

Abstract
Tumor suppressors negatively regulate angiogenesis, an essential step in tumor progression. Together, HPV 16 E6 and E7 proteins, which target p53 and pRb family members, respectively, for degradation, increase the expression of two angiogenic inducers, VEGF and IL-8, in primary foreskin keratinocytes (HFKs). Conditioned media from such cells are sufficient to alter endothelial cell behavior. Here, the individual contribution of E6 and E7 to angiogenesis was investigated. E7 and, to a lesser extent E6, increased expression of VEGF and IL-8. Nevertheless, neither conditioned media from HPV 16 E6 nor E7-expressing HFKs were sufficient to induce migration of endothelial cells. Conditioned media from HFKs expressing the HPV 16 E6 and the E7 mutant E7C24G, which can target p107 and p130 but not pRb for degradation, contained increased levels of VEGF and IL-8. The results suggest that the mechanism of HPV 16 E7-mediated increased levels of VEGF is pRb-independent.
Keywords: Human papillomaviruses, pRb, p107, p130, angiogenesis, VEGF, IL-8, human foreskin keratinocytes, human microvascular endothelial cells
Introduction
The establishment and maintenance of a vascular supply is required for growth of malignant tissue. Consistent with this, progression of the pre-malignant stages to invasive cervical cancer is associated with the development of new blood vessels, a process called angiogenesis (Bouck, 1996; Folkman, 2002). Angiogenesis is crucial for a developing tumor to acquire nutrients and oxygen for sustained growth and expansion (Bouck, 1996). Angiogenesis is tightly regulated by a number of secreted factors. According to the current model of angiogenesis, the vessels seen in normal tissue are quiescent due to the presence in the microenvironment of elevated levels of angiogenic inhibitors such as thrombospondin-1 and maspin, relative to levels of angiogenic inducers, such as VEGF and IL-8 (Bouck, 1996; Hanahan et al., 1996). As cells progress towards tumorigenicity, the balance between angiogenic inducers and angiogenic inhibitors is switched, such that angiogenic inducers dominate.
In tumor cells, a link between the functions of tumor suppressors and angiogenesis has long been established (Gomez-Manzano et al., 2003; Lee et al., 2003). For example, the tumor suppressor p53 positively regulates the expression of angiogenic inhibitors and negatively regulates the expression of angiogenic inducers (Volpert et al., 1997). Similarly, pRb family members regulate expression of angiogenic factors to maintain a quiescent environment (Gabellini et al., 2006).
Human papillomaviruses (HPVs) are small double-stranded DNA viruses that infect mucosal and cutaneous stratified squamous epithelia generally of the anogenital tract, head and neck and hands or feet (Alani and Munger, 1998; Longworth and Laimins, 2004). There are over 200 genotypes of HPVs. These viruses can cause a wide range of diseases from benign genital warts to invasive cancer. Approximately 40 types infect the mucosa and based on their oncogenic potential these viruses can be divided into two groups: high-risk HPVs and low-risk HPVs (Munger, 2002). The high-risk HPV types such as HPV 16, 18, 31, 33 and 45 are mainly associated with the development of high-grade intraepithelial neoplasias of the anogenital tract, whereas low-risk HPV types such as HPV 6 and 11 are mainly associated with low-grade abnormalities such as benign condyloma acuminatum or anogenital warts. The high-risk HPV type 16 and 18 are responsible for more than 70% of cervical cancer cases (Munoz et al., 2003). Persistent infection with these high-risk types is thought to be a precursor to neoplastic transformation (Munoz et al., 2003).
HPV 16-mediated tumorigenicity is dependent on the activity of two early proteins, E6 and E7. Both in vitro and in vivo studies show that the functions of E6 and E7, of high-risk types, are necessary for the induction and maintenance of the transformed phenotype (Goodwin and DiMaio, 2000; Hawley-Nelson et al., 1989; Kaur et al., 1989; Munger et al., 1989a; Wells et al., 2000). HPV 16 E6 and E7 contribute to cellular transformation, in part, by binding to and targeting p53 and pRb family members, respectively, for degradation (Boyer et al., 1996; Crook et al., 1988; Jones et al., 1997; Scheffner et al., 1990; zur Hausen, 2002). Degradation of p53 mediated by E6 blocks apoptosis to allow continued proliferation. Degradation of pRb by E7 results in the activation of E2F transcription factors inducing cellular genes responsible for the S phase of the cell cycle.
The contribution of HPV 16 E6 and E7 expression to the early stages of recruitment of a blood supply to an HPV lesion has been modeled by analysis of primary human foreskin keratinocytes (HFKs) shortly after transduction with HPV 16 E6 and E7 retrovirus or transfection with the intact HPV 16 genome. These studies show that expression of HPV 16 E6 and E7 together in HFKs increases the level of two angiogenic inducers VEGF and IL-8 (Toussaint-Smith et al., 2004). Further, conditioned media from cells expressing HPV E6 and E7 are sufficient to alter endothelial cell behavior (Chen et al., 2007). These studies, however, did not establish the individual contribution of HPV 16 E6 and E7 to this angiogenic response. To examine the role that E6 and E7 each plays, translational termination linkers (TTLs) were introduced into the coding region of E6 or E7. The ability of E7 in the context of the E6TTL mutation (E6TTLE7) and E6 in the context of the E7TTL mutation (E6E7TTL) to induce VEGF and IL-8 in HFKs was determined. Both VEGF and IL-8 were significantly enhanced in HFKs expressing either HPV 16E6TTLE7 or HPV 16E6E7TTL. However, migration assays revealed that conditioned media from neither HPV 16 E6 nor E7-expressing HFKs were able to induce migration of human microvascular endothelial cells (HMVECs). Immunological depletion experiments showed that VEGF, but not IL-8, was required for HPV 16 E6 and E7 together to induce HMVEC migration. Further experiments showed that the ability of HPV 16 E7 to induce VEGF may be dependent upon degradation of pRb family members p107 and/or p130.
Results
Conditioned media from human foreskin keratinocytes (HFKs) transduced with HPV 16E6TTLE7 or HPV 16E6E7TTL have elevated levels of IL-8 and VEGF
Retroviral transduction of HFKs with the HPV 16E6E7 cassette expressing HPV 16 E6 and E7 (Halbert et al., 1991) results in the increased expression of angiogenic inducers, IL-8 and VEGF (Toussaint-Smith et al., 2004). To determine whether E6 and E7 each contributes to the increased levels of IL-8 and VEGF, a translation termination linker was placed into either the E6 or the E7 gene within the HPV 16E6E7 cassette. Pooled HFKs were transduced with parental retrovirus LXSN or retrovirus encoding HPV 16E6E7, HPV 16E6TTLE7 (expressing E7 in the context of the HPV 16E6TTL) or HPV 16E6E7TTL (expressing E6 in the context of the HPV 16E7TTL) and the levels of VEGF and IL-8 in the conditioned media from the transduced HFKs were determined by ELISA. Consistent with previous results (Chen et al., 2007; Toussaint-Smith et al., 2004), relative to HFKs transduced with LXSN, the level of IL-8 was increased 8-fold in conditioned media from cells expressing HPV 16 E6 and E7 (HPV 16E6E7). The level of secreted IL-8 in conditioned media from cells expressing HPV 16E6E7TTL was increased 2.9-fold compared to parental retrovirus LXSN (p< 0.001) (Figure 1A). A greater increase, approximately 11.7-fold, was observed in the conditioned media from cells expressing HPV 16E6TTLE7 compared to parental retrovirus LXSN (p< 0.0001) (Figure 1A). Also consistent with previous results, the level of VEGF was increased 1.5-fold in conditioned media from cells expressing HPV 16E6E7 compared to parental retrovirus LXSN (p< 0.001). The level of secreted VEGF in conditioned media from cells expressing HPV 16E6E7TTL was increased approximately 2-fold compared to parental retrovirus LXSN (p< 0.001) (Figure 1B). A greater increase, approximately 4-fold, was observed from cells expressing HPV 16E6TTLE7 compared to parental retrovirus LXSN (p< 0.0001) (Figure 1B). Therefore, E6 and E7 each significantly increased the levels of IL-8 and VEGF relative to control, LXSN. However, when compared to HPV 16E6E7, E6 caused a lesser increase in IL-8 and minimal increase in VEGF; E7 caused a greater increase in both angiogenic inducers.
Figure 1. Level of IL-8 and VEGF in conditioned media was increased in cells expressing either E6 or E7.
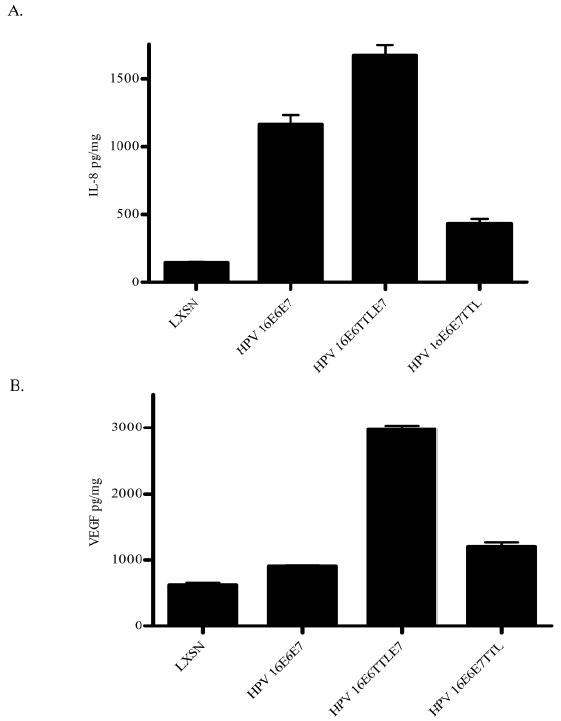
(A) The quantity of IL-8 was determined by ELISA and normalized to total protein in the conditioned media. The data represent mean ± SEM and are representative of three experiments each conducted in triplicate. p < 0.0001 for LXSN vs HPV 16E6E7 and LXSN vs HPV 16E6TTLE7; p< 0.001 for LXSN vs HPV 16E6E7TTL; p< 0.001 for HPV 16E6E7 vs HPV 16E6TTLE7 and p< 0.0005 for HPV 16E6E7 vs HPV 16E6E7TTL. (B) The quantity of VEGF was determined by ELISA and normalized to total protein in the conditioned media. The data represent mean ± SEM and are representative of three experiments each conducted in triplicate. p < 0.001 for LXSN vs HPV 16E6E7; p< 0.0001 for LXSN vs HPV 16E6TTLE7; p< 0.001 for LXSN vs HPV 16E6E7TTL; p< 0.0001 for HPV 16E6E7 vs HPV 16E6TTLE7 and p< 0.001 for HPV 16E6E7 vs HPV 16E6E7TTL.
Conditioned media from HFKs transduced with HPV 16E6TTLE7 or HPV 16E6E7TTL do not induce migration of human microvascular endothelial cells
Conditioned media from HFKs transduced with HPV 16E6E7 increase the migration of human microvascular endothelial cells (HMVECs) (Chen et al., 2007). This assay was, therefore, used to determine whether conditioned media from HFKs transduced with E6 or E7 alone were sufficient to alter endothelial cell behavior. HMVECs were placed in the top of a transwell plate, conditioned media from control HFKs or HFKs expressing HPV gene products were placed in the bottom well and migration was monitored. Consistent with previous results (Chen et al., 2007) conditioned media from HPV 16E6E7-expressing cells increased the migration of HMVECs (p<0.005) (Figure 2). In contrast, conditioned media from retrovirally transduced cells expressing HPV 16E6TTLE7 or HPV 16E6E7TTL did not induce migration of HMVECs relative to LXSN (p< 0.5 and p< 0.3, respectively) (Figure 2). Consistent with these observations, the level of migration of endothelial cells seen in the presence of conditioned media from HPV 16E6E7-expressing cells was statistically different from that seen with conditioned media from either HPV 16E6TTLE7 or HPV 16E6E7TTL (p< 0.02). These results indicate that both E6 and E7 are required to alter endothelial cell behavior. Together the data indicate that although expression of E7 is sufficient to increase the levels of VEGF and IL-8 in conditioned media, additional E6-mediated contributions to the conditioned media are needed to induce endothelial cell migration.
Figure 2. Conditioned media from HFKs transduced with either E6 or E7 did not induce migration of HMVECs in vitro.
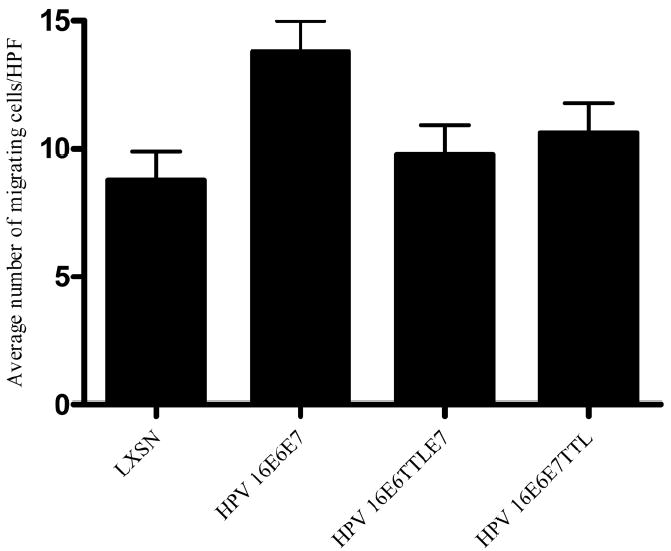
HMVECs were plated on collagen-coated transwell membranes. Conditioned media from HFKs retroviral transduced with LXSN, HPV 16E6E7, HPV 16E6TTLE7 and HPV 16E6E7TTL were added to the bottom chamber. After 3 hrs, 6 high power fields (HPF) of cells on the bottom were counted. The results (mean number of migrated cells per high power field (HPF) ± SEM) are representative of three experiments each conducted in triplicate. p< 0.005 for LXSN vs HPV 16E6E7 and p< 0.02 for HPV 16E6E7 and HPV 16E6TTLE7 or HPV 16E6E7TTL.
VEGF, but not IL-8, is required for the increased migration of HMVECs in vitro
Angiogenesis involves activation of endothelial cells. IL-8 and VEGF have been reported to regulate angiogenesis and affect endothelial cell migration and proliferation (Heidemann et al., 2003; Olsson et al., 2006; Rousseau et al., 1997). VEGF is elevated in cervical carcinomas (Smith-McCune et al., 1997) and IL-8 is elevated in oral squamous cell carcinomas (Lingen et al., 1996a; b 1996). Data presented in Figure 2 indicate that an increase in these two inducers in the conditioned media from HPV E6 or E7-expressing HFKs is insufficient to induce endothelial cell migration. The requirement for IL-8 and/or VEGF, however, in HPV-mediated angiogenesis remains unclear. To determine the necessity of IL-8 and/or VEGF for increased migration of endothelial cells seen when HMVECs are exposed to conditioned media from HPV 16E6E7-expressing cells, neutralizing Abs directed against IL-8 or VEGF or isotype controls (mouse IgG1 or mouse IgG2B, respectively) were used to deplete conditioned media of IL-8 or VEGF. If IL-8 is successfully depleted and the depletion is specific, the level of IL-8 following incubation with antibodies to IL-8 should decrease but the level of VEGF should not. Similarly, if VEGF is successfully depleted and the depletion is specific, the level of VEGF should decrease following incubation with antibodies to VEGF but the level of IL-8 should not. The level of neither IL-8 nor VEGF should decrease following incubation with isotype controls. VEGF or IL-8 was specifically immunodepleted from the conditioned media (Figure 3A and B, respectively). Consistent with the literature (Chen et al., 2007) and as in Figure 2, conditioned media from cells expressing HPV 16E6E7 induced migration of HMVECS relative to LXSN, whether the media were untreated or incubated with the isotype control immunoglobulins (Figure 3C). IL-8 depleted conditioned media from cells expressing HPV 16E6E7 retained the ability to increase migration of HMVECs when compared to IL-8 depleted conditioned media from LXSN transduced cells. However, VEGF-depleted conditioned media from cells expressing HPV 16E6E7 did not induce migration of HMVECs when compared to VEGF-depleted conditioned media from LXSN transduced cells (Figure 3C). These results indicate that VEGF, but not IL-8, is required to increase endothelial cell migration in response to conditioned media from early passage HPV 16E6E7-expressing cells.
Figure 3. VEGF, but not IL-8, is required for the increased migration of HMVECs in vitro.
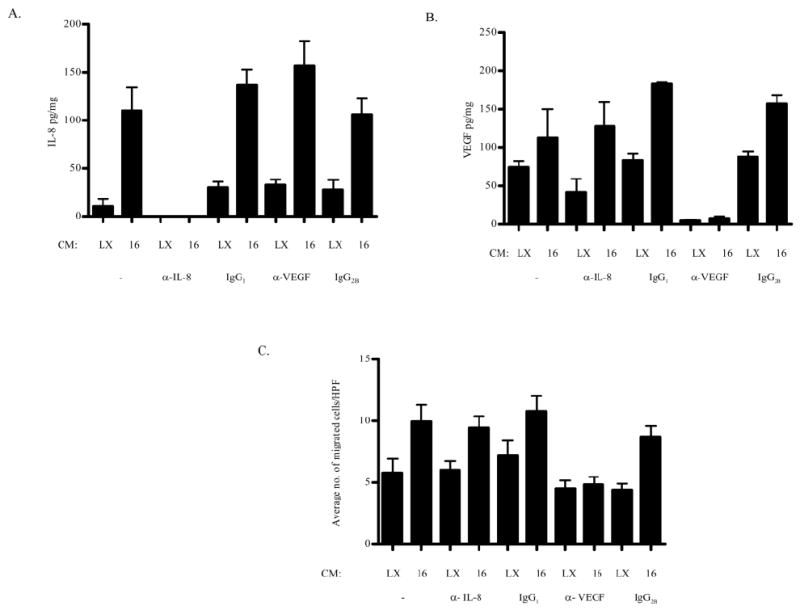
Conditioned media from HFKs retrovirally transduced with LXSN and HPV 16E6E7 depleted of (A) IL-8 or (B) VEGF were (C) tested for the ability to induce migration of endothelial cells as in Figure 2. The results (mean number of migrated cells per high power field (HPF) ± SEM) are representative of three experiments each conducted in triplicate. p< 0.0001 for LXSN vs HPV 16E6E7 (control isotype IgG2B); p< 0.002 for LXSN vs HPV 16E6E7 (depleted IL-8) and LXSN vs HPV 16E6E7 (control isotype IgG1).
pRb degradation is not required for the enhanced expression of VEGF
Data in Figure 1 indicate that expression of HPV 16 E7 is required for the enhanced levels of IL-8 and VEGF, equivalent to or greater than the levels seen with HPV 16E6E7. The LXCXE motif (aa 22-26) of HPV 16 E7 interacts with pRb as well as its other family members, p107 and p130 (Dyson et al., 1992; Munger et al., 1989b). By changing the cysteine at amino acid 24 to a glycine (C24G), HPV 16 E7 loses affinity for pRb and the ability to target pRb for degradation (Jones et al., 1997). Therefore, pooled HFKs were transduced with retrovirus expressing HPV 16E6E7C24G to test the hypothesis that pRb binding and degradation is required for increased levels of VEGF and IL-8. Consistent with the results in Figure 1, expression of HPV 16E6E7 significantly increased the levels of both angiogenic inducers relative to LXSN (Figure 4A). Conditioned media from cells expressing HPV 16E6E7C24G caused a 2.2-fold increase in the level of secreted IL-8 compared to retrovirus control LXSN (p< 0.0001), although not to the same extent as seen with conditioned media from HPV 16E6E7-expressing cells (Figure 4A). In contrast, VEGF expression was increased 8.7-fold (p< 0.0001) in cells expressing HPV 16E6E7C24G compared to retrovirus control LXSN (p< 0.0001) (Figure 4B) and to a greater extent than conditioned media from HPV 16E6E7-expressing cells (Figure 4B). Thus, in the presence of HPV 16 E6, HPV 16 E7 degradation of pRb is not required for enhanced IL-8 and VEGF expression.
Figure 4. Levels of IL-8 and VEGF in conditioned media were increased in cells expressing HPV 16E6E7C24G.
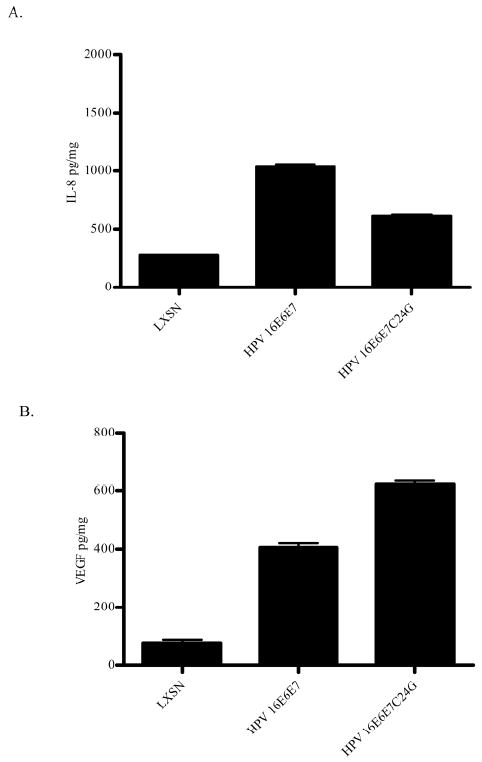
The experiment was conducted as in Figure 1. (A) The quantity of IL-8 was determined by ELISA and normalized to total protein in the conditioned media. The data represent mean ± SEM and are representative of three experiments each conducted in triplicate. p < 0.0001 for LXSN vs HPV 16E6E7; LXSN vs HPV 16E6E7C24G and p< 0.0002 for HPV 16E6E7 vs HPV 16E6E7C24G. (B) The quantity of VEGF was determined by ELISA and normalized to total protein in the conditioned media. The data represent mean ± SEM and are representative of three experiments each conducted in triplicate. p < 0.001 for LXSN vs HPV 16E6E7; LXSN vs HPV 16E6E7C24G and p< 0.0003 for HPV 16E6E7C24G vs HPV 16E6E7.
HPV 16E6E7C24G retains its ability to target p130 for degradation
VEGF and p130 expression are inversely correlated (Sanseverino et al., 2006). Although the HPV 16 E7C24G mutant has been reported to retain the ability to target another pRb family member, p107, for degradation (Gonzalez et al., 2001), the ability of this mutant to target p130 for degradation has not been reported. Therefore, the effect of HPV 16E6E7C24G on the steady state level of p130 was determined by Western Blot analysis. As expected, in cells expressing wild-type E7, the levels of hypophosphorylated pRb and of p130 in HPV 16E6E7-expressing cells were decreased relative to LXSN (Figure 5A,B). The steady state level of p130, but not pRb, was also decreased in cells expressing HPV 16E6E7C24G relative to LXSN (Figure 5A,B). As has been reported when HPV 16 E6 alone is expressed (Malanchi et al., 2004), relative to LXSN, the steady state level of pRb was increased in cells infected with HPV 16E6E7C24G (Figure 5A). Relative to LXSN, as expected, p53 expression was decreased in cells infected with HPV 16E6E7C24G (Scheffner et al., 1990), similarly to the decrease seen with HPV 16E6E7 (Figure 5C). These results demonstrate that HPV 16E6E7C24G, which does not destabilize pRb (Jones et al., 1997) still retained the ability to decrease the steady state level of p130.
Figure 5. HPV 16E6E7C24G, which does not associate with pRb, is still capable of targeting p130 for degradation.
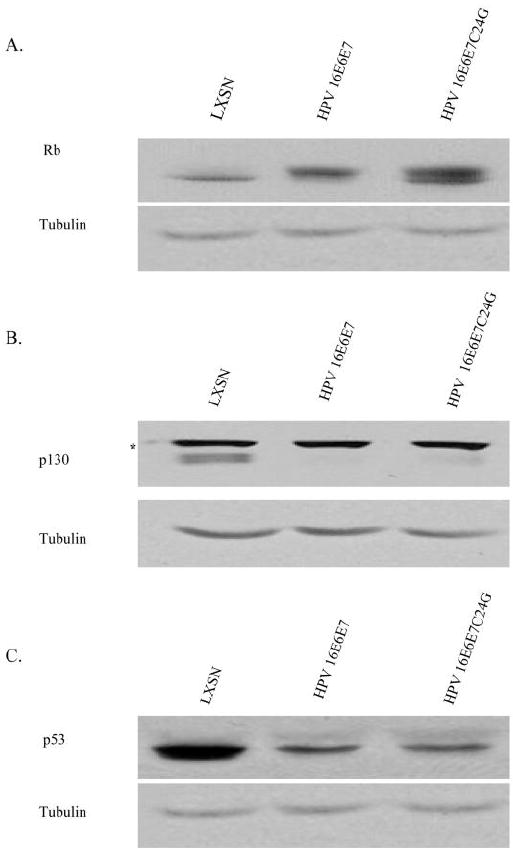
Whole cell lysates from HFKs transduced with LXSN, HPV 16E6E7 and HPV 16E6E7C24G, were analyzed by Western Blot to determine the steady state levels of (A) pRb, (B) p130, (C) p53 expression. Tubulin serves as a loading control. * = unknown cross-reacting protein.
Discussion
Increased angiogenesis has been correlated with progression of HPV lesions from premalignant dysplasia to malignancy. The onset of angiogenesis, both in an HPV 16 early region transgenic mouse model and in HPV 16-positive human cervices, is seen early in this process (Smith-McCune et al., 1997). Further, the direct contribution of HPV 16 E6 and E7 to angiogenesis has been demonstrated. Human foreskin keratinocytes expressing HPV 16 E6 and E7 contain higher levels of VEGF and IL-8 mRNA (Toussaint-Smith et al., 2004). Conditioned media taken from primary foreskin keratinocytes soon after their transduction with HPV 16 E6 and E7 contain increased levels of VEGF and IL-8 and enhance angiogenesis when introduced into mice in a Matrigel plug (Bequet-Romero and Lopez-Ocejo, 2000; Chen et al., 2007; Toussaint-Smith et al., 2004). In the present study, the individual contributions of HPV 16 E6 and E7 oncoproteins in regulating the angiogenic response seen in the early stages of cervical cancer were elucidated. To do so, translation termination linkers were introduced into the coding regions of HPV 16 E6 or E7. E6 in the context of the E7TTL mutation (E6E7TTL) and E7 in the context of the E6TTL mutation (E6TTLE7) in human foreskin keratinocytes led to a significant increase in VEGF and IL-8 secretion (Figure 1).
Several mechanisms could account for how HPV 16 E6 and E7 each increase the levels of IL-8 and VEGF. One possibility is that E6 and/or E7 increases IL-8 and VEGF protein levels by affecting transcription factors that bind cognate sites located on the promoters of their respective genes. IL-8 transcription is regulated through cooperation with three inducible transcription factors, NFκB, AP-1 and C/EBP (Hoffmann et al., 2002). HPV 16 E6 is able to transactivate promoters containing NF B sites (Desaintes et al., 1992; James et al., 2006; Nees et al., 2001). HPV 16 E7 is able to transactivate promoters containing AP-1 sites (Antinore et al., 1996) as well as promoters containing C/EBP responsive elements (Muller et al., 1999). E6 can stimulate several promoters through their Sp-1 responsive elements (Desaintes et al., 1992) and specifically stimulates the VEGF promoter through Sp-1 sites via a p53 independent mechanism (Lopez-Ocejo et al., 2000). E7 may also transactivate AP-1 sites on the VEGF promoter (Antinore et al., 1996). Alternatively E6 and E7 may stimulate transcription of IL-8 and VEGF through interactions with the basal transcription machinery (Desaintes et al., 1992; Massimi et al., 1996; Phillips and Vousden, 1997). Finally, E7 may alter expression of these angiogenic factors by causing chromosomal remodeling by interacting with histone deacetylases (Bernat et al., 2003; Brehm et al., 1999).
Transcription of VEGF is also upregulated by hypoxia inducible factor (HIF-1) (Bardos and Ashcroft, 2005; Brat et al., 2003). Under normoxic conditions, HIF-1α is hydoxylated by prolyl hydroxylases and targeted for degradation by the VHL E3 ubiquitin ligase complex (Epstein et al., 2001). HPV 16 E7 appears to replace VHL in this complex to target pRb for degradation (Huh et al., 2007). This suggests that another mechanism by which E7 could enhance the level of VEGF would be through increasing the level of HIF-1 in normoxic conditions. However, neither HIF-1α nor HIF-2α (another inducer of VEGF) was elevated in any of the transduced HFKs under normoxic conditions (data not shown).
Data shown in Figure 1 suggest that E7 is the major contributor to the increased expression of VEGF and IL-8. While this could be because E7 has a greater ability to alter the level of transcription of these two angiogenic inducers, another possibility could relate to the level of E7 versus E6 produced by the different HPV 16E6E7 wild-type or mutated cassettes. Expression of E6E7 within the retroviral vector favors unspliced bicistronic mRNA and translation of E6 (Zheng et al., 2004) (Tang et al., 2006). Premature termination of E6, brought about by splicing of the bicistronic mRNA within E6, favors translation of E7 (Tang et al., 2006). This suggests that placement of the TTL within E6 (at nt 280) would not only result in premature termination of E6 but would similarly enhance translation of E7. Further, we noted that the introduction of the TTL into E6 enhanced the level of splicing (data not shown), again enhancing E7 translation. Thus, the quantity of E7 in the HPV 16E6TTLE7 recombinant retrovirus may be greater than that produced by the HPV 16E6E7 recombinant retrovirus.
Functionally, conditioned media from either HPV 16 E6 or E7-expressing cells did not induce migration of human microvascular endothelial cells (HMVECs) (Figure 2). This is in marked contrast to the ability of E6 and E7 together to induce HMVEC migration. These results indicate that the increased levels of VEGF and IL-8 seen in the presence of E7 are insufficient to induce an angiogenic response. Since angiogenesis requires a shift in the ratio of angiogenic inhibitors to inducers, to favor the latter, the data suggest that other components in the conditioned media must be altered by HPV 16 E6 expression within HFKs for this switch to occur. In an HPV 16 transgenic mouse model of estrogen-dependent cervical carcinogenesis, E6 expression does not cause malignancy while E7 expression gives rise to cervical tumors which are smaller than those seen with E6 and E7 (Riley et al., 2003). While the underlying mechanisms responsible for these results may be many, based on our data one contributor may be the limited ability of either oncogene alone to provide the appropriate microenvironment for maximal tumor growth. It will be informative to determine whether combining conditioned media from E6-expressing cells with conditioned media from E7-expressing cells results in media competent to alter HMVEC migration.
Data further show that the ability of HPV 16 E7 to induce VEGF is pRb-independent (Figures 4 and 5). One possible mechanism for the increase in VEGF expression is through degradation of pRb family member p130. Sanseverino et al 2006 found that VEGF and p130 expression were inversely correlated in endometrial cancers (Sanseverino et al., 2006). Specifically, immunohistochemical staining showed low VEGF staining correlated with high expression of p130 while higher levels of VEGF correlated with low expression of p130 (Sanseverino et al., 2006). Alternatively, the pRb-independent activity of E7 could be due to degradation of p107 (Gonzalez et al., 2001) or the interactions, for example with AP-1or histone deacetylases, which are retained with the HPV 16 E7C24G mutant (Antinore et al., 1996; Brehm et al., 1999). A functional analysis of the conditioned media from cells expressing HPV 16 E6E7C24G will establish whether, in the context of E6, pRb-independent activities of E7 are sufficient to the altered endothelial cell behavior.
Although the level of IL-8 is greater in conditioned media from cells expressing HPV 16E6E7C24G when compared to conditioned media from control (LXSN) cells, the level of IL-8 is not as great as that produced by HFKs expressing wild type E7 (HPV 16 E6E7, Figure 4). The fold increase in IL-8 with HPV 16E6E7C24G is similar to that seen with HPV 16E6E7TTL (compare Figure 4 to Figure 1). This suggests that pRb-dependent functions of E7 contribute to the increased expression of IL-8.
Using specific blocking antibodies to IL-8 or VEGF, we showed that VEGF was required for HPV 16 E6 and E7 together to induce HMVEC migration (Figure 3). Thus blockade of either E6 or E7 and VEGF expression may result in suppression of an angiogenic response in vivo.
Finally, although this research focused on the HPV 16 E6 and E7 proteins, conditioned media from cervical keratinocytes stably transfected with the intact HPV 16 genome also increase endothelial cell proliferation and migration in vitro and recruit a blood supply in vivo (Chen et al., 2007). To understand the role that altered expression of angiogenic factors plays in the viral life cycle, it will be important to determine the ability of the low-risk HPV genome and E6 and/or E7 to alter expression of these factors.
In summary, our findings suggest the following: (i) HPV 16 E6 and E7 each upregulates the level of VEGF and IL-8 in conditioned media; (ii) neither conditioned media from HPV 16 E6 nor E7-expressing HFKs induce migration of HMVECs; (iii) VEGF, but not IL-8, is required for HPV 16 E6 and E7 together to induce HMVEC migration; and (iv) the increased level of VEGF induced by HPV 16 E7 is independent of pRb binding and degradation.
Materials and Methods
Cell culture and Retroviral Infection
Human foreskin keratinocytes (HFKs) were prepared as previously described (Chen et al., 2007; Toussaint-Smith et al., 2004; Wu et al., 1982) and cultured in complete keratinocyte serum free media (C-KSFM) supplemented with human recombinant epidermal growth factor (GIBCO/BRL) and bovine pituitary extract (GIBCO/BRL). Pooled HFKs were grown to about 40% confluence and infected with 5 ml of the recombinant retrovirus encoding HPV 16E6E7, HPV 16E6TTLE7, HPV 16E6E7TTL, HPV 16E6E7C24G or parental virus LXSN and 8 μg polybrene/ml. As a control, mock infected cells was generated by incubating HFKs with 5 ml of DMEM + 10% FCS and 8 μg/ml polybrene. After 6 hrs the culture media was aspirated and fresh C-KSFM added. The neomycin resistance gene expressed within the vector, allow selection of infected cells with 200 μg/ml G418 for 3 days. Selected cells were expanded and used for further experiments.
Human dermal microvascular endothelial cells (HMVECS) (Cambrex) were obtained at passage 7 or 8. Cells were seeded in tissue culture flasks coated with type 1 rat tail collagen (BD Biosciences) in complete endothelial cell growth medium-2 (EGM-2) (Lonza) supplemented with 10% FBS (Hyclone) and 1% antibiotic/antimycotic (100 U/ml penicillin/streptomycin and 25 μg of amphotericin B) (Invitrogen). Cells were collected for passage with trypsin (Invitrogen), and centrifuged at 500 × g for 5 minutes. Cells were resuspended in 5 ml EBM-2 and live cells counted by trypan blue exclusion on a hemocytometer. Three to 5 × 105 live cells were seeded in 75 cm2 flasks pre-coated with type 1 rat-tail collagen.
Mutational Analysis
HPV 16E6E7 was generated similarly to the HPV 16E6E7 cassette previously cloned into the retrovirus LXSN by the Galloway laboratory (Halbert et al., 1991). The original cloning strategy, however, did not allow manipulation of the cassette. Briefly, a DNA fragment 56 to 883, encompassing E6 and E7 was PCR amplified with the following primers containing BamHI and EcoRI sites: 5’-CGGAATTCACCGGTTAGTATAAAAGC-3’ and 5’-CGCGGATCCG GATCAGCCATGGTAGATTATGG- 3’. The HPV-16 E6 and E7 mutants, HPV 16E6TTLE7 and HPV 16E6E7TTL, were generated using the Quick-Change Site-Directed Mutagenesis kit (Stratagene) according to manufacturers instructions. Oligonucleotides for HPV 16E6TTLE7 (5’-GTATATAGAGATGGGAATCCTTAGTTAACTAAATATGCTGTATGTG-3’ and 5’-CA CATACAGCATATTTAGTTAACTAAGGATTCCCATCTCTATATAC-3; HPV 16E6E7TTL 5’-CCGGACAGAGCCTTAGTTAACTAACATTACAATATTGTAACC-3’ and 5’-GGTTACA ATATTGTAATGTTAGTTAACTAAGGCTC GTCCGG-3’) were designed based on the published sequence for HPV 16 (Seedorf et al., 1985) with the translation termination linker (TTL) sequence and placement taken from Munger et al., 1989a, to preclude expression of E6 or E7. The presence of TTLs in the E6 and E7 ORFs was verified by presence of an engineered HpaI site within the linker. The HPV 16E6E7C24G pRb-binding deficient mutant was generated by site-directed mutagenesis. Site-directed mutagenesis was performed using the Quick-Change II XL Site-Directed Mutagenesis Kit (Stratagene) according to manufacturers instructions. The following oligonucleotides (Invitrogen) were used to introduce a single amino acid substitution at residue 24: 5’-CAGAGACAACTGATCTCTACGGTTATGAGCAATTAAATGAC- 3’ and 5’-GTCATTTAATTGCTCATAACCGTAGAGATCAGTTGTCTCTG- 3’) based on the published sequence (Seedorf et al., 1985). The plasmid DNAs were subcloned into the BamHI (Invitrogen) and EcoRI (Invitrogen) sites of the pLXSN vector. Plasmids were sequenced to confirm the presence of corresponding mutation.
Collection of Condition Media
Following selection, retrovirally transduced HFKs were transferred 1:4 onto mitomycin-C (4ug/ml in ddH2O, Sigma) treated NIH-3T3 J2 fibroblasts and grown in complete E media until 80% confluence (passage 2). At passage 3, 2.5×106 cells were plated in E media in 10 cm plates with 2×105 feeder cells for 24 hours. Serum-free conditioned media (Lingen et al., 1996b) was generated by rinsing the cells three times with 4 ml of basal media (BM) (3 parts DMEM/1 part Ham’s F-12 with 100 U/ml of penicillin and 100 μg/ml of streptomycin), incubating the cells for 4 hours in BM, replacing this media with 8 ml of the same media and harvesting CM 24 hours later. For functional assays, CM was concentrated 30x using 15 ml Amicon Ultracel (Millipore) centrifugal filter devices (10000 MWCO) at 4°C for 45 minutes at 3000 rpms. Protein concentrations were determined using the Bio-Rad DC protein assay kit.
Western Blot Analysis
Whole cell lysates were prepared by using 1x lysis buffer (20 mM Tris, pH 6.8, 1% sodium dodecyl sulfate (SDS), 1 mM EDTA) plus protease and phosphatase inhibitor cocktail (20 μM Na3VO4 and 100 mM NaF, Sigma) and protein concentrations were determined by using the Bio-Rad (Bradford) protein assay. SDS-PAGE and Western Blotting were performed as previously described using 50 μg of protein (Chen et al., 2007). Primary antibodies were diluted in TBS-t supplemented with 5% nonfat dry milk used at the following dilutions as previously described (Chen et al., 2007; Toussaint-Smith et al., 2004; Zhang et al., 2006): p53 (DO-1 Sc-126, Santa Cruz) 1:1000, pRb (BD Pharminogen) 1:1000; Rb2 (p130, BD Bioscience) 1:1000; and loading controls GAPDH (MAB 374, Chemicon) 1:30,000 and Tubulin (Sigma) 1:3000. Secondary goat anti-mouse horseradish peroxidase (HRP) (BioRad) was diluted 1:5000 into TBS-t for 1 hr. The Pierce ECL Western Blotting substrate (Thermo Scientific) protein detection system was used to visualize proteins. Densitometry was performed using Quality One software (BioRad).
Depletion of IL8 or VEGF from Conditioned Media
Concentrated CM (500 μg) from LXSN and HPV 16E6E7 were incubated with 10 μg of mouse monoclonal neutralizing antibodies (Abs) directed against IL-8 (R&D Systems, MAB208), VEGF (R&D Systems, MAB 293), or isotype controls (IgG1 for IL-8, R&D Systems, MAB002 or IgG2B for VEGF R&D Systems, MAB 004) and rotated for 2 hours at 4°C. Twenty μl of 50% vol/vol of protein G sepharose beads (Delorme et al., 2001) in 1x phosphate buffered saline (PBS) was added and rotated for 2 hours. The beads were then centrifuged at 4,000 rpm for 30 seconds and the supernatants were collected into microfuge tubes. The protein concentration of the supernatants was determined using the Bio-Rad DC protein assay kit and used for ELISA and migration assays.
ELISA Analysis
VEGF165 secretion was detected in unconcentrated cell supernatants using the Quantikine Human VEGF ELISA kit (R&D systems) according to the manufacturer’s protocol. IL-8 secretion was detected in unconcentrated cell supernatants using the Quantikine Human CXCL8/IL-8 ELISA kit (R&D systems).
In vitro Endothelial Cell Migration Assay
The endothelial cell migration assay was performed as previously described (Chen et al., 2007) with minor modifications. Briefly, the upper and lower chamber of the transwell inserts (8 μm pore filter; Costar) were coated with 60 μl of type I rat tail collagen 50 μg/ml (BD Pharmingen) and incubated for no more than 90 minutes at 37°C, 5% CO2. The collagen was aspirated and the transwell inserts were placed into individual wells of a 24 well tissue culture plate. Human dermal microvascular endothelial cells (HMVECs) (2 ×104 cells) were resuspended in 100 μl of EBM-2 (Cambrex) supplemented with 0.5% BSA and added to the top of each transwell. Six hundred microliters of EBM-2 supplemented with 0.5% BSA, VEGF (50 ng/ml, Peprotech) or conditioned media (50 μg) from retrovirally transduced HFKs was added to the lower chamber of each transwell. After 3.5 hrs of incubation at 37°C, non-migrating cells were removed using a cotton swab. The migrating cells that attached to the bottom surface of the membrane were fixed with 600 μl of methanol and stained with Diff-quick kit (Invitrogen). The number of migrated cells per membrane was determined by counting 6 high power fields (HPFs) on the bottom of the membrane with an inverted microscope at 20x magnification. Assays were performed in triplicate.
Statistical analysis
Student unpaired two tailed t-test (Prism GraphPad 4.0) was used for statistical analysis. A probability value of less than 0.05 was considered significant.
Acknowledgments
This work was supported by NIH R21CA124314 (AR), NIH T32 CA111198 (JW). We thank the National Gene Vector Biorepository P40 RR024928 for providing AmphoPheonix cells. We thank Maureen Harrington, Harikrishna Nakshatri, Kenneth Cornetta, Mark Goebl and D. Wade Clapp for helpful discussions, Lisa Barrow-Laing for alerting us to the relationship of VEGF to p130 and Mark Goebl and Lisa Barrow-Laing for critically reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alani RM, Munger K. Human papillomaviruses and associated malignancies. J Clin Oncol. 1998;16:330–337. doi: 10.1200/JCO.1998.16.1.330. [DOI] [PubMed] [Google Scholar]
- Antinore MJ, Birrer MJ, Patel D, Nader L, McCance DJ. The human papillomavirus type 16 E7 gene product interacts with and trans-activates the AP1 family of transcription factors. EMBO J. 1996;15:1950–1960. [PMC free article] [PubMed] [Google Scholar]
- Bardos JI, Ashcroft M. Negative and positive regulation of HIF-1: a complex network. Biochim Biophys Acta. 2005;1755:107–120. doi: 10.1016/j.bbcan.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Bequet-Romero M, Lopez-Ocejo O. Angiogenesis modulators expression in culture cell lines positives for HPV-16 oncoproteins. Biochem Biophys Res Commun. 2000;277:55–61. doi: 10.1006/bbrc.2000.3628. [DOI] [PubMed] [Google Scholar]
- Bernat A, Avvakumov N, Mymryk JS, Banks L. Interaction between the HPV E7 oncoprotein and the transcriptional coactivator p300. Oncogene. 2003;22:7871–7881. doi: 10.1038/sj.onc.1206896. [DOI] [PubMed] [Google Scholar]
- Bouck N. P53 and angiogenesis. Biochim Biophys Acta. 1996;1287:63–66. doi: 10.1016/0304-419x(96)00005-4. [DOI] [PubMed] [Google Scholar]
- Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- Brat DJ, Kaur B, Van Meir EG. Genetic modulation of hypoxia induced gene expression and angiogenesis: relevance to brain tumors. Front Biosci. 2003;8:d100–116. doi: 10.2741/942. [DOI] [PubMed] [Google Scholar]
- Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999;18:2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Li F, Mead L, White H, Walker J, Ingram DA, Roman A. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology. 2007;367:168–174. doi: 10.1016/j.virol.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook T, Storey A, Almond N, Osborn K, Crawford L. Human papillomavirus type 16 cooperates with activated ras and fos oncogenes in the hormone-dependent transformation of primary mouse cells. Proc Natl Acad Sci U S A. 1988;85:8820–8824. doi: 10.1073/pnas.85.23.8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme C, Brussow H, Sidoti J, Roche N, Karlsson KA, Neeser JR, Teneberg S. Glycosphingolipid binding specificities of rotavirus: identification of a sialic acid-binding epitope. J Virol. 2001;75:2276–2287. doi: 10.1128/JVI.75.5.2276-2287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaintes C, Hallez S, Van Alphen P, Burny A. Transcriptional activation of several heterologous promoters by the E6 protein of human papillomavirus type 16. J Virol. 1992;66:325–333. doi: 10.1128/jvi.66.1.325-333.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66:6893–6902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- Gabellini C, Del Bufalo D, Zupi G. Involvement of RB gene family in tumor angiogenesis. Oncogene. 2006;25:5326–5332. doi: 10.1038/sj.onc.1209631. [DOI] [PubMed] [Google Scholar]
- Gomez-Manzano C, Fueyo J, Jiang H, Glass TL, Lee HY, Hu M, Liu JL, Jasti SL, Liu TJ, Conrad CA, Yung WK. Mechanisms underlying PTEN regulation of vascular endothelial growth factor and angiogenesis. Ann Neurol. 2003;53:109–117. doi: 10.1002/ana.10396. [DOI] [PubMed] [Google Scholar]
- Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75:7583–7591. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin EC, DiMaio D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A. 2000;97:12513–12518. doi: 10.1073/pnas.97.23.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Christofori G, Naik P, Arbeit J. Transgenic mouse models of tumour angiogenesis: the angiogenic switch, its molecular controls, and prospects for preclinical therapeutic models. Eur J Cancer. 1996;32A:2386–2393. doi: 10.1016/s0959-8049(96)00401-7. [DOI] [PubMed] [Google Scholar]
- Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989;8:3905–3910. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidemann J, Ogawa H, Dwinell MB, Rafiee P, Maaser C, Gockel HR, Otterson MF, Ota DM, Lugering N, Domschke W, Binion DG. Angiogenic effects of interleukin 8 (CXCL8) in human intestinal microvascular endothelial cells are mediated by CXCR2. J Biol Chem. 2003;278:8508–8515. doi: 10.1074/jbc.M208231200. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002;72:847–855. [PubMed] [Google Scholar]
- Huh K, Zhou X, Hayakawa H, Cho JY, Libermann TA, Jin J, Harper JW, Munger K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J Virol. 2007;81:9737–9747. doi: 10.1128/JVI.00881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MA, Lee JH, Klingelhutz AJ. Human papillomavirus type 16 E6 activates NF-kappaB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding motif-dependent manner. J Virol. 2006;80:5301–5307. doi: 10.1128/JVI.01942-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DL, Thompson DA, Munger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- Kaur P, McDougall JK, Cone R. Immortalization of primary human epithelial cells by cloned cervical carcinoma DNA containing human papillomavirus type 16 E6/E7 open reading frames. J Gen Virol. 1989;70(Pt 5):1261–1266. doi: 10.1099/0022-1317-70-5-1261. [DOI] [PubMed] [Google Scholar]
- Lee HY, Srinivas H, Xia D, Lu Y, Superty R, LaPushin R, Gomez-Manzano C, Gal AM, Walsh GL, Force T, Ueki K, Mills GB, Kurie JM. Evidence that phosphatidylinositol 3-kinase- and mitogen-activated protein kinase kinase-4/c-Jun NH2-terminal kinase-dependent Pathways cooperate to maintain lung cancer cell survival. J Biol Chem. 2003;278:23630–23638. doi: 10.1074/jbc.M300997200. [DOI] [PubMed] [Google Scholar]
- Lingen MW, Polverini PJ, Bouck NP. Inhibition of squamous cell carcinoma angiogenesis by direct interaction of retinoic acid with endothelial cells. Lab Invest. 1996a;74:476–483. [PubMed] [Google Scholar]
- Lingen MW, Polverini PJ, Bouck NP. Retinoic acid induces cells cultured from oral squamous cell carcinomas to become anti-angiogenic. Am J Pathol. 1996b;149:247–258. [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA. Pathogenesis of human papillomaviruses in differentiating epithelia. Microbiol Mol Biol Rev. 2004;68:362–372. doi: 10.1128/MMBR.68.2.362-372.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Ocejo O, Viloria-Petit A, Bequet-Romero M, Mukhopadhyay D, Rak J, Kerbel RS. Oncogenes and tumor angiogenesis: the HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene. 2000;19:4611–4620. doi: 10.1038/sj.onc.1203817. [DOI] [PubMed] [Google Scholar]
- Malanchi I, Accardi R, Diehl F, Smet A, Androphy E, Hoheisel J, Tommasino M. Human papillomavirus type 16 E6 promotes retinoblastoma protein phosphorylation and cell cycle progression. J Virol. 2004;78:13769–13778. doi: 10.1128/JVI.78.24.13769-13778.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massimi P, Pim D, Storey A, Banks L. HPV-16 E7 and adenovirus E1a complex formation with TATA box binding protein is enhanced by casein kinase II phosphorylation. Oncogene. 1996;12:2325–2330. [PubMed] [Google Scholar]
- Muller C, Alunni-Fabbroni M, Kowenz-Leutz E, Mo X, Tommasino M, Leutz A. Separation of C/EBPalpha-mediated proliferation arrest and differentiation pathways. Proc Natl Acad Sci U S A. 1999;96:7276–7281. doi: 10.1073/pnas.96.13.7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K. The role of human papillomaviruses in human cancers. Front Biosci. 2002;7:d641–649. doi: 10.2741/a800. [DOI] [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989a;63:4417–4421. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Werness BA, Dyson N, Phelps WC, Harlow E, Howley PM. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989b;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive genes and upregulate proliferation-associated and NF-kappaB-responsive genes in cervical keratinocytes. J Virol. 2001;75:4283–4296. doi: 10.1128/JVI.75.9.4283-4296.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson R, Maxhuni A, Carlsson PO. Revascularization of transplanted pancreatic islets following culture with stimulators of angiogenesis. Transplantation. 2006;82:340–347. doi: 10.1097/01.tp.0000229418.60236.87. [DOI] [PubMed] [Google Scholar]
- Phillips AC, Vousden KH. Analysis of the interaction between human papillomavirus type 16 E7 and the TATA-binding protein, TBP. J Gen Virol. 1997;78(Pt 4):905–909. doi: 10.1099/0022-1317-78-4-905. [DOI] [PubMed] [Google Scholar]
- Riley RR, Duensing S, Brake T, Munger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–4871. [PubMed] [Google Scholar]
- Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- Sanseverino F, Santopietro R, Torricelli M, D’Andrilli G, Russo G, Cevenini G, Bovicelli A, Leoncini L, Scambia G, Petraglia F, Claudio PP, Giordano A. pRb2/p130 and VEGF expression in endometrial carcinoma in relation to angiogenesis and histopathologic tumor grade. Cancer Biol Ther. 2006;5:84–88. doi: 10.4161/cbt.5.1.2345. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Seedorf K, Krammer G, Durst M, Suhai S, Rowekamp WG. Human papillomavirus type 16 DNA sequence. Virology. 1985;145:181–185. doi: 10.1016/0042-6822(85)90214-4. [DOI] [PubMed] [Google Scholar]
- Smith-McCune K, Zhu YH, Hanahan D, Arbeit J. Cross-species comparison of angiogenesis during the premalignant stages of squamous carcinogenesis in the human cervix and K14-HPV16 transgenic mice. Cancer Res. 1997;57:1294–1300. [PubMed] [Google Scholar]
- Tang S, Tao M, McCoy JP, Jr, Zheng ZM. The E7 oncoprotein is translated from spliced E6*I transcripts in high-risk human papillomavirus type 16- or type 18-positive cervical cancer cell lines via translation reinitiation. J Virol. 2006;80:4249–4263. doi: 10.1128/JVI.80.9.4249-4263.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toussaint-Smith E, Donner DB, Roman A. Expression of human papillomavirus type 16 E6 and E7 oncoproteins in primary foreskin keratinocytes is sufficient to alter the expression of angiogenic factors. Oncogene. 2004;23:2988–2995. doi: 10.1038/sj.onc.1207442. [DOI] [PubMed] [Google Scholar]
- Volpert OV, Dameron KM, Bouck N. Sequential development of an angiogenic phenotype by human fibroblasts progressing to tumorigenicity. Oncogene. 1997;14:1495–1502. doi: 10.1038/sj.onc.1200977. [DOI] [PubMed] [Google Scholar]
- Wells SI, Francis DA, Karpova AY, Dowhanick JJ, Benson JD, Howley PM. Papillomavirus E2 induces senescence in HPV-positive cells via pRB- and p21(CIP)-dependent pathways. EMBO J. 2000;19:5762–5771. doi: 10.1093/emboj/19.21.5762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YJ, Parker LM, Binder NE, Beckett MA, Sinard JH, Griffiths CT, Rheinwald JG. The mesothelial keratins: a new family of cytoskeletal proteins identified in cultured mesothelial cells and nonkeratinizing epithelia. Cell. 1982;31:693–703. doi: 10.1016/0092-8674(82)90324-5. [DOI] [PubMed] [Google Scholar]
- Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci U S A. 2006;103:437–442. doi: 10.1073/pnas.0510012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZM, Tao M, Yamanegi K, Bodaghi S, Xiao W. Splicing of a cap-proximal human Papillomavirus 16 E6E7 intron promotes E7 expression, but can be restrained by distance of the intron from its RNA 5’ cap. J Mol Biol. 2004;337:1091–1108. doi: 10.1016/j.jmb.2004.02.023. [DOI] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat Rev Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]