvis to the bladder is visible for all contrast agents, supporting their clearance via renal filtration. The relatively high concentration of PEG1000-GDCP and GDCP in the first 30 min post-injection (Figure 8) results in excellent contrast enhancement with an MRA pulse sequence requiring a relatively long acquisition time.
Figure 11.
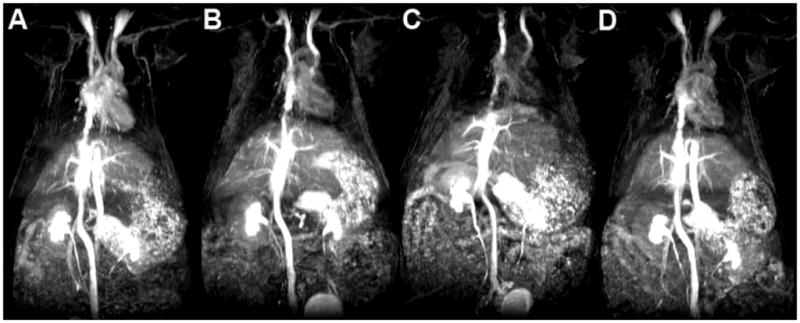
3D maximum intensity projection (MIP) images of high-resolution TOF-MRA images of PEG1000H-GDCP (A), PEG1000M-GDCP (B), PEG1000L-GDCP (C), and GDCP (D). Each contrast agent was administered intravenously at a dose of 0.03 mmol-Gd/kg.
The polydisulfide Gd(III) complexes are effective for contrast enhanced tumor imaging in animal models. Figure 12 shows the T1-weighted 2D axial spin-echo images of tumor tissues before and after injection of GDCC, GDCP and GDGP with different sizes. All agents result in substantial enhancement in the tumor periphery, the region rich with angiogenic microvessels, for at least 60 minutes [30]. The size and degradability of the agents did not significantly affect the tumor enhancement. The biodegradable macromolecular agents result in 30–100% tumor enhancement, which should be sufficient to detect tumor tissue for diagnostic purpose.
Figure 12.

Two dimensional T1 weighted MR images of mice before injection (a) and 2 (b), 5 (c), 10 (d), 15 (e), 30 (f) and 60 (g) minutes after injection of GDCC (23 kDa, A; 43 kDa, B; 113 kDa, C), GDCP (23 kDa, D; 43 kDa, E; 109 kDa, F) and GDGP (21 kDa, G; 43 kDa, H; 108 kDa, I) at a dose of 0.1 mmol-Gd/kg via a tail vein.
Dynamic contrast enhanced MRI of tumor with the biodegradable macromolecular contrast agents in mice
Dynamic contrast enhanced MRI (DCE-MRI) measures the kinetics of the uptake of contrast agents in the tissue of interest and can be used for non-invasive characterization of tumor angiogenesis and vascular permeability, which are inversely correlated to cancer patients’ survival [34, 35]. DCE-MRI acquires MR images repetitively and the dynamic change of the concentration of contrast agents can be correlated with the change of signal intensity in the tissue of interest. Tumor vascular parameters, including endothelial transfer coefficient (Ktrans) and fractional tumor plasma volume (fPV), can be calculated from the DCE-MRI data [36–40]. These quantitative parameters can be correlated with tumor vascularity and angiogenesis, and can be used for assessing the efficacy of anticancer therapies, including antiangiogenic therapy [17, 40, 41]. It has been reported that macromolecular MRI contrast agents provides more accurate determination of tumor vascular hyperpermeability than small molecular contrast agents [36, 42].
Polydisulfide-based biodegradable macromolecular contrast agents are effective for evaluating tumor angiogenesis in DCE-MRI [39]. Figure 13 shows the representative MR signal intensity-time courses in both PC-3 and MDA PCa 2b tumor tissues in mice for GDCC and GDCP of two different molecular weights [39]. The tumor uptake kinetics varies with the tumor tissues and the size and degradability of the contrast agents. After the initial SI rise, the permeation of the contrast agents into the extracellular space of tumor tissues is controlled by the contrast agent concentration in the blood and the permeability of the tumor microvessels. The MDA-PCa-2b tumor has more rapid uptake kinetics than the PC-3 tumor for the contrast agents. For the same tumor, contrast uptake kinetics slows down with increasing molecular weight and decreasing degradability of the agents. GDCP has a slower degradation rate than GDCC and shows slower tumor uptake than GDCC with the same molecular weight.
Figure 13.

MR signal intensity (ΔSI) time curves for the whole tumors of PC-3 and MDA PCa 2b xenografts enhanced by GDCC-20, GDCP-20, GDCC-70, GDCP-70.
Figure 14 plots the values of the vascular parameter Ktrans of two tumors and different agents calculated from the DCE-MRI data using a two-compartment model [39]. The values of endothelial transfer coefficient (Ktrans) depend on the size and degradability of the contrast agents. High Ktrans values are obtained for the agents of the low molecular weight in both tumor models because of the rapid diffusion and permeation of the agents. The measured Ktrans values decrease with the increase of the size of the contrast agents because of limited diffusion of the large molecules into tumor tissue. GDCP with slow degradability results in smaller Ktrans values than GDCC of similar molecular weight. With the same high molecular weight, GDCC-70 resulted in higher Ktrans values than GDCP-70 in both tumors due to high degradability of the former. However, the Ktrans values measured by GDCC-20 and GDCP-20 of similar low molecular weights are not significantly different, possibly due to their relatively small size and high diffusion rate from plasma to tumor extracellular space. The Ktrans values of MDA-PCa-2b tumor are higher than those of PC-3 tumor measured by the same contrast agents.
Figure 14.

Comparison of Ktrans of PC-3 and MDA PCa 2b tumor xenografts estimated by GDCC-20, GDCP-20, GDCC-70, and GDCP-70, respectively. * (P < 0.05), ** (P < 0.01).
Noninvasive evaluation of the efficacy of cancer therapy by DCE-MRI with GDCC
DCE-MRI is an effective approach for non-invasive evaluation of the efficacy of cancer therapy based on the change of tumor vascular parameters [35, 43]. DCE-MRI with biodegradable macromolecular contrast agent GDCC has a potential to non-invasively assess early tumor response to antiangiogenic therapy [40]. Figure 15 shows the representative arterial input function and signal enhancement (ΔSI)–time courses in the tumor periphery for GDCC-40 (40 kDa) and Gd(DTPA-BMA) before, and at 36 hours and 7 days after the first administration of an antiangiogenic drug Avastin®. The arterial input function shows that both agents have a high initial blood concentration after the injection. The low molecular weight Gd(DTPA-BMA) is then rapidly cleared from blood, while GDCC-40 is cleared at a relatively slow rate. DCE-MRI with GDCC-40 is more sensitive for assessing the change of tumor vascular permeability than that with the low molecular weight contrast agent. The tumor uptake of GDCC-40 decreases significantly after the treatment with Avastin®, while there is no significant change of tumor uptake of Gd(DTPA-BMA) before and after the treatment.
Figure 15.
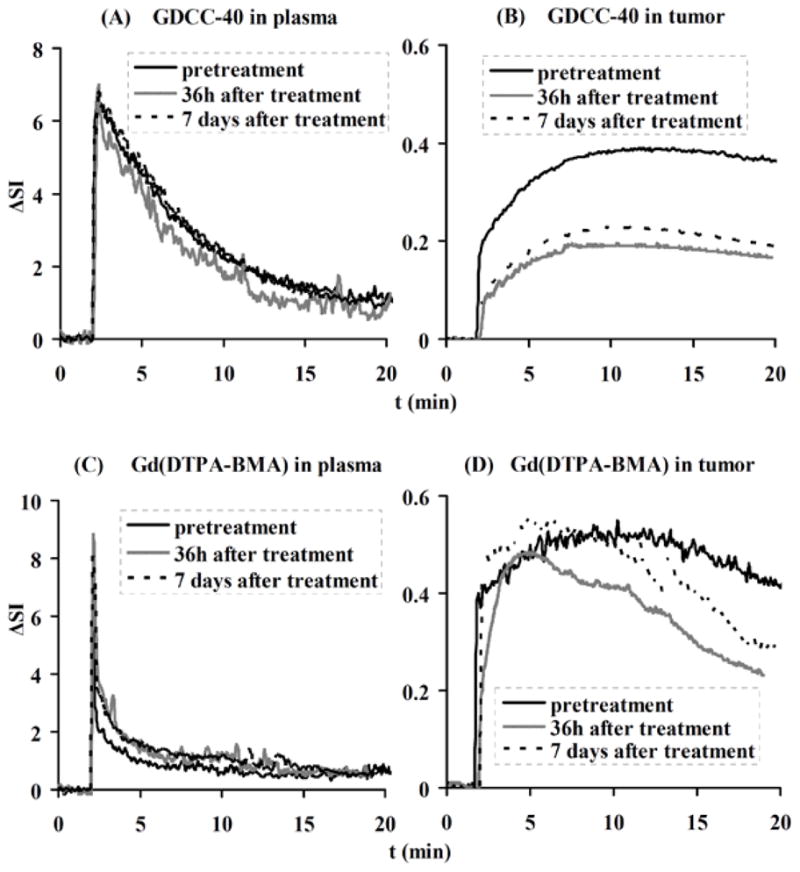
Representative contrast enhancement-time curves in plasma and tumor for GDCC-40 (A, B) and Gd(DTPA-BMA) (C, D) before, 36 h and 7 days after the first administration of Avastin®.
DCE-MRI with GDCC-40 is able to measure the changes of tumor vascular parameters, vascular permeability (Ktrans) and fractional tumor plasma volume (fPV), in correlation with therapeutic efficacy of Avastin® in the animal tumor model [40]. Figure 16 illustrates the average Ktrans and fPV values of tumor tissues before and after treatment calculated from the DCE-MRI data using a two-compartment model. Tumor periphery has higher Ktrans and fPV values than the inner tumor tissues in all cases. Both Ktrans and fPV values in tumor periphery determined by DCE-MRI with GDCC-40 significantly decreases at 36 hours after the first treatment as compared to the pretreatment values (p < 0.05). The values are increased at 7 days after three treatments and are similar to the pretreatment values (p > 0.05), indicating the recovery of peripheral vascular permeability after the initial therapeutic response. The average Ktrans in the tumor core does not change significantly after the treatment as compared to the pretreatment values. The average fPV in the tumor core after the treatment decreases significantly from the pretreatment values. In comparison, the Ktrans and fPV values in both tumor periphery and tumor core calculated from DCE-MRI with Gd(DTPA-BMA) after the treatments are not significantly different from the pretreatment values (p > 0.05). The Ktrans and fPV values estimated from Gd(DTPA-BMA) are much higher than those estimated from GDCC-40 due to the diffusion of the contrast agents into tumor tissue. DCE-MRI with the biodegradable macromolecular MRI contrast agents is more sensitive for measuring tumor response to anti-angiogenic therapy than that with the low molecular weight contrast agents. No significant changes are observed for the vascular parameters in tumor periphery estimated by DCE-MRI with Gd(DTPA-BMA) before and after the treatment with Avastin®. The biodegradable macromolecular contrast agents are effective for evaluating the efficacy of anti-angiogenic therapy in DCE-MRI.
Figure 16.

Graphs showing change in Ktrans (A) and fPV (B) for GDCC-40 and Gd(DTPA-BMA) before and after Avastin administration. *: P<0.05. Ktrans = endothelial transfer coefficient (mL of plasma/mL of tissue/min), fPV = fractional vascular volume (mL of plasma/mL of tissue).
Challenges and future directions
The polydisulfide Gd(III) complexes have shown promising features as biodegradable macromolecular MRI contrast agents. These agents overcome the limitations of the slow excretion of macromolecular MRI contrast agents. However, detailed toxicological evaluations are needed to demonstrate the safety of these new contrast agents in order to proceed to clinical development. Although the polydisulfide Gd(III) complexes have demonstrated similar kinetic stability against transmetallation as the clinically used MRI contrast agents with linear ligands, it will be ideal to design and prepare biodegradable macromolecular MRI contrast agents with improved kinetic stability to eliminate potential toxic side effects related to the release of toxic Gd(III) ions due to transmetallation. The biodegradable macromolecular contrast agents prepared by condensation polymerization have a broad molecular weight distribution, which is acceptable for contrast enhanced diagnostic imaging. However, the broad molecular weight distribution may not be suitable for quantitative DCE-MRI studies, because the agents with different molecular weights would result in a broad range of vascular parameters, which would affect the accuracy of characterizing tumor vascular property. Biodegradable macromolecular contrast agents with narrow molecular weight distribution would be suitable for DCE-MRI.
The future study on biodegradable macromolecular contrast agents should be focused on design and development of the agents with high kinetic stability. Macrocyclic Gd(III) complexes, e.g. Gd-DOTA and Gd(DO3A-HP), have high kinetic stability and can be completely excreted with little transmetallation with endogenous metal ions. The macrocyclic agents can be used in designing the new generation of biodegradable macromolecular contrast agents. The design should also consider the structures with the best balance of the degradability, pharmacokinetics and relaxivity in order to achieve the best possible contrast enhancement at low doses. Detailed studies are needed to further demonstrate the effectiveness of the biodegradable macromolecular MRI contrast agents in tumor characterization and therapeutic response evaluation with DCE-MRI at substantially reduced doses. Effective contrast enhanced MRI at low doses can reduce dose related toxic side effects. Target-specific agents can be conjugated to the biodegradable macromolecular contrast agents to achieve target-specific contrast enhancement.
Conclusion
Polydisulfide Gd(III) complexes have been designed and developed as biodegradable macromolecular MRI contrast agents. The polydisulfides can be readily reduced by the endogenous thiols in the plasma into small Gd(III) complexes, which can be rapidly excreted via renal filtration. The structure of the polydisulfides can be modified to alter their degradability and in vivo pharmacokinetics to achieve the best contrast enhancement and minimal long-term tissue accumulation. Introduction of steric hindrance around the disulfide bonds decreases the degradability of the biodegradable macromolecular contrast agents and increases vascular circulation of the agents. The polydisulfide Gd(III) complexes initially behave as macromolecular contrast agents, resulting in significant contrast enhancement in the blood pool, and then gradually degrade and excrete with minimal tissue retention. The biodegradable macromolecular contrast agents are effective for cardiovascular and cancer MR imaging, and for non-invasive evaluation of tumor angiogenesis and assessment of therapeutic response to antiangiogenic therapy. The polydisulfide Gd(III) complexes are promising for clinical applications as safe and effective biodegradable macromolecular MRI contrast agents.
Acknowledgments
This work was supported in part by the NIH grants R01 EB00489 and R33 CA095873.
References
- 1.Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Chemical Reviews. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- 2.Magerstadt M, Gansow OA, Brechbiel MW, Colcher D, Baltzer L, Knop RH, Girton ME, Naegele M. Magnetic Resonance in Medicine. 1986;3:808–812. doi: 10.1002/mrm.1910030517. [DOI] [PubMed] [Google Scholar]
- 3.Weinmann HJ, Brasch RC, Press WR, Wesbey GE. American Journal of Roentgenology. 1984;142:619–624. doi: 10.2214/ajr.142.3.619. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi H, Brechbiel MW. Advanced Drug Delivery Reviews. 2005;57:2271–2286. doi: 10.1016/j.addr.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Ladd DL, Hollister R, Peng X, Wei D, Wu G, Delecki D, Snow RA, Toner JL, Kellar K, Eck J, Desai VC, Raymond G, Kinter LB, Desser TS, Rubin DL. Bioconjugate Chemistry. 1999;10:361–370. doi: 10.1021/bc980086+. [DOI] [PubMed] [Google Scholar]
- 6.Barrett T, Kobayashi H, Brechbiel M, Choyke PL. European Journal of Radiology. 2006;60:353–366. doi: 10.1016/j.ejrad.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 7.Langereis S, de Lussanet QG, van Genderen MHP, Backes WH, Meijer EW. Macromolecules. 2004;37:3084–3091. [Google Scholar]
- 8.Kaneshiro TL, Jeong EK, Morrell G, Parker DL, Lu ZR. Biomacromolecules. 2008;9:2742–2748. doi: 10.1021/bm800486c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ogan MD, Schmiedl U, Moseley ME, Grodd W, Paajanen H, Brasch RC. Investigative Radiology. 1987;22:665–671. [PubMed] [Google Scholar]
- 10.Fritz T, Unger E, Wilsonsanders S, Ahkong QF, Tilcock C. Investigative Radiology. 1991;26:960–968. doi: 10.1097/00004424-199111000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Wang SC, Wikstrom MG, White DL, Klaveness J, Holtz E, Rongved P, Moseley ME, Brasch RC. Radiology. 1990;175:483–488. doi: 10.1148/radiology.175.2.1691513. [DOI] [PubMed] [Google Scholar]
- 12.Schmiedl U, Ogan M, Paajanen H, Marotti M, Crooks LE, Brito AC, Brasch RC. Radiology. 1987;162:205–210. doi: 10.1148/radiology.162.1.3786763. [DOI] [PubMed] [Google Scholar]
- 13.Kellar KE, Henrichs PM, Hollister R, Koenig SH, Eck T, Wei D. Magnetic Resonance in Medicine. 1997;38:712–716. doi: 10.1002/mrm.1910380506. [DOI] [PubMed] [Google Scholar]
- 14.Duarte MG, Gil MH, Peters JA, Colet JM, Vander Elst L, Muller RN, Geraldes CFGC. Bioconjugate Chemistry. 2001;12:170–177. doi: 10.1021/bc000065r. [DOI] [PubMed] [Google Scholar]
- 15.Gerlowski LE, Jain RK. Microvascular Research. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- 16.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:4607–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gossmann A, Okuhata Y, Shames DM, Helbich TH, Roberts TPL, Wendland MF, Huber S, Brasch RC. Radiology. 1999;213:265–272. doi: 10.1148/radiology.213.1.r99oc43265. [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi H, Kawamoto S, Jo SK, Bryant HL, Brechbiel MW, Star RA. Bioconjugate Chemistry. 2003;14:388–394. doi: 10.1021/bc025633c. [DOI] [PubMed] [Google Scholar]
- 19.Cacheris WP, Quay SC, Rocklage SM. Magnetic Resonance Imaging. 1990;8:467–481. doi: 10.1016/0730-725x(90)90055-7. [DOI] [PubMed] [Google Scholar]
- 20.Helbich TH, Gossman A, Mareski PA, Raduchel B, Roberts TPL, Shames DM, Muhler M, Turetschek K, Brasch RC. Journal of Magnetic Resonance Imaging. 2000;11:694–701. doi: 10.1002/1522-2586(200006)11:6<694::aid-jmri17>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 21.Wang SJ, Brechbiel M, Wiener EC. Investigative Radiology. 2003;38:662–668. doi: 10.1097/01.rli.0000084887.47427.75. [DOI] [PubMed] [Google Scholar]
- 22.Lu ZR, Parker DL, Goodrich KC, Wang XH, Dalle JG, Buswell HR. Magnetic Resonance in Medicine. 2004;51:27–34. doi: 10.1002/mrm.10656. [DOI] [PubMed] [Google Scholar]
- 23.Andersson A, Lindgren A, Hultberg B. Clinical Chemistry. 1995;41:361–366. [PubMed] [Google Scholar]
- 24.Deneke SM. Current Topics in Cellular Regulation. 2000;36:151–180. doi: 10.1016/s0070-2137(01)80007-8. [DOI] [PubMed] [Google Scholar]
- 25.Zong YD, Wang XH, Goodrich KC, Mohs AM, Parker DL, Lu ZR. Magnetic Resonance in Medicine. 2005;53:835–842. doi: 10.1002/mrm.20402. [DOI] [PubMed] [Google Scholar]
- 26.Mohs AM, Wang XH, Goodrich KC, Zong YD, Parker DL, Lu ZR. Bioconjugate Chemistry. 2004;15:1424–1430. doi: 10.1021/bc049828r. [DOI] [PubMed] [Google Scholar]
- 27.Mohs AM, Zong YD, Guo JY, Parker DL, Lu ZR. Biomacromolecules. 2005;6:2305–2311. doi: 10.1021/bm050194g. [DOI] [PubMed] [Google Scholar]
- 28.Mohs AM, Nguyen T, Jeong EK, Feng Y, Emerson L, Zong YD, Parker DL, Lu ZR. Magnetic Resonance in Medicine. 2007;58:110–118. doi: 10.1002/mrm.21270. [DOI] [PubMed] [Google Scholar]
- 29.Kaneshiro TL, Ke T, Jeong EK, Parker DL, Lu ZR. Pharmaceutical Research. 2006;23:1285–1294. doi: 10.1007/s11095-006-0024-0. [DOI] [PubMed] [Google Scholar]
- 30.Zong YD, Wang XL, Jeong EK, Parker DL, Lu ZR. Magnetic Resonance Imaging. 2009;27:503–511. doi: 10.1016/j.mri.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu XM, Zong YD, Zhen Y, Lu ZR. Pharmaceutical Research. 2010 doi: 10.1007/s11095-010-0131-9. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang XH, Feng Y, Ke TY, Schabel M, Lu ZR. Pharmaceutical Research. 2005;22:596–602. doi: 10.1007/s11095-005-2489-7. [DOI] [PubMed] [Google Scholar]
- 33.Feng Y, Zong YD, Ke TY, Jeong EK, Parker DL, Lu ZR. Pharmaceutical Research. 2006;23:1736–1742. doi: 10.1007/s11095-006-9028-z. [DOI] [PubMed] [Google Scholar]
- 34.Su MY, Cheung YC, Fruehauf JP, Yu H, Nalcioglu O, Mechetner E, Kyshtoobayeva A, Chen SC, Hsueh S, McLaren CE, Wan YL. Journal of Magnetic Resonance Imaging. 2003;18:467–477. doi: 10.1002/jmri.10380. [DOI] [PubMed] [Google Scholar]
- 35.Raatschen HJ, Simon GH, Fu YJ, Sennino B, Shames DM, Wendland MF, McDonald DM, Brasch RC. Radiology. 2008;247:391–399. doi: 10.1148/radiol.2472070363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daldrup H, Shames DM, Wendland M, Okuhata Y, Link TM, Rosenau W, Lu Y, Brasch RC. American Journal of Roentgenology. 1998;171:941–949. doi: 10.2214/ajr.171.4.9762973. [DOI] [PubMed] [Google Scholar]
- 37.Roberts HC, Roberts TPL, Bollen AW, Ley S, Brasch RC, Dillon WP. Academic Radiology. 2001;8:384–391. doi: 10.1016/S1076-6332(03)80545-7. [DOI] [PubMed] [Google Scholar]
- 38.Tofts PS, Brix G, Buckley DL, Evelhoch JL, Henderson E, Knopp M, Larsson HBW, Lee TY, Mayr NA, Parker GJM, Port RE, Taylor J, Weisskoff RM. Journal of Magnetic Resonance Imaging. 1999;10:223–232. doi: 10.1002/(sici)1522-2586(199909)10:3<223::aid-jmri2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 39.Wu XM, Feng Y, Jeong EK, Emerson L, Lu ZR. Pharmaceutical Research. 2009;26:2202–2208. doi: 10.1007/s11095-009-9935-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu XM, Jeong EK, Emerson L, Hoffman J, Parker DL, Lu ZR. Molecular Pharmaceutics. 2010;7:41–48. doi: 10.1021/mp900153f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jordan BF, Runquist M, Raghunand N, Gillies RJ, Tate WR, Powis G, Baker AF. Clinical Cancer Research. 2005;11:529–536. [PubMed] [Google Scholar]
- 42.de Lussanet QG, Langereis S, Beets-Tan RGH, van Genderen MHP, Griffioen AW, van Engelshoven JMA, Backes WH. Radiology. 2005;235:65–72. doi: 10.1148/radiol.2351040411. [DOI] [PubMed] [Google Scholar]
- 43.Feng Y, Emerson L, Jeong EK, Parker DL, Lu ZR. Journal of Magnetic Resonance Imaging. 2009;30:401–406. doi: 10.1002/jmri.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]