

Abstract
In an effort to develop novel covalent modifiers of dimethylarginine dimethylaminohydrolase (DDAH) that are useful for biological applications, a set of “fragment”-sized inhibitors that were identified using a high-throughput screen are tested for time-dependent inhibition. One structural class of inactivators, 4-halopyridines, show time- and concentration-dependent inactivation of DDAH and the inactivation mechanism of one example, 4-bromo-2-methylpyridine (1), is characterized in detail. The neutral form of halopyridines is not very reactive with excess glutathione. However, 1 readily reacts, with loss of its halide, in a selective, covalent and irreversible manner with the active-site Cys249 of DDAH. This active-site Cys is not particularly reactive (pKa ca. 8.8) and 1 does not inactivate papain (Cys pKa ca. ≤ 4), suggesting that, unlike many reagents, Cys nucleophilicity is not a predominating factor in selectivity. Rather, binding and stabilization of the more reactive pyridinium form of the inactivator by a second moiety, Asp66, is required for facile reaction. This constraint imparts a unique selectivity profile to these inactivators. To our knowledge, halopyridines have not previously been reported as protein modifiers, and therefore represent a first-in-class example of a novel type of quiescent affinity label.
INTRODUCTION
Human isoforms of dimethylarginine dimethylaminohydrolase (DDAH; EC 3.5.3.18) regulate nitric oxide production by catabolizing Nω, Nω-dimethyl-L-arginine, an endogenous inhibitor of nitric oxide synthases.1 Development of activity-based probes for this enzyme and other putative drug targets in the pentein superfamily of guanidine-modifying enzymes2,3 is of interest for both inhibitor design and for biological studies because the activity levels of these enzymes do not always correspond to their cellular abundance.4 Prior activity-based probes for this superfamily have relied on an α-haloacetamidine core5,6 that resembles the well-known α-halo ketone motif of many cysteine protease inactivators in structure and mechanism.7 The similarities between the amidine inactivators and the guanidine substrates contribute to the target selectivity of these probes, but may limit their broader application in profiling the reactivity of proteomic mixtures. In general, many well-studied affinity labels have been used as the reactive groups of activity-based protein profiling reagents.8 However, as the methodology for reactivity-based proteomic screening evolves, it will become increasingly important to develop novel reactive groups with different selectivity profiles that will allow access to different subsets of the proteome. To address these issues, we explored “fragment”-sized inhibitors9 of DDAH, derived from a high-throughput screen, as a source for potentially novel covalent modifiers. Herein we describe the discovery of 4-halopyridines as quiescent affinity labels of DDAH. The mechanism of 4-bromo-2-methylpyridine is characterized as a paradigm case. To our knowledge, halopyridines have not previously been reported as covalent protein modifiers, and therefore represent a novel class.
RESULTS AND DISCUSSION
4-Halopyridines are time-dependent irreversible inhibitors
In an effort to discover structurally diverse inhibitors of DDAH, we performed a high-throughput screen of 4000 “fragment”-sized compounds from a commercial library (Chembridge, San Diego, CA, full description to be published elsewhere). A number of structurally divergent DDAH inhibitors were identified. Among these hits, we sought to distinguish the rapidly-reversible inhibitors from the time-dependent inhibitors. A selection of compounds with apparent IC50 values < 120 μM were assayed for time-dependent inhibition of DDAH that could not be readily reversed upon dilution into excess substrate. Three compounds were found to inhibit DDAH in a time-dependent manner: 4-bromo-2-methylpyridine (1), 4-chloro-2-hydroxymethylpyridine (2) and 4-(2-quinolinyl)-3-buten-2-ol (3) (Figure 1A-C). Notably, each of these compounds contains a pyridine moiety, and a quinoline-5,8 dione-containing compound has recently been reported as an inactivator of the related enzyme protein arginine deiminase-4.10
Figure 1.

Time-dependent inhibition of DDAH by inactivators 1-3. Time-dependent loss in DDAH (29 μM) activity (λ, solid line) is observed after incubation with 1 (300 μM) (A), 2 (1.2 mM) (B), or 3 (1 mM) (C), each in K2HPO4 buffer (100 mM), KCl (100 mM) and EDTA (0.05 mM) at pH 7.5, 25 °C. Inactivation experiments are also repeated when preceded by an incubation (1 h) of each inactivator with excess glutathione (5 mM) (ν, dashed lines); (D) Inactivated enzyme was dialyzed overnight and assayed for recovery of activity (“E“ is an enzyme-only control; “E dialysis” is enzyme-only after dialysis; “E+I” is inactivated enzyme; “E+I dialysis” is inactivated enzyme after dialysis).
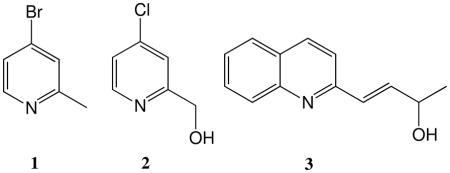
To determine if these inactivators are just reactive species that indiscriminately modify biological nucleophiles, we preincubated each with an excess of glutathione and then retested for time-dependent inhibition of DDAH (Figure 1A-C). Subsequent to its preincubation with glutathione, 3 lost all ability to inactivate DDAH. This compound was not considered further because reactivity with glutathione would likely preclude its use in many biological applications. In contrast, the remaining two compounds, both 4-halopyridines, did not show any reduction in their ability to inactivate DDAH after preincubation with glutathione, indicating that they are not “reactive” compounds as defined by these experimental conditions.
To further characterize DDAH inactivation by 4-halopyridines, we focused our mechanistic studies on 1. To determine whether the observed time-dependent inhibition is irreversible or just slowly reversible, DDAH was inactivated by 1 and then subjected to extensive dialysis. Control reactions that omit the inactivator retained > 90% of the initial activity, but inactivated samples recovered < 3% of their initial activity (Figure 1D). These results are consistent with an irreversible inactivation mechanism.
4-Halopyridines act at the active site
The concentration dependence of DDAH inactivation by 1 was measured to determine whether a reversible EI complex forms preceding inactivation. Inactivation rates do not display saturation kinetics, but instead show a linear dependence on inhibitor concentration that can be fitted by a second order inactivation rate constant of 4.8 ± 0.3 M-1s-1 (Figure 2). The lack of observed saturation kinetics suggests several possibilities: that 1 can react with DDAH purely by diffusion encounters, that an EI complex does form but the dissociation rate is much slower than the inactivation rate, or, most likely, that the concentration used here is much less than the inactivator’s KI value. In the absence of kinetic evidence for the formation of an EI complex, we sought to determine whether the addition of a DDAH substrate could compete for binding at the active site and slow the inactivation rate. Inactivation by 1 is slowed by the presence of excess substrate (Figure 3A), which is consistent with active-site binding. This hypothesis is further supported by additional experiments described below.
Figure 2.

Time- and concentration-dependent inactivation of DDAH by 1 at pH 7.5 (A) Exponential fits to the observed inactivation of DDAH at pH 7.5 and 25 °C by different concentrations of inhibitor: 0 (○), 83 μM (λ), 140 μM (□), 330 μM (ν), 600 μM (△), 1 mM (▲), 2 mM (◇), and 3 mM (◆). (B) Concentration dependence of the pseudo-first-order kobs values give a second order rate constant of 4.8 ± 0.3 M−1 s−1 for inactivation.
Figure 3.

Substrate protection and stoichiometry of bromide ion release. (A) Enzyme pre-incubated with substrate (Nω, Nω-dimethyl-L-arginine (3 mM, λ), 1 (300 μM, ν) and both substrate and 1 (■). (B) The molar equivalents ([Br−] / [enzyme], λ) of bromide ion release were monitored at various time points during enzyme inactivation, resulting in total release of approximately 1.2 equiv Br− ion.
Inactivation proceeds by covalent bond formation and loss of bromide
We performed two experiments to characterize the stoichiometry and covalent nature of inactivation. First, ESI-MS was used to compare uninhibited and fully inactivated DDAH. Treatment of DDAH with 1 leads to a mass addition of + 91 ± 10 Da (Table 1). The calculated monoisotopic mass is 170.97 Da for 1, and is 92.05 Da for 1 without the bromide. Therefore, the observed mass increase upon inactivation is consistent with that calculated for covalent attachment of one methylpyridine adduct to each DDAH monomer, and loss of the bromide ion from the inactivator and one proton from the enzyme (91.04 Da). Second, ion chromatography was used to quantify bromide ion release that occurs during the inactivation process. As inactivation of DDAH progressed to completion, 1.2 ± 0.1 equiv of bromide were released, consistent with a turnover number of approximately one inactivator per inactivation event (Figure 3B). These results support a mechanism in which one inactivator molecule forms a 1:1 covalent adduct with DDAH, with elimination of its bromide ion.
Table 1.
Summary of deconvoluted protein masses observed in ESI-MS spectra of control and inactivated DDAH
| DDAH preparation | Theoretical calc’d mass | Control incubations with no inactivator | Incubations with 1 | Mass difference | Incubations with 11 | Mass difference |
|---|---|---|---|---|---|---|
| (Da) | observed mass (Da)a | observed mass (Da) a | (Da) | observed mass (Da) | (Da) | |
| Wild-type | 30, 503 | 30, 498 | 30, 588 | 91 | 30,604 | 106 |
| Denatured b | 30, 503 | NDc | 30, 497 | 1 | ND | ND |
| C249S | 30,487 | 30, 481 | 30, 481 | 0 | ND | ND |
| H162G | 30, 423 | 30, 418 | 30, 508 | 90 | ND | ND |
| D66N | 30, 502 | 30, 496 | 30, 496 d | 0 | 30,604 | 108 |
| 30, 586 d | 90 | |||||
| E65Q | 30, 502 | 30, 496 | 30,498 | 2 | 30, 496 | 0 |
| D244N | 30, 502 | 30, 497 | 30, 588 | 91 | 30, 603 | 106 |
The masses obtained from deconvoluted ESI-MS spectra are reported with errors of ± 10 Da.
Heat denatured wild-type DDAH
Not determined
The modified peak occurs at an approximate 2:1 peak intensity ratio with the unmodified peak.
Inactivation modifies the active-site Cys residue
The point of attachment of the inactivator was mapped, and a more precise mass of the adduct was obtained by digestion of uninhibited and fully inactivated DDAH with V8 endoprotease and characterization of the resulting peptide fragments in each digest by MALDI-TOF/TOF MS. Twenty six peptides were identified by comparison with masses calculated for a theoretical digest (Table 2). These peptides cover approximately 53% of the primary sequence and include the active-site Cys249 and His162 residues. Two ions with m/z of 2353.14 and 1821.92 do not correspond to predicted peptides, but do match the calculated masses for the 235-254 and 240-254 peptides with each bearing an extra 91.06 ± 0.05 Da mass addition. A third signal with m/z at 1837.91 matches that of the 240-254 peptide bearing a 91.05 Da adduct plus an additional oxidation (16 Da) totaling 107.05 ± 0.05 Da. The control digest of uninhibited DDAH does not contain any signal at these m/z values, but instead contains the unmodified 240-254 peptide at m/z 1730.90. Thus, three peptide-adduct species are detected in MALDI-MS of inactivated DDAH digests. Their masses are consistent with addition of a methylpyridine adduct, which gives a theoretical mass addition of 91.04 Da, and with the whole protein ESI-MS mass measurement wheich yielded a mass addition of 91 ± 10 Da after inactivation.
Table 2.
Summary of proteolytic cleavage of inactivated DDAH with endoprotease V8a
| DDAH sequence positions | Calc’d peptide | Observed peptide |
|---|---|---|
| Plus leader (−18 to 0) | Mass (Da) | Mass (Da)b |
| 95-105 | 1322.70 | 1322.72 |
| 95-114 | 2275.20 | 2275.22 |
| 95-129 | 3910.90 | 3910.92 |
| 106-114 | 970.51 | 970.53 |
| 106-129 | 2606.21 | 2606.24 |
| 115-129 | 1653.72 | 1653.72 |
| 115-129 + Oxc | 1669.71 | 1669.72 |
| 115-129 + 2 Ox | 1685.71 | 1685.71 |
| 130-146 | 1829.94 | 1829.96 |
| 130-146 + Ox | 1845.94 | 1845.95 |
| 137-146 | 1100.60 | 1100.61 |
| 137 + 146 + Ox | 1116.60 | 1116.60 |
| 147-158 | 1280.72 | 1280.74 |
| 147-171 | 2746.59 | 2746.60 |
| 159-171 | 1483.88 | 1483.88 |
| 159-180 | 2403.32 | 2403.35 |
| 159-186 | 3090.69 | 3090.72 |
| 172-186 | 1624.82 | 1624.83 |
| 211-223 | 1544.82 | 1544.84 |
| 211-223 + Ox | 1560.82 | 1560.82 |
| 211-234 | 2843.61 | 2843.63 |
| 224-234 | 1316.79 | 1316.81 |
| 224-239 | 1848.01 | 1847.98 |
| 235-254 + Me-Pyrd | 2353.12 | 2353.14 |
| 240-254 + Me-Pyr | 1821.90 | 1821.92 |
| 240-254 + Me-Pyr + Ox | 1837.90 | 1837.91 |
| 240-254e | 1730.86 | 1730.90 |
Comparison of average calculated peptide masses for up to two missed cleavage sites and two oxidations. Amino acid numbering is assigned to match that of Protein Data Bank entry 1H70.
Masses are reported with errors of ± 0.05 Da.
Ox stands for addition of +16 Da for one calculated oxidation.
Me-Pyr stands for the 2-methyl-pyridine adduct (10) shown in Scheme 3.
Peptide mass is taken from a digest done in parallel using an uninhibited DDAH sample.
Ions from the MALDI-TOF/TOF MS fragmentation spectra of the smallest modified peptide identified above, 240-254, reveal a more precise location of the covalent adduct (Table 3). The b5 and y5 daughter ions match those expected from an unmodified peptide. However, the b14*, y7*, y9*, y10*, y11* and y13* ions all have been shifted to higher mass by the addition of the adduct (Figure 4). These results localize the inactivator attachment site to either of two residues: Ser248 or Cys249. Notably, Cys249 is the active-site nucleophile in the normal catalytic mechanism of DDAH.11
Table 3.
Summary of major ions observed in MALDI-TOF/TOF fragmentation spectra from the m/z 1821 parent ion (YRKIDGGVCSMSLRF + Me-Pyr).a
| ion | Calcd m/z | Observed m/zb |
|---|---|---|
| y5 | 653.3 | 653.3 |
| y7* + Me-Pyr | 934.4 | 934.4 |
| y9* + Me-Pyr | 1090.5 | 1090.5 |
| y10* + Me-Pyr | 1147.5 | 1147.6 |
| y11* + Me-Pyr | 1262.6 | 1262.6 |
| y13* + Me-Pyr | 1503.7 | 1503.8 |
| b5 | 676.4 | 676.4 |
| b14* + Me-Pyr | 1657.8 | 1657.8 |
Me-Pyr stands for the 2-methylpyridine adduct (10) shown in Scheme 3.
The masses are reported with errors of ± 0.2 Da.
Figure 4.

Summary of MALDI-TOF/TOF MS fragmentation pattern obtained for modified peptide 240-254. The 91 Da mass addition is mapped to residues Ser248 or Cys249.
The adduct site was also mapped using site-directed mutagenesis and ESI-MS. Previously, two active-site residues, Cys249 and His162, have been shown to be susceptible to covalent modification.5,12 Although a peptide containing unmodified His162 was observed (Table 2), this position was still included in our mutational analysis. We purified the catalytically inactive C249S and H162G variants of DDAH and subjected each to incubation with 1, followed by ESI-MS analysis. As expected, the H162G DDAH variant still shows a 90 ± 10 Da mass addition upon treatment with the inactivator (Table 1). However, the C249S variant did not show any significant mass difference after incubation, even though this DDAH variant still contains four other Cys residues and the unmodified Ser249. Taken together, these data support a mechanism in which 1 reacts covalently with the active-site Cys249 of DDAH.
Inactivation requires the halogen substituent and does not occur through a metabolically-activated species
Because the observed adduct lacks bromide, we tested whether the 4-bromo substituent was required for inactivation. Incubation of DDAH with 2-methylpyridine does not result in time-dependent loss of activity, indicating that the halogen is required for inactivation (Supporting Information, Figure S1). We also considered the possibility that 1 is metabolized by DDAH to produce a reactive product that is released from the active site, and subsequently leads to DDAH inactivation. To rule out this possibility, DDAH was fully inactivated and a fresh aliquot of enzyme was then added to the same preincubation mixture. The observed inactivation rate for the second aliquot of enzyme is not faster than that of the initial inactivation rate, indicating that 1 is not a metabolically activated inhibitor (Supporting Information, Figure S2). This conclusion is also consistent with the low inactivation turnover number, and the observation that excess glutathione, which would react with any released reactive metabolites, does not prevent inactivation.
Proposed inactivation mechanism
We examined three possible mechanisms leading to covalent bond formation with DDAH. First we considered the possibility that a minor portion of the stock solution of 1 may have become oxidized to the N-oxide (4) during storage and handling (Scheme 1). Pyridine N-oxides are more reactive than their parent pyridine compounds13 and could possibly serve as the inactivating species. The resulting enzyme adduct (5) should reflect a 107.04 Da mass increase. This value is close to, but slightly above that seen in ESI-MS characterization of the undigested inactivated enzyme (91 ± 10 Da). More accurate masses were determined in the MALDI-TOF/TOF MS analysis of peptide digests. One peptide (m/z 1837.91) contains mass addition(s) that total the predicted 107 Da mass, but there are two other peptides that contain mass additions that are 16 Da less than this value. These smaller masses are inconsistent with the mechanism in Scheme 1. These results indicate that N-oxidation of the inactivator is not obligatory for covalent bond formation. The observed oxidation of the m/z 1837.91 peptide may occur on Met250 rather than on the inactivator, and likely explains the observed 16 Da mass addition in the larger peptides.
Scheme 1.

Proposed inactivation mechanism by N-oxidized inactivator
The second mechanism that we considered was hydrolysis (possibly DDAH-catalyzed) of 1 and tautomerization of the product (6) to a pyridone species (7) that could react with a nucleophile on the enzyme (Scheme 2). However, the resulting adduct (8) should reflect a 108.04 Da mass increase, which is not observed in any of the peptides bearing adducts. Also, significant background hydrolysis was not observed in the bromide quantification experiment (Figure 3B), and incubations of DDAH with 6 do not result in enzyme inactivation (Supporting Information, Figure S3). These observations do not support the mechanism shown in Scheme 2.
Scheme 2.

Proposed inactivation mechanism by hydrolyzed inactivator
The third inactivation mechanism that we considered is attack of the active-site Cys249 on the pyridinium form of 1, formation of a σ-complex (9), followed by elimination of bromide to result in a stable 2-methylpyridine-Cys249 adduct (10, Scheme 3). This mechanism is most consistent with the experimental results described herein. This mechanism is dependent on enzyme stabilization of the pyridinium form of 1 to enhance its reactivity.
Scheme 3.

Proposed inactivation mechanism by the pyridinium form of the inactivator
The pyridinium form is likely the reactive species
Typically, 4-halopyridines are not very sensitive to nucleophilic aromatic substitution while in their neutral forms. However, formation of the positive pyridinium ion, often by N-oxidation or N-methylation, greatly increases reaction rates because of the increased ability of the positive charge to stabilize σ-complex formation upon attack by a nucelophile. For example, methanolysis of 4-chloropyridine by methoxide/methanol at 50 °C shows dramatic rate enhancements of 1.1 × 103 and 5.7 × 109 when the nitrogen is derivatized to the N-oxide or N-methyl, respectively.13 Additionally, nucleophilic aromatic substitution of halopyridines has been shown to be acid-catalyzed through protonation of the heterocyclic nitrogen.14 However, the calculated15 pKa of 1 is 4.74, indicating that only a small amount of the more reactive pyridinium form (e.g. 1.7 μM in a 1 mM solution) is present at the incubation pH value of 7.5. Enzyme inactivation by this minor form may explain why saturating inactivation kinetics was not observed at pH 7.5 (Figure 2). We repeated these experiments at pH 5.0 to increase the protonated fraction of 1. Saturating kinetics were now observed at high inhibitor concentrations, and could be fit to give KI (2.6 ± 0.8 mM) and kinact (0.18 ± 0.03 min−1) values (Figure 5). This result is consistent with a mechanism in which the positively charged pyridinium ion is the inhibitory species that binds and inactivates DDAH. Although the KI value is large, its magnitude is not unexpected due to the small “fragment” size of the inactivator. These values compare quite favorably with other “warheads” that have been derivatized into potent biologically useful tools. For example, the protein arginine deiminase-4 inactivator fragment 2-chloroacetamidine (KI = 20 mM, kinact = 0.7 min−1)5 is greatly enhanced by incorporation into a peptidyl arginine analog scaffold (KI = 180 μM, kinact = 2.4 min-1).16 Derivatives of 1 will likely increase potency as well and are a subject of current work.
Figure 5.

Time- and concentration-dependent inactivation of DDAH by 1 at pH 5.0. (A) Exponential fits to the observed inactivation of DDAH (29 μM) by different concentrations of inhibitor: 0 (○), 330 μM (λ), 500 μM (□), 1 mM (ν), 1.5 mM (△), or 3 mM (▲), each in MES buffer (100 mM), KCl (100 mM), EDTA (0.05 mM) at pH 5.0, 25 °C. (B) Concentration dependence of the pseudo-first-order kobs values shows saturation kinetics and is fitted by KI (2.6 ± 0.8 mM) and kinact (0.18 ± 0.03 min−1) values for inactivation.
To maximize the fraction of the inactivator’s pyridinium form without adjusting the pH of the reaction solution, we synthesized 1, 2-dimethyl-4-bromopyridinium (11).

This compound readily causes time-dependent inactivation of DDAH that cannot be reversed upon dilution into excess substrate. The observed inactivation rate of 11 is faster than that of a similar concentration of 1 (Figure 6). However, preincubation of 11 with excess glutathione completely blocks its ability to inactivate DDAH (Figure 6). Apparently, installing a permanent charge on the inactivator increases its reactivity to an extent that the inhibitor becomes non-selectively reactive, thereby losing much of its biological utility. These results also suggest that the enzyme may play an analogous role in inactivation.
Figure 6.

Time-dependent inactivation of DDAH by 11 and protection by glutathione. The observed DDAH inactivation rate by 11 (300 μM, λ) is faster than that of 1 (300 μM, ▲, taken from Figure 1). However, preincubation of 11 with glutathione (5 mM) prevents any time-dependent inhibition of DDAH (ν, dashed line).
The active-site environment promotes inactivation
Two ways that DDAH might participate in the proposed inactivation mechanism are 1) by providing a good active-site nucleophile with a low pKa, and 2) by stabilizing the protonated, more reactive form of the inactivator. However, DDAH (as well as other enzymes in the pentein superfamily)2 is unusual in that the active-site Cys249 has a high pKa (ca. 8.8)17 in the enzyme’s resting state. This value is less than the pKa value of glutathione’s thiol (ca. 9.2)18, but would still be predominantly protonated and presumably less reactive at most physiological pH values (e.g. > 95% protonated at pH 7.5). Therefore, it appears that the inactivator’s selective reaction with the Cys249 of DDAH is not because the enzyme provides an exceptionally good nucleophile. To further illustrate this point, we tested inactivation of a different hydrolase, papain. Papain has a very nucelophilic active-site Cys (pKa ≤ 4)19 that can readily react with thiol-modifying reagents. However, preincubation of papain with 1 does not result in any time-dependent inactivation (Figure 7). In contrast, the chemically-activated N-methylated inactivator 11 does cause inactivation. These results illustrate the need for a pyridinium-stabilizing group in the target enzyme, and that the selectivity of this halopyridine is not solely due to its reaction with good nucelophiles.
Figure 7.

Time-dependent inactivation of papain by 1 and 11. Time-dependent inactivation by 11 (1 mM, ν) is observed. No inactivation by 1 (1 mM, λ) is observed relative to a no-inactivator control (○). Prior to the assays, papain is activated by incubation with reduced L-cysteine (500 μM). Papain samples that are subsequently depeleted of free L-cysteine do not lose activity over the experimental timeframe (data not shown).
Inspection of the structures of DDAH substrates and the 4-halopyridines suggest some similarities; both contain flat moieties that bear positive charges (guanidinium and pyridinium ions, respectively). The positive charge of the substrate binds to the carboxylate-rich active site of DDAH. Therefore, we considered the possibility that one or more of these carboxylates might also help to bind and stabilize the pyridinium ion of the inactivator, thereby facilitating inactivation. We prepared site-directed mutants of three carboxylates that are located within 8 Å of Cys249: Glu65, Asp66 and Asp244. E65Q and D66N are catalytically inactive. D244 retains catalytic activity toward Nω, Nω-dimethyl-L-arginine (kcat = 1.32 ± 0.06 s-1; KM = 700 ± 100 μM at pH 7.5, 25 ºC) with steady-state rate constants similar to wild-type enzyme (kcat = 1.27 s−1; KM = 39 μM).17 These DDAH variants were each incubated with 1 under the same conditions that led to complete inactivation of wild-type DDAH (Table 1). The D244N mutant behaves the same as wild-type DDAH, modifying all of the enzyme with a 91 Da mass addition. Incubation with the D66N mutant, however, does not fully modify the enzyme, but rather results in mass addition (90 Da) to only a fraction of the enzyme, with a modified : unmodified peak ratio of 1:2.1 This is consistent with a mechanism in which Asp66 promotes formation of the covalent adduct. Treatment of the D66N mutant with the N-methylated inactivator 11, results in complete modification. Therefore, chemical activation of the pyridine is able to “rescue” the impaired D66N mutant with respect to covalent modification. The inactive E65Q mutant completely blocks all covalent adduct formation, but also can not be modified by 11, suggesting that this mutation may block inactivation by deformation of the active-site rather than by destabilizing pyridinium binding. In addition to these three mutants, we also found that incubation of the inactivator with heat-denatured wild-type DDAH does not result in any covalent adduct formation. Taken together, these results demonstrate that the active-site carboxylate Asp66 promotes covalent inactivation, likely by facilitating the binding and stabilization of the reactive pyridinium form of the inactivator.
Inhibitor classification and precedence
In classifying the 4-halopyridine inhibitors, we first established that they are not rapidly reversible inhibitors and that their time-dependent irreversible inhibition leads to covalent bond formation at the active site. This inactivation mechanism also does not include release of a reactive species, ruling out metabolic activation.20 Because the 4-halopyridines do not readily react with glutathione, they are not considered to be intrinsically reactive compounds and so can not be classified as classic affinity labels.21 Taking into account the proposed inactivation mechanism (Scheme 3), the 4-halopyridines are also not classified as mechanism-based inactivators because the enzyme does not use a step of the normal catalytic mechanism to unmask a latent reactivity.20 Therefore, considering all of the evidence to date, the most fitting classification for 4-halopyridine inactivators is that of quiescent affinity labels.22 This term was first introduced by Krantz and co-workers to describe acyloxymethylketone inhibitors of cysteine proteases.23,24 This type of inhibitor readily reacts when bound in an appropriate enzyme site, but is “quiescent” in the presence of other biologically-relevant nucelophiles that do not present a suitable complementary surface.
In general, there are some similarities in our proposed inactivation mechanism with other known enzyme inactivators. For example, some inactivation mechanisms use nucleophilic aromatic substitution,25 and others use nucleophilic attack on a dihydropyridinium metabolite.26 There are also similarities in catalytic mechanisms. Some pyridoxal phosphate-dependent enzymes use a carboxylate to stabilize the pyridinium form of the cofactor to facilitate reaction.27 However, to our knowledge, there have been no prior reports of halopyridines used as covalent protein modifiers. One possible exception is a mention of preliminary results showing irreversible inhibition of glucose transport in human erythrocytes by two highly derivatized halopyridines.28 However, no experimental details were given and it is unclear if the observed inhibition is covalent in nature.
Conclusions
4-Bromo-2-methylpyridine (1) is a quiescent affinity label of DDAH that specifically and covalently modifies the active-site Cys residue. To our knowledge, 4-halopyridine (or the similarly reactive 2-halopyridine) moieties have not previously been identified as covalent modifiers of DDAH or any other proteins. Although the affinity of these reagents for DDAH is weak, they can likely be used as the “warheads” of more elaborate inhibitors with increased potency and selectivity. Although more detailed characterization will be required, selective protein modification by these halopyridines appears to require a binding site with a nucelophilic residue placed at a suitable distance and orientation from a second moiety that binds and stabilizes the more reactive pyridinium form. These constraints confer a unique selectivity profile to this novel class of quiescent affinity label.
EXPERIMENTAL SECTION
Materials
Unless noted otherwise, all chemicals are from Sigma-Aldrich Chemical Co. (St. Louis, MO). All DDAH enzymes, including wild-type Pseudomonas aeruginosa DDAH and the mutants C249S and H162G were purified and assayed as described previously.11 Synthetic DNA primers are from Sigma-Genosys (The Woodlands, TX) and Invitrogen (Carlsbad, CA). Rapid plasmid isolation kits are from Invitrogen. V8 (Glu-C) endoprotease is from Roche (Indianapolis, IN). 2-Methyl-4-bromopyridine•HBr is from ChemBridge (San Diego, CA) and 2-methyl-4-bromopyridine (the free base form) is from Sigma-Aldrich. Nα-Benzoyl-L-arginine p-nitroanilide•HCl is from Bachem Americas (Torrance, CA). 4-Hydroxy-2-methylpyridine is from Ark Pharm (Libertyville, IL).
Construction of vector for E65Q, D66N and D244N DDAH, protein expression and purification
Two complementary mutagenic oligonucleotides, (forward) 5′-TCGGTGTTCGTCcagGACCCGGTGC-3′ and (reverse) 5′-GCACCGGGTCctgGACGAACACCGA-3′, were designed to introduce a mutant codon (lowercase) encoding an E65Q mutation in the DDAH sequence by Quikchange mutagenesis. Two complementary mutagenic oligonucleotides, (forward) 5′-GGTGTTCGTCGAGaacCCGGTGCTCTG-3′ and (reverse) 5′-CAGAGCACCGGgttCTCGACGAACACC-3′, were designed to introduce a mutant codon (lowercase) encoding a D66N mutation in the DDAH sequence by Quikchange mutagenesis. Two complementary mutagenic oligonucleotides, (forward) 5′-CCGAATATCGCAAGATCaacGGCGGCGTCA-3′ and (reverse) 5′-TGACGCCgttGATGAACGCTATTTCGG-3′, were designed to introduce a mutant codon (lowercase) encoding a D244N mutation in the DDAH sequence by Quikchange mutagenesis. Briefly, a PCR mixture using a pET28a vector containing P. aeruginosa DDAH as a template, the mutagenic primers, a dNTP mixture and Pfu DNA polymerase in the manufacturer’s buffer (Stratagene, La Jolla, CA) was run using a temperature program of 95 ºC for 1 min, followed by 25 cycles of 95 ºC for 30 s, 45–60 ºC for 1 min, and 68 ºC for 12 min. Dpn I was then added to the cooled reaction mixture to digest the methylated parent plasmid. After incubation at 37 ºC for 1 h, the mixture was transformed into electrocompetent DH5α Escherichia coli cells and selected on LB agar plates supplemented with kanamycin (30 μg / mL). The final plasmid was purified from an overnight culture and the gene insert was fully sequenced (DNA Facility, University of Texas at Austin) to verify the desired sequence. Expression and purification of mutant D66N, E65Q and D244N DDAH was completed as described previously.11
Time-dependent inactivation of DDAH
Purified recombinant DDAH (29 μM) was incubated with KCl (100 mM) and 1 (0.150-3 mM) in K2HPO4 buffer (100 mM) and EDTA (0.05 mM) at pH 7.5 and 25 ºC. Some experiments also included an additional incubation (1 h) of 1 with excess glutathione (5 mM) before addition of enzyme to initiate the inactivation. Substrate protection experiments included excess Nω,Nω-dimethyl-L-arginine (3 mM) in the preincubation mixture. Aliquots were removed from these incubations at various time points (0-80 min) and diluted into an assay solution containing excess (1 mM) substrate Nω, Nω-dimethyl-L-arginine. Remaining enzyme activity was assayed as described previously, using a color-developing reagent (COLDER) to derivatize the urea group of the L-citrulline product.11,29 Briefly, COLDER (200 μL) was added to each 100 μL reaction in a clear polystyrene 96-well plate (Nunc, Rochester, NY) and incubated for 15 min at 95 ºC. Samples were cooled for 20 min at 25 ºC and the absorbance at 540 nm determined for each well using a Perkin-Elmer (Waltham, MA) VICTOR3V 1420 multi-label counter plate reader. The observed inactivation rates (kobs) were determined by fitting the percent remaining activity over time to a single exponential equation. The resulting kobs values were then plotted against inactivator concentration and fitted to kobs = (kinact[I])/(KI + [I]) to obtain KI and kinact values.20 All fits were calculated using KaleidaGraph (Synergy Software, Reading, PA). To further study irreversibility, fully inhibited DDAH was dialyzed overnight at 4 ºC against 1000 volumes of phosphate buffer (10 mM) and KCl (100 mM) at pH 7.5, and the resulting activity was assayed as described above. Control reactions were conducted in parallel to ensure that uninhibited DDAH retained activity under these conditions.
Mass spectral analysis of inactivated DDAH and DDAH mutants
To characterize covalent adducts formed during enzyme inactivation, 90 min incubations of 1 (300 μM) with DDAH were carried out at 25 ºC under the same conditions used in the preincubations described above and quenched with triflouroacetic acid (TFA, 6N). Similar incubations were carried out using the C249S, H162G, D66N, D244N and E65Q DDAH enzymes, along with wild-type DDAH that had previously been heat denatured. Samples were analyzed by ESI-MS on a ThermoFinnigan LCQ (San Jose, CA) ion trap mass spectrometer as described previously.11
Bromide quantification by ion chromatography
To further investigate the mechanism of inactivation of DDAH, 110 μM enzyme and 300 μM 1 (free base form) were incubated at 25 ºC and acid quenched at varied time points. The enzyme was removed from solution using a Microcon centrifugal filter device with a 3000 Da molecular weight cutoff (MWCO). Samples were centrifuged for 45 min at 11,750 × g at 25 ºC. The flow-through was then diluted 1:20 in sodium bicarbonate buffer (20 mM) at pH 7. Inactivated products were analyzed using a Dionex Ion Chromatography System (ICS-1500, Dionex Corp) equipped with an AG12A pre-column (4 × 40 mm), an IonPac AS12A column (4 × 200 mm), an anion self-regenerating suppressor (ASRS ULTRA II, 4 mm), and an AS40 autosampler. Anion detection was performed by suppressed electrical conductivity using an electrochemical detector with an applied current of 25 mA. A solution of 2.7 mM Na2CO3 / 0.3 mM NaHCO3 at pH 9.6 was used as the eluent at a flow rate of 1.5 mL / min. Bromide was quantified by use of a calibration curve made with serial dilutions of a NaBr bromide standard (Bromide IC Standard Solution, 1 g / L, 1000 ppm bromide ion (Fluka)) in 20 mM NaHCO3 buffer, pH 9.0. Calibration curve solutions included bromide concentrations: 10 ppm, 2.5 ppm, 1.25 ppm, 0.625 ppm, 0.3125 ppm, 0.0156 ppm, and 0 ppm, all in 20 mM NaHCO3 buffer at pH 9.0.
Identification of a covalently modified DDAH peptide
To determine the amino acid that is modified by inactivator, fully inactivated DDAH was exchanged into 50 mM ammonium acetate buffer (pH 4) by overnight dialysis. Samples were then digested with V8 endoprotease, and the products were analyzed using MALDI-TOF/TOF MS on an Applied Biosystems 4700 Proteomics Analyzer.
Activity and inhibition assays for papain
Papain from papaya latex was from Sigma-Aldrich. The activity assay for papain was as previously reported,30 using Nα-benzoyl-L-arginine p-nitroanilide as the substrate. Briefly, papain was activated by incubating (1 h) with reduced L-cysteine (500 μM) in Na3PO4 (2 mM) and EDTA (0.1 mM). Activity was measured by following the release of p-nitroanilide at 410 nm (ε410 = 8800 M-1cm-1) using a Cary 50 UV-Vis spectrophotometer (Varian, Inc., Walnut Creek, CA). As a control, activated papain was subsequently depleted of free L-cysteine (depletion was achieved by multiple washes in a 3000 Da MWCO centrifugal filter) in order to demonstrate that any loss of activity in the experimental assays was not due to a loss of free L-cysteine in the reaction mixture (through reaction with the inactivators).
Synthesis of 1,2-dimethyl-4-bromopyridine (11)
A solution of 1 (0.206 g, 1.19 mmol) in 3.0 mL of CH2Cl2 was treated with CH3I (0.498 g, 3.50 mmol) and the reaction mixture was stirred overnight at 25º C. The resulting white precipitate was collected by vacuum filtration, washed with CH2Cl2 (30 mL), and dried under high vacuum to yield 11. (0.302 g, 0.962 mmol, 81%). 1H NMR (400 MHz, DMSO-d6) δ 8.87 (d, J = 6.7 Hz, 1H, Ar-H), 8.46 (d, J = 2.3 Hz, 1H, Ar-H), 8.29 (dd, J = 6.7 Hz, J = 2.3 Hz, 1H, Ar-H), 4.15 (s, 3H, N-CH3), 2.74 (s, 3H, CH3); 13C NMR (100 MHz, DMSO-d6) δ 156.80, 146.44, 141.20, 131.89, 128.33, 45.14, 19.75.; HRMS (CI) (m/z): M+ calcd for C7H9BrN+ 185.99129, found 185.99129.
Supplementary Material
Acknowledgments
We thank Gottfried Schroeder (University of Texas, Austin) for assistance with Figure 3B. This work was supported in part by grants from the National Institutes of Health (GM069754), the Robert A. Welch foundation (F-1572) and a seed grant from the Texas Institute for Drug and Diagnostic Development (Welch Foundation Grant H-F-0032).
Footnotes
Comparison of peak intensities assumes similar ionization efficiencies for the unmodified enzyme and enzyme-inactivator adducts.
SUPPORTING INFORMATION AVAILABLE Three figures showing time dependent inhibition assays of 2-methylpyridine, 1 after pretreatement with enzyme, and 6.
References
- 1.Pope AJ, Karuppiah K, Kearns PN, Xia Y, Cardounel AJ. J Biol Chem. 2009;284:35338. doi: 10.1074/jbc.M109.037036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Linsky TW, Fast W. In: Comprehensive Natural Products II Chemistry and Biology. Mander L, Liu HW, editors. Vol. 8. Elsevier; Oxford: 2010. p. 125. [Google Scholar]
- 3.Linsky T, Fast W. Biochim Biophys Acta. 2010;1804:1943. doi: 10.1016/j.bbapap.2010.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stuhlinger MC, Tsao PS, Her JH, Kimoto M, Balint RF, Cooke JP. Circulation. 2001;104:2569. doi: 10.1161/hc4601.098514. [DOI] [PubMed] [Google Scholar]
- 5.Stone EM, Schaller TH, Bianchi H, Person MD, Fast W. Biochemistry. 2005;44:13744. doi: 10.1021/bi051341y. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. J Am Chem Soc. 2006;128:1092. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 8.Cravatt BF, Wright AT, Kozarich JW. Annu Rev Biochem. 2008;77:383. doi: 10.1146/annurev.biochem.75.101304.124125. [DOI] [PubMed] [Google Scholar]
- 9.Congreve M, Carr R, Murray C, Jhoti H. Drug Discov Today. 2003;8:876. doi: 10.1016/s1359-6446(03)02831-9. [DOI] [PubMed] [Google Scholar]
- 10.Knuckley B, Jones JE, Bachovchin DA, Slack J, Causey CP, Brown SJ, Rosen H, Cravatt BF, Thompson PR. Chem Commun (Camb) 2010;46:7175. doi: 10.1039/c0cc02634d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stone EM, Person MD, Costello NJ, Fast W. Biochemistry. 2005;44:7069. doi: 10.1021/bi047407r. [DOI] [PubMed] [Google Scholar]
- 12.Forbes SP, Druhan LJ, Guzman JE, Parinandi N, Zhang L, Green-Church KB, Cardounel AJ. Biochemistry. 2008;47:1819. doi: 10.1021/bi701659n. [DOI] [PubMed] [Google Scholar]
- 13.Livers M, Miller J. J Chem Soc. 1963:3486. [Google Scholar]
- 14.Reinheimer JD, Gerig JT, Garst R, Schrier B. J Am Chem Soc. 1962;84:2770. [Google Scholar]
- 15.MARVIN 5.2.0, ChemAxon. 2009 http://www.chemaxon.com.
- 16.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Biochemistry. 2006;45:11727. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stone EM, Costello AL, Tierney DL, Fast W. Biochemistry. 2006;45:5618. doi: 10.1021/bi052595m. [DOI] [PubMed] [Google Scholar]
- 18.Tajc SG, Tolbert BS, Basavappa R, Miller BL. J Am Chem Soc. 2004;126:10508. doi: 10.1021/ja047929u. [DOI] [PubMed] [Google Scholar]
- 19.Pinitglang S, Watts AB, Patel M, Reid JD, Noble MA, Gul S, Bokth A, Naeem A, Patel H, Thomas EW, Sreedharan SK, Verma C, Brocklehurst K. Biochemistry. 1997;36:9968. doi: 10.1021/bi9705974. [DOI] [PubMed] [Google Scholar]
- 20.Silverman RB. Methods Enzymol. 1995;249:240. doi: 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- 21.Krantz A. Bioorg Med Chem Lett. 1992;2:1327. [Google Scholar]
- 22.Krantz A. Adv Med Chem. 1992;1:235. [Google Scholar]
- 23.Krantz A, Copp LJ, Coles PJ, Smith RA, Heard SB. Biochemistry. 1991;30:4678. doi: 10.1021/bi00233a007. [DOI] [PubMed] [Google Scholar]
- 24.Smith RA, Copp LJ, Coles PJ, Pauls HW, Robinson VJ, Spencer RW, Heard SB, Krantz A. J Am Chem Soc. 1988;110:4429. [Google Scholar]
- 25.Boger DL, Marsilje TH, Castro RA, Hedrick MP, Jin Q, Baker SJ, Shim JH, Benkovic SJ. Bioorg Med Chem Lett. 2000;10:1471. doi: 10.1016/s0960-894x(00)00271-7. [DOI] [PubMed] [Google Scholar]
- 26.Hiebert CK, Sayre LM, Silverman RB. J Biol Chem. 1989;264:21516. [PubMed] [Google Scholar]
- 27.Eliot AC, Kirsch JF. Annu Rev Biochem. 2004;73:383. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 28.Hershfield R, Richards FM. J Biol Chem. 1976;251:5141. [PubMed] [Google Scholar]
- 29.Knipp M, Vasak M. Anal Biochem. 2000;286:257. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- 30.Evans BL, Knopp JA, Horton HR. Arch Biochem Biophys. 1981;206:362. doi: 10.1016/0003-9861(81)90103-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.