

Abstract
Anti-GM1 antibodies are present in some patients with autoimmune neurological disorders. These antibodies are most frequently associated with acute immune neuropathy called Guillain-Barré syndrome (GBS). Some clinical studies associate the presence of these antibodies with poor recovery in GBS. The patients with incomplete recovery have failure of nerve repair, particularly axon regeneration. Our previous work indicates that monoclonal antibodies can inhibit axon regeneration by engaging cell surface gangliosides (Lehmann et al., 2007). We asked whether passive transfer of human anti-GM1 antibodies from patients with GBS modulate axon regeneration in an animal model. Human anti-GM1 antibodies were compared with other GM1 ligands, cholera toxin B subunit and a monoclonal anti-GM1 antibody. Our results show that patient derived anti-GM1 antibodies and cholera toxin β subunit impair axon regeneration/repair after PNS injury in mice. Comparative studies indicated that the antibody/ligand-mediated inhibition of axon regeneration is dependent on antibody/ligand characteristics such as affinity-avidity and fine specificity. These data indicate that circulating immune effectors such as human autoantibodies, which are exogenous to the nervous system, can modulate axon regeneration/nerve repair in autoimmune neurological disorders such as GBS.
Introduction
Anti-glycolipid antibodies (Abs) of various specificities have been described in association with several autoimmune disorders of PNS and CNS including immune neuropathies and multiple sclerosis (Cross et al., 2001; Willison and Yuki, 2002). In neuroimmunological disorders autoantibodies against GM1 [a major ganglioside of vertebrate PNS and CNS (Svennerholm et al., 1992)] are frequently reported (Ogawara et al., 2000; Kanter et al., 2006). Anti-GM1 Abs have strongest association with axonal forms of Guillain-Barré syndrome (GBS) (Yuki et al., 1990; Ho et al., 1995; Hadden et al., 1998; Ogawara et al., 2000), which is now the commonest cause of acute flaccid paralysis worldwide. GBS comprises a group of clinically and pathophysiologically related, acute monophasic demyelinating and axonal neuropathic disorders of autoimmune origin (Willison and Yuki, 2002; Hughes and Cornblath, 2005). There is strong evidence for postinfectious molecular mimicry as a mechanism for the induction of anti-ganglioside (including anti-GM1) Abs in GBS (Yuki et al., 1992; Aspinall et al., 1994; Jacobs et al., 1997; Sheikh et al., 1998). Some clinical studies indicate that anti-GM1 Abs in adult patient groups with GBS are associated with poor prognosis and/or incomplete recovery (Ilyas et al., 1992; Gregson et al., 1993; Simone et al., 1993; Jacobs et al., 1996; Bech et al., 1997; Kuwabara et al., 1998a,b; Carpo et al., 1999; Press et al., 2001; Annunziata et al., 2003; Koga et al., 2003). The patients with incomplete recovery almost always have some degree of failure of nerve repair/axon regeneration and target reinnervation (Brown and Feasby, 1984). These clinical observations raise the possibility that anti-GM1 Abs can adversely affect the nerve repair process in this disease and potentially in other disorders associated with anti-GM1 antibodies.
To test this hypothesis we examined the effects of IgG anti-GM1 antibodies present in patients with axonal forms of GBS in a peripheral nerve injury and repair paradigm, described previously (Lehmann et al., 2007). Further, as a proof of concept, we studied the effects of two different GM1 ligands, namely, Cholera toxin β subunit (CTB) and a-specific IgG anti-GM1 monoclonal antibody (mAb) on nerve repair. Our data provide evidence that engagement of GM1-like epitopes by autoimmune Abs could be a mechanism that impairs axon regeneration. An implication of this finding is that circulating immune factors, including autoantibodies, can inhibit axonal regeneration/neural repair; an effect that is mostly attributed to endogenous regeneration inhibitors in CNS.
Materials and Methods
Patient sera.
Plasma from one patient with acute motor sensory axonal neuropathy (AMSAN, JHH-9) and one patient with acute motor axonal neuropathy (AMAN, patient 98-7) with high titers of IgG anti-GM1 Abs was collected during the acute phase of the disease from plasma exchange (PE) performed as part of their treatment. Plasma was later dialyzed against PBS to remove anticoagulants, filtered and stored at −20°C until use. Serum from a normal healthy volunteer without reactivity against GM1 was used as negative control.
IgG fractions and affinity purified anti-GM1 Abs were prepared from the serum of patient JHH-9. The IgG fractions from sera were prepared using a Protein G Sepharose column (GE Healthcare) according to the manufacturer's instructions. Anti-GM1 Abs from serum (JHH-9) were purified by affinity chromatography using GM1 ganglioside according to the method described by Hirabayasi et al. (1983). The purified Abs were stored at −20°C until use.
GM1 ligands.
For comparison to the patient derived antibodies a non-antibody GM1 ligand, i.e., CTB) (List Biologicals) was used in animal studies. A previously well characterized IgG2b mAb specific against GM1 (GM1-2b) was also included for passive transfer studies. The generation, specificity, and production of this mAb were reported previously (Gong et al., 2002; Schnaar et al., 2002). In the present study, GM1-2b mAb hollow fiber supernatant was used for animal studies.
Sciatic nerve crush model.
All studies were done on 12- to 16-week-old wild-type (C57BL/6) mice. Experimental procedures were approved by the institutional Animal Care and Use Committee. A standardized mouse sciatic nerve crush model was used (Lehmann et al., 2007). Sciatic nerves were crushed 35 mm rostral to the middle toe for 30 s with fine forceps on day 0, either on left side or bilaterally. Separation of proximal and distal endoneurial contents indicated complete crush. Animals injected with human sera were pretreated with 100 mg/kg cyclophosphamide intraperitoneally 2 d before the nerve crush to minimize the immune response (serum sickness) to human proteins, as described previously (Toyka et al., 1977). These wild-type mice received daily intraperitoneal injections of 1 ml of sera from patient JHH-9 (n = 12 nerves), patient 98-7 (n = 4 nerves), or control human sera (n = 8 nerves) for 15 d. Further passive transfer studies were done with the JHH-9 IgG fractions (n = 4 nerves) or affinity purified polyclonal IgG anti-GM1 Abs (n = 4 nerves). For the studies with the patient IgGs, human intravenous Ig (IVIg) (Carimune NF, ZLB Bioplasma) was used as control (n = 4 nerves).
The effects of JHH-9 serum were also examined on the regeneration of sensory (sural) nerves. For these studies wild-type mice were pretreated with 100 mg/kg cyclophosphamide intraperitoneally 2 d before surgeries. Sural nerves were crushed 22 mm rostral to the middle toe for 30 s with fine forceps on day 0, on left side. These animals received 10 daily intraperitoneal injections of 1 ml of JHH-9 serum (n = 3) or healthy control serum without anti-ganglioside reactivity (n = 3) and nerves were harvested on day 11 after crush for morphological studies.
In another set of studies CTB or GM1-2b was administered to wild-type mice. These animals did not receive cyclophosphamide pretreatment because serum sickness was not anticipated with these ligands. CTB or vehicle control (1 ml total volume) was administered to mice (n = 8 nerves each) intraperitoneally, 0.5 mg on day 0 and 0.25 mg on days 3, 5, and 7 after the crush. Another group of animals was administered five doses of GM1-2b mAb (hollow fiber supernatant containing 2 mg/ml) or control IgG (2 mg/ ml) intraperitoneally on days 0, 3, 5, 7, and 9 after surgery intraperitoneally (n = 4 nerves each).
At day 15 or 16 after surgery, a magnetic resonance imaging (MRI) of the calf muscles and/or electrophysiology were done in a selected group of animals and mice were killed on day 16 or 17 after nerve crush and tissues and sera were harvested for morphological and serological analysis.
Morphometry.
Mice were perfused with a mixture of 3% glutaraldehyde and 4% paraformaldehyde. Sciatic and tibial nerves were harvested and immersion-fixed overnight before processing. For morphology, three segments of the crushed sciatic nerves were analyzed (Fig. 1A): S1, 10 mm proximal to the crush site; S2, 10 mm distal to the crush site (sciatic nerve segment) and S3, 20 mm distal to the crush site (tibial nerve segment). For sural nerve studies a segment of the nerve 12 mm distal to the crush site was collected for analysis. These segments were embedded in Epon, and 1 μm cross sections were stained with toluidine blue as described previously (Sheikh et al., 1999b, 2004). Sections from nerve segments were used for quantification at light level (40× lens) by a motorized stage and stereotactic imaging software (Stereo Investigator, version 5). We counted all myelinated regenerating sprouts in a single whole cross section of the nerve. Sciatic nerve cross sections (S2) were used for quantifying mean caliber of regenerating axons/sprouts in S2 segments as well for electron microscopy in studies with CTB. Caliber was calculated from the diameter of a circle with an area equivalent to that of each axon/sprout, as described previously (Sheikh et al., 2004).
Figure 1.

GBS patient sera-mediated inhibition of peripheral nerve repair. A, Schematic diagram showing the site of nerve crush and its relationship to different nerve segments (S1–S3), as outlined in the text. B, C, Micrographs from control and GBS sera-treated S3 nerve segments showing fewer regenerating fibers in GBS sera-treated nerve. Scale bar, 25 μm. D, E, Significant decrease in number of regenerating axons at sciatic (S2; D) and tibial (S3; E) nerve levels in GBS (AMSAN and AMAN)-treated nerves compared with control serum-treated nerves. *p < 0.05.
Electrophysiology.
These studies were done in selected groups of animals. Sciatic nerves were stimulated at the sciatic notch and compound muscle action potential (CMAP) amplitudes were recorded with a PowerLab signal acquisition set-up (ADInstruments) in the hindpaws as described previously (Lehmann et al., 2007).
Magnetic resonance imaging.
Live anesthetized animals were imaged by an 11.7 Tesla nuclear magnetic resonance spectrometer (Bruker Biospec) and processed, as described previously (Zhang et al., 2008). From 3D images of the leg, volumetric measurements of all muscle groups were obtained (Lehmann et al., 2007).
Immunostaining.
Sciatic nerves were harvested on day 16–17 after nerve crush and fixed with 4% paraformaldehyde for 24 h. These nerves were cryoprotected with sucrose, longitudinally sectioned (10 μm) and immunostained with anti-human IgG γ chain-specific Abs conjugated to Cy3 (Jackson Immunoresearch Laboratories) and analyzed by fluorescent microscopy, as described previously (Lehmann et al., 2007).
Determination of antibody titer.
The titers of circulating IgG anti-GM1 Abs in mouse sera, collected at the time of tissue harvesting, was determined by ELISA, as described previously (Lopez et al., 2000).
Determination of affinity.
The affinity of GBS patient sera (patients JHH-9 and 98-7) and a mAb (GM1-2b) used in our studies was determined with soluble GM1-oligosaccharide in a soluble binding inhibition assay, as described previously (Lopez et al., 2002). GM1-oligosaccharide was used as a competing antigen because it is soluble and the values obtained can be considered as monovalent affinity. GM1-oligosaccharides were generated as described previously (Lopez et al., 2008). GM1-oligosaccharides from GM1 ganglioside were generated by a specific ceramide glycanase (1 U/mg ganglioside) as suggested by the manufacturer's instructions (Calbiochem) that catalyzes removal of ceramide moieties from gangliosides, and their identity was confirmed by mass spectrometry through a core facility at Johns Hopkins University. Different concentrations of soluble sugars (10−4–10−7 m) were used to determine the amount of oligosaccharide required for 50% inhibition of Ab binding to GM1 (IC50%).
Immunostaining of ventral and dorsal roots.
Affinity purified anti-GM1 antibodies from patient JHH-9 were used for these studies. Cauda equina collected from adult 8- to 12-week-old Sprague Dawley rats were snap-frozen in isopentane at −70°C and cryosectioned. These sections were immunostained with purified IgG anti-GM1 Abs from patient JHH-9 diluted (1:20), as described previously (Lopez et al., 2008). Isolectin B4 (IB4) (Sigma) was used to mark Remak bundles in sensory roots in cauda equina, as described previously (Gong et al., 2002).
Preparation of GM1-derivatives.
GM1-derivatives were synthesized to examine the fine specificity of different GM1 ligands used in this study. Sialic acid on GM1 was modified to obtain GM1 NeuAc 1-amide, GM1 NeuAc 1-alcohol, GM1 NeuAc 1-methyl ester, and GM1 NeuAc 7-aldehyde, as described previously (Lopez et al., 2008). GM1-de-N-acetyl was derived by removal of the N-acetyl group of GM1 sialic acid. This was achieved by incubating 2 mg of GM1 in 0.4 ml of DMSO, 3.2 ml of water and 0.4 ml of 10 m KOH at 100°C for 4 h as described previously (Nores et al., 1988). Products were analyzed by thin-layer chromatography (TLC) and fast atom bombardment-mass spectrometry (Dell et al., 1994). at the Middle Atlantic Mass Spectrometry Laboratory.
Binding of GM1 ligands to GM1-derivatives.
Binding of GM1 ligands (JHH-9, 98-7, CTB, and GM1-2b) to GM1 and GM1-derivatives was determined by either ELISA or TLC-immunoverlay and quantified, as described previously (Lopez et al., 2000, 2008). These studies were repeated at least once with all ligands.
Statistical analysis.
All numerical results are presented as mean ± SEM. Differences between groups were determined using ANOVA with corrections for multiple comparisons or Student's t test, p values <0.05 were considered statistically significant.
Results
Passive transfer of GBS sera containing high titers of circulating IgG anti-GM1 ganglioside Abs impairs nerve repair in the PNS
To study the effect of circulating IgG anti-GM1 Abs on nerve repair, we passively transferred sera containing high titers of these Abs in a mouse model of nerve regeneration described previously (Lehmann et al., 2007). In this model (Fig. 1A) the distal stump undergoing Wallerian degeneration has breakdown of blood-nerve-barrier allowing circulating Abs to access injured nerve undergoing repair. We found that sera from GBS patients decreased the number of regenerating axons in sciatic and tibial nerve segments (S2 and S3 segments, respectively) compared with those in controls (sham Ab-treated regenerating nerves) (Fig. 1B,C). Morphometric analysis showed that compared with controls the numbers of regenerating myelinated fibers (MFs) in the S2 segment were decreased by ∼39% and 32% with the administration of AMSAN (JHH-9) and AMAN (98-7) sera, respectively (Fig. 1D). The inhibitory effect was more pronounced at the tibial (S3 segment) level and numbers of MFs at this level were decreased by ∼70%, 42% with the administration of AMSAN (JHH-9) and AMAN (98-7) respectively (Fig. 1E). Nerve segments above the crush site (S1 segments) did not show any antibody related injury (data not shown), consistent with our previous results (Lehmann et al., 2007).
Passive transfer of IgG anti-GM1 Abs from AMSAN serum inhibits axon regeneration in the PNS
These studies were restricted to serum from patient with AMSAN (JHH-9) because sufficient quantities were not available to perform these studies with AMAN sera (98-7). The goal of these studies was to directly link IgG anti-GM1 Abs with inhibition of axon regeneration. IgG fractions were isolated by protein-G affinity chromatography and administered to animals with sciatic nerve crush injury, as described in Materials and Methods. We found that IgG fractions from AMSAN serum decreased the number of regenerating MFs by ∼22% and 67% in sciatic (S2) and tibial (S3) nerves, respectively (Fig. 2A,B). Further, affinity purified IgG anti-GM1 Abs from AMSAN serum decreased the number of regenerating MFs by ∼28% and 40% in sciatic (S2) and tibial (S3) nerves, respectively (Fig. 2A,B). The inhibition induced by affinity purified IgG anti-GM1 Abs was less than the corresponding serum and IgG fractions. This could reflect less activity of the purified anti-GM1 Abs transferred passively due to differences in pharmacokinetics of purified Abs versus whole serum. Alternatively, GBS serum contained inhibitory factors other than anti-GM1 Abs. Overall, these sequential studies directly link the inhibitory effects on nerve repair to IgG fractions and IgG anti-GM1 Abs in the AMSAN serum.
Figure 2.
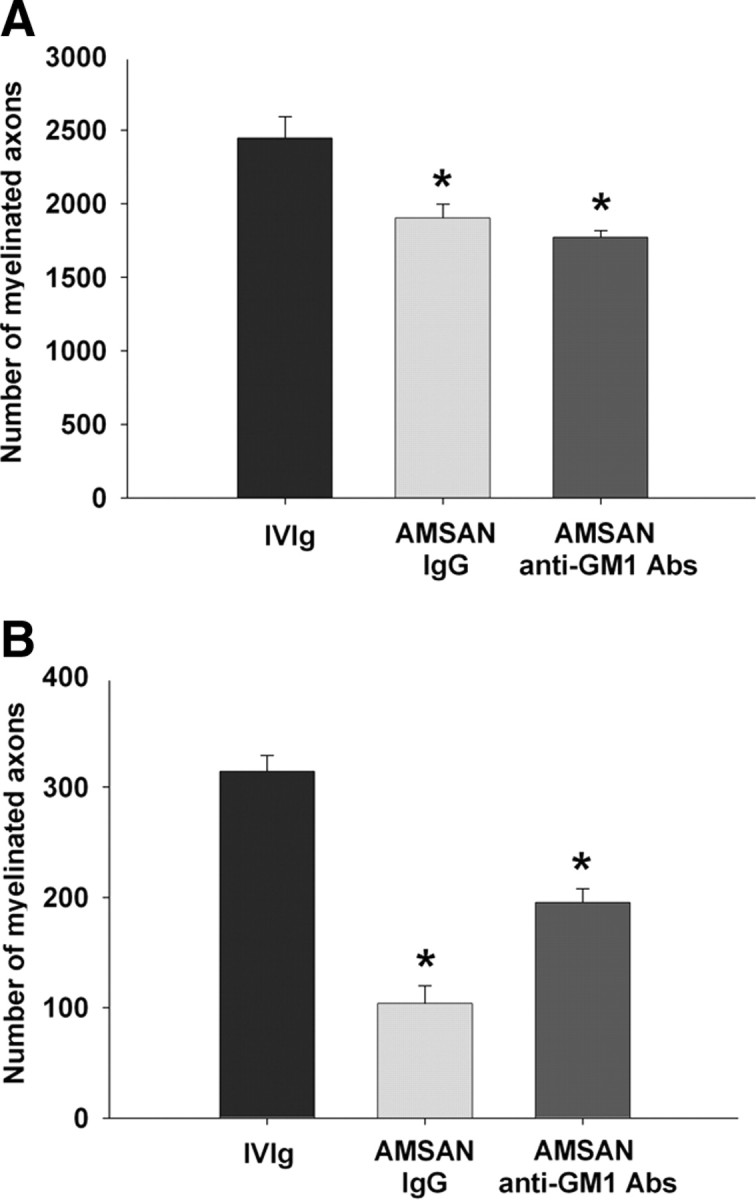
IgG fractions and affinity-purified IgG anti-GM1 antibodies from AMSAN serum inhibit nerve repair. A, B, Significant decrease in number of regenerating axons at sciatic (S2; A) and tibial (S3; B) nerve levels in animals treated with IgG fractions or IgG anti-GM1 antibodies compared with control animals treated with IVIg. *p < 0.05.
Human IgG antibodies access injured peripheral nerves
To demonstrate that passively transferred human IgG circulating antibodies accessed the injured nerve; we analyzed the presence of IgG in sciatic nerve sections by immunocytochemistry. Figure 3 shows that immunoreactivity for human IgG was seen in the endoneurial compartment of injured nerves in both AMSAN serum and control IgG-administered animals. Quantification showed significantly more (∼40%; p < 0.05) immunoreactivity (pixel intensity) for human IgG in nerves treated with AMSAN serum (62 ± 21) compared with controls (40 ± 18). These results are consistent with our previous findings that with nerve injury both specific and nonspecific immunoglobulins are recruited to the endoneurium but more immunoglobulins are retained in the anti-ganglioside antibody-treated nerves (Lehmann et al., 2007).
Figure 3.

A, B, Human IgG accumulates in injured nerves. Compared with control nerves (A) there is a significant increase in IgG accumulation in AMSAN sera-treated nerves (B). Scale bar, 1 mm.
Passive transfer of AMSAN serum impairs sensory nerve regeneration
These studies were done to evaluate the effects of AMSAN serum on sensory nerve regeneration. JHH-9-treated nerves had almost no regenerating fibers compared with control serum-treated nerves (Fig. 4A,B). Analysis of sural nerves showed a significant (p ≤ 0.05) reduction in the regeneration of the MFs in animals treated with JHH-9 serum (1.3 ± 0.7) compared with control serum-treated mice (52.3 ± 18.9). This finding would suggest that these antibodies predominantly inhibit sensory fiber regeneration in mice. Our finding that immunostaining of rodent cauda equina using affinity purified IgG anti-GM1 Abs from AMSAN serum showed a preferential staining of sensory fibers (Fig. 4C) would be consistent with inhibition of sensory fiber regeneration.
Figure 4.

AMSAN serum inhibits regeneration in sensory nerves. A, B, Representative micrographs showing regenerating nerve fibers in control serum-treated nerve (A) compared with JHH-9 serum-treated nerve (B), which hardly has any regenerating fibers. Scale bar, 20 μm. C, Affinity purified IgG anti-GM1 antibodies from AMSAN serum preferentially bind to dorsal roots (red, left panel). IB4 staining (green) on the corresponding field is shown in the right panel to delineate ventral (VR) and dorsal roots (DR). Scale bar, 50 μm.
Passive transfer of AMSAN serum does not impair motor reinnervation
We used two measures of motor reinnervation in sciatic nerve crush model, these include sciatic nerve conductions and volumetric magnetic resonance imaging (MRI) of the calf muscles. We found that compound motor axonal potential (CMAP) amplitudes in hindpaw were delayed and dispersed (consistent with regeneration) in both AMSAN and control sera-treated animals (Fig. 5A) with no quantitative differences between two groups (data not shown). Volumetric MRI also showed that there was no significant difference in the volumes of calf muscles in AMSAN (142 ± 5 mm3) and control (143 ± 3 mm3) sera-treated animals (Fig. 5B). These observations suggest that anti-GM1 Abs in AMSAN serum did not induce inhibition of motor nerve regeneration by the measures of motor reinnervation used in this study.
Figure 5.

AMSAN serum does not affect motor nerve regeneration. A, Representative micrographs showing comparable evoked motor amplitudes in AMSAN- and control serum-treated animals. B, Control- and AMSAN serum-treated nerves do not show significant differences in calf muscle volumes determined by MRI.
Passive transfer of CTB impairs nerve repair in the PNS
We next examined the effects of CTB, a specific ligand of ganglioside GM1, to determine whether other GM1 ligands have inhibitory effects on nerve repair. We found that CTB-treated nerves had a significant increase in large diameter unmyelinated axons resembling dystrophic sprouts in the S2 segment of the nerve compared with vehicle-treated controls (Fig. 6A,B). Electron microscopy confirmed that these large dystrophic axons remained unmyelinated and were almost always associated with Schwann cells (Fig. 6D). Histograms of the axon calibers showed that the distribution of MFs was similar in both CTB- and vehicle-treated mice but the distribution of unmyelinated fibers showed a significant rightward shift due to dystrophic change in CTB-treated animals (Fig. 6E,F). Dystrophic sprouts were restricted to the S2 (sciatic) segment and were not seen in S3 (tibial) segment of the CTB-treated nerves. Morphometry showed that the total number of regenerating myelinated fibers was significantly (p < 0.05) decreased in CTB-treated nerves at sciatic (1264 ± 86) and tibial (135 ± 5) levels (S2 and S3 segments, respectively) compared with vehicle-treated controls (2309 ± 349 and 350 ± 23 in sciatic and tibial segments, respectively).
Figure 6.
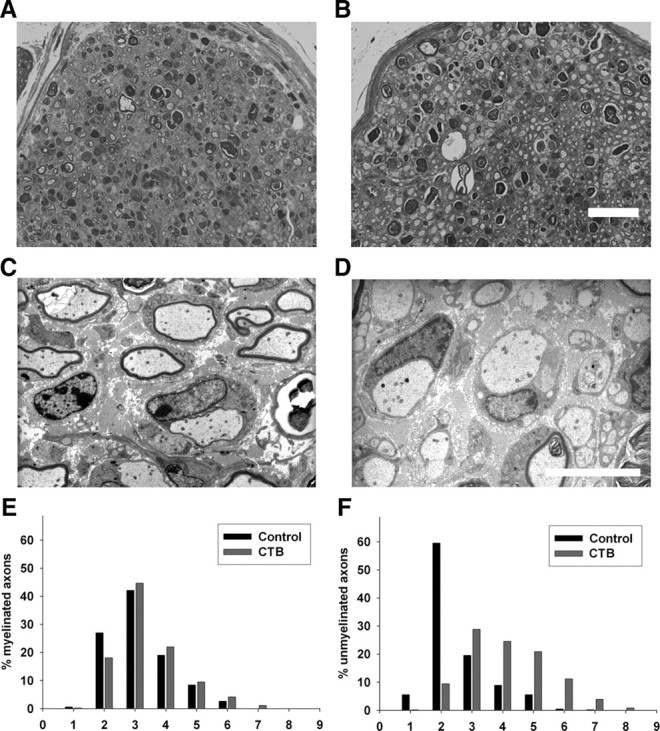
Cholera toxin B-mediated inhibition of peripheral nerve repair. A–D, Light (A, B) and electron (C, D) micrographs of sciatic nerve S2 segments. A, B, Many regenerating fibers are present in both vehicle (A)- and CTB (B)-treated nerves. Scale bar, 2 μm. C, D, Normally myelinating fibers are present in the vehicle-treated nerves (C), whereas large dystrophic-appearing axons without myelination are commonly present in the CTB-treated nerves (D) at the sciatic level (S2). Scale bar, 2 μm. E, F, Histograms showing distribution of myelinated (E) and unmyelinated dystrophic fibers (F) in vehicle (control)- and CTB-treated nerves at sciatic (S2) level; a marked rightward shift in the distribution of unmyelinated dystrophic axons is seen with CTB treatment (F).
Motor reinnervation was assessed in mice treated with CTB. We found that CMAP amplitudes (recorded in hindpaws; Fig. 7) were significantly (p < 0.05) smaller in the CTB-treated group (0.6 ± 0.25 mV) compared with controls (1.5 ± 0.3 mV).
Figure 7.

Motor reinnervation is reduced in CTB-treated animals. A, B, Representative tracings recorded in hindpaws of mice showing decreased CMAP amplitudes in CTB-treated mice (B) compared with vehicle-treated controls (A).
Passive transfer of IgG anti-GM1 mAb does not impair axon regeneration
We then examined the effects of an IgG anti-GM1 mAb on nerve repair and found that this mAb did not induce inhibition of axon regeneration in wild-type mice at sciatic (2658 ± 97) and tibial (656 ± 34) levels (S2 and S3 segments, respectively) compared with control IgG-treated animals (2723 ± 146 and 644 ± 42 in sciatic and tibial segments, respectively).
Determination of titer and affinity of anti-GM1 antibodies used for passive transfer
To examine factors that could influence anti-GM1 Ab-mediated impairment of axon regeneration, we analyzed the titer of circulating anti-GM1 Abs in mice receiving GBS sera. Table 1 shows that mice treated with patient sera have an antibody titer that ranges from 1/3200 to 1/12,800 and animals treated with GM1-2b mAb had titers up to 1/409,600. We found that affinity values (IC50) for JHH-9 and 98-7 were 8 × 10−6 m and 1 × 10−4 m, respectively (Table 1). IC50 could not be determined for GM1-2b mAb with highest concentration tested (1 × 10−4 m). These results indicate that the antibody affinity but not the titers correlate with inhibition.
Table 1.
Circulating titers and affinity (IC50) of patient sera and anti-GM1 mAb
| GBS patient/mAb |
|||
|---|---|---|---|
| JHH-9 | 98-7 | IgG GM1-2b | |
| Circulating Ab titer | 6400–12,800 | 3200 | 204,800–409,600 |
| IC50 (oligo-GM1) | 8 × 10−6 m | 1 × 10−4 m | >1 × 10−4 m |
GM1 ligands have distinct patterns of binding to GM1-derivatives
Figure 8A shows the structures of the sialic-acid derivatives used in this study. The structural requirements of GM1 ligands were evaluated by examining their reactivities to different derivatives of GM1 bearing chemical substitutions on the sialic acid (Fig. 8B). We observed that modifications of the carboxyl group dramatically affect the reactivity of CTB and GM1-2b and 98-7 Abs in contrast to the reactivity of JHH-9 Abs that was much less affected. It is noteworthy that CTB and 98-7 react with one of the three derivatives targeting the carboxyl group (GM1-alcohol and GM1-methylester, respectively) supporting the notion that the interaction of the carboxyl group with these ligands is different. Modification of the glycerol chain (GM1-7-aldehyde) abolished the reactivity of 98-7 and GM1-2b Abs and CTB but did not affect the reactivity of JHH-9 Abs. Finally elimination of the N-acetyl group dramatically reduced the reactivity of 98-7 Abs and CTB but did not affect the binding of JHH-9 and GM1-2b Abs (Fig. 8B). In summary, the individual GM1 ligands used in this study display unique structural requirements on the sialic acid of GM1 ganglioside.
Figure 8.

A, GM1 derivatives. Positions of the chemical substitutions made on the N-acetylneuraminic acid residue of GM1 according to description under Materials and Methods. B, Effect of sialic acid modifications on binding of GM1 ligands. Ligand reactivity to GM1 and GM1-derivatives was analyzed by solid phase assays and quantified as described in Materials and Methods.
Discussion
Our results show that passive transfer of sera and/or IgG fractions and affinity purified IgG anti-GM1 Abs from two patients with GBS impaired axon regeneration/nerve repair in an animal model. We found that these inhibitory effects could also be reproduced by CTB, which is a specific ligand for ganglioside GM1. In contrast, a low affinity mAb GM1-2b did not inhibit regeneration in wild-type animals. These results indicate that ligand related factors such as affinity and fine specificity are important determinants for antibody/ligand-mediated inhibition of nerve repair. These results support the notion that patient IgG anti-GM1 Abs can impair axon regeneration and modulate repair process in the peripheral nerves.
This study included a patient who had severe form of GBS with motor and sensory axon involvement, IgG anti-GM1 Abs, and poor recovery (JHH-9). We show that these patient derived IgG fractions and affinity purified IgG anti-GM1 Abs inhibit nerve repair. These findings demonstrate that patient derived autoimmune Abs targeting GM1-like epitopes inhibit nerve repair in a preclinical animal model. High titers of IgG anti-GM1 Abs have been strongly associated with GBS particularly with axonal forms of this disorder (Yuki et al., 1990) and with poor prognosis and/or incomplete recovery (Ilyas et al., 1992; Gregson et al., 1993; Simone et al., 1993; Jacobs et al., 1996; Bech et al., 1997; Kuwabara et al., 1998a,b; Carpo et al., 1999; Hadden et al., 2001; Press et al., 2001; Annunziata et al., 2003; Koga et al., 2003). The current study provides one potential explanation for poor recovery and associated impairment in nerve repair in GBS patients with high titers of anti-GM1 Abs.
Comparative studies with Abs and CTB, in addition to providing “proof of concept,” also provide important insights into the properties of the ligands that determine the selectivity of the fiber type affected. We observed preferential inhibition of sensory fiber regeneration with GBS sera whereas CTB-induced inhibition of motor and likely sensory fibers. The inhibitory effects of the JHH-9 and CTB correlate with the nerve fiber binding patterns of these ligands. In this study we show that JHH-9 Abs preferentially stain sensory fibers and inhibit regeneration of sensory fibers. We have reported that CTB binds to both motor and sensory fibers (Sheikh et al., 1999a), which is consistent with the CTB-mediated inhibition of motor and sensory nerve fibers in this study. GM1-2b mAb did not induce inhibitory effects likely due to its affinity-avidity (see below) despite its reported binding to motor and sensory nerve fibers (Gong et al., 2002). The immunocytochemical studies [which require affinity purified anti-GM1 Abs because normal human serum can have anti-neurofilament Abs and axonal binding (Stefansson et al., 1985)] with 98-7 Abs could not be performed because of limited quantity of this serum was available. Fine specificity experiments indicate that GM1 ligands used in this study have distinct reactivities to GM1-derivatives and this could be one factor that influences selective nerve fiber/target recognition, as was suggested previously (O'Hanlon et al., 1996; Lopez et al., 2008). In addition to fine specificity of the Abs, accessibility of glycolipid antigens to Abs is also very important. Willison's group in Glasgow has recently demonstrated that just a very small fraction of all the GM1 epitopes recognized by Abs may be accessible for binding on mouse motor nerves under physiological conditions (Greenshields et al., 2009) providing another explanation for selective sensory fiber involvement with passive transfer of anti-GM1 Abs in GBS sera in our animal studies. Further, CTB has been shown to cross-link cell surface GM1 and induce calcium influx via L-type voltage gated calcium channels or TRPC5 channels (Buckley et al., 1995; Wang et al., 2009), whereas IgG anti-GM1 Abs in one study did not induce calcium influx presumably due to binding without cross-linking of GM1 (Quattrini et al., 2001). Whether GM1 cross-linking by CTB and engagement without cross-linking by IgG anti-GM1 Abs relate to differences in inhibitory effects is not possible to address in these animal studies and requires separate examination of this issue.
Our findings indicate that beside fine specificity of Abs affinity-avidity of the ligands may also relevant to the inhibition of nerve fiber regeneration. CTB is an oligomeric protein complex ring containing five β-subunits, each of them able to bind GM1 ganglioside. The high affinity interaction of each individual subunit with GM1 plus the pentameric nature of the β-subunit makes CTB one of the most powerful ligands for GM1 (Schön and Freire, 1989; MacKenzie et al., 1997). On the other hand, the affinity of IgG anti-GM1 Abs from GBS sera and experimental mAb used in this study is diverse. For instance, serum JHH-9 has an affinity of 8 × 10−6 m comparable to single β-subunit of CTB (1 × 10−6 m) (Schön and Freire, 1989), and serum 98-7 showed relatively lower affinity anti-GM1 Abs (10−4 m). GM1-2b mAb with the lowest affinity (>10−4 m) did not produce inhibition. Noticeably, the inhibition with human anti-GM1 Abs (low to moderate affinity) is not that different from that of highest affinity ligand CTB (Figs. 1, 6). The major explanation for this is most probably due to differences in the kinetics and distribution of CTB and Abs in the injured nerves. It is established that CTB is retrogradely transported by intact or injured nerve fibers after systemic or local intraneural administration (Tong et al., 1999; Alisky et al., 2002; Sántha and Jancsó, 2003). In our animal model, it is very likely that CTB is internalized and retrogradely transported by injured axons, thereby, terminating CTB's interaction with surface glycolipids of injured axonal tips. Further, we have recently found that expression of Fc-gamma (Fcγ) receptors on injured Schwann cells is a prerequisite for anti-ganglioside antibody-mediated inhibition of axon regeneration (Sheikh et al., 2009) suggesting that these receptors anchor IgG antibodies via their Fc fragments for interaction with adjacent regenerating axons. This Fc-Fcγ binding is likely to influence the operative affinity/avidity of antibody-antigen interactions and also prevent internalization of the Abs thus enhancing their effective half-life and pathogenicity in the injured nerves.
The current study directly links patient-derived IgG poly/oligoclonal anti-GM1 Abs to inhibition of axon regeneration and extends our previous findings showing that a GD1a/GT1b-reactive mAb induced severe inhibition of axon regeneration in mice (Lehmann et al., 2007). The GD1a/GT1b-reactive mAb induced more inhibition of both sensory and motor nerve fibers compared with the GBS sera or CTB, however, the two studies are not directly comparable because the specificity of anti-ganglioside Abs, origin of Abs/ligands, and dose of anti-ganglioside Abs/ligands used in these studies are very different. Our results are consistent with previous findings indicating that Abs directed against GM1 or other gangliosides can inhibit regeneration in nonmammalian neurons (Schwartz and Spirman, 1982; Spirman et al., 1982; Sparrow et al., 1984; Spoerri et al., 1988); current study extends this finding to mammalian neurons.
There are two clinical studies that have noted that not all patients with GM1 Abs and GBS have adverse outcome or poor recovery (Enders et al., 1993; Vriesendorp et al., 1995). The pathogenicity of anti-ganglioside antibody-mediated nerve injury is a complex multifactorial issue (Sheikh and Zhang, 2010). Neurobiological factors such as extent and site of axonal injury and age-related regenerative capacity of the host (Black and Lasek, 1979) are important factors on the one hand. On the other hand are immunological factors related to antibody-antigen characteristics such as antibody affinity and fine specificity (highlighted in this study), antigen density (Goodfellow et al., 2005), isotype of the antibody which affects the half-life of circulating Abs (Koga et al., 2003), and as yet other undefined factors. Serological studies primarily defining the specificity of anti-ganglioside Abs in patients with GBS and correlating this with clinical recovery are likely to have low predictability because solid phase assays commonly used to determine the presence of Abs do not provide any information about these immunological factors related to antibody-mediated adverse effects on nerve repair (Sheikh and Zhang, 2010).
Our findings have important implications for neural repair in autoimmune neurological disorders. We show that circulating immune effectors can access injured nerves and adversely affect nerve repair. This is particularly relevant to immune neuropathies such as GBS and chronic inflammatory demyelinating polyneuropathy in which recovery relates to the extent of axon injury (Asbury et al., 1969; Brown and Feasby, 1984; Feasby et al., 1986; Bouchard et al., 1999). Recovery in these situations require axon regeneration and target reinnervation and the presence of autoantibodies against gangliosides can foil the injured axons attempt to regenerate. These observations raise the possibility that immune effectors (including Abs) may also impair the neural repair in CNS disorders such as multiple sclerosis in which autoantibodies including those against glycolipids are increasingly recognized (Cross et al., 2001; Kanter et al., 2006). Modulation of CNS repair by immune effectors is an important issue that requires further studies.
Footnotes
This study was supported by National Institutes of Health–National Institute of Neurological Disorders and Stroke (Grants NS42888 and NS54962) and the Guillain-Barré Syndrome/Chronic Inflammatory Demyelinating Polyneuropathy Foundation.
References
- Alisky JM, van de Wetering CI, Davidson BL. Widespread dispersal of cholera toxin subunit b to brain and spinal cord neurons following systemic delivery. Exp Neurol. 2002;178:139–146. doi: 10.1006/exnr.2002.8031. [DOI] [PubMed] [Google Scholar]
- Annunziata P, Figura N, Galli R, Mugnaini F, Lenzi C. Association of anti-GM1 antibodies but not of anti-cytomegalovirus, Campylobacter jejuni and Helicobacter pylori IgG, with a poor outcome in Guillain-Barre syndrome. J Neurol Sci. 2003;213:55–60. doi: 10.1016/s0022-510x(03)00149-7. [DOI] [PubMed] [Google Scholar]
- Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. Medicine. 1969;48:173–215. doi: 10.1097/00005792-196905000-00001. [DOI] [PubMed] [Google Scholar]
- Aspinall GO, Fujimoto S, McDonald AG, Pang H, Kurjanczyk LA, Penner JL. Lipopolysaccharides from Campylobacter jejuni associated with Guillain-Barre syndrome patients mimic human gangliosides in structure. Infect Immun. 1994;62:2122–2125. doi: 10.1128/iai.62.5.2122-2125.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bech E, Orntoft TF, Andersen LP, Skinhøj P, Jakobsen J. IgM anti-GM1 antibodies in the Guillain-Barre syndrome: a serological predictor of the clinical course. J Neuroimmunol. 1997;72:59–66. doi: 10.1016/s0165-5728(96)00145-2. [DOI] [PubMed] [Google Scholar]
- Black MM, Lasek RJ. Slowing of the rate of axonal regeneration during growth and maturation. Exp Neurol. 1979;63:108–119. doi: 10.1016/0014-4886(79)90188-2. [DOI] [PubMed] [Google Scholar]
- Bouchard C, Lacroix C, Planté V, Adams D, Chedru F, Guglielmi JM, Said G. Clinicopathologic findings and prognosis of chronic inflammatory demyelinating polyneuropathy. Neurology. 1999;52:498–503. doi: 10.1212/wnl.52.3.498. [DOI] [PubMed] [Google Scholar]
- Brown WF, Feasby TE. Conduction block and denervation in Guillain-Barre polyneuropathy. Brain. 1984;107:219–239. doi: 10.1093/brain/107.1.219. [DOI] [PubMed] [Google Scholar]
- Buckley NE, Su Y, Milstien S, Spiegel S. The role of calcium influx in cellular proliferation induced by interaction of endogenous ganglioside GM1 with the B subunit of cholera toxin. Biochim Biophys Acta. 1995;1256:275–283. doi: 10.1016/0005-2760(95)00030-g. [DOI] [PubMed] [Google Scholar]
- Carpo M, Pedotti R, Allaria S, Lolli F, Matà S, Cavaletti G, Protti A, Pomati S, Scarlato G, Nobile-Orazio E. Clinical presentation and outcome of Guillain-Barre and related syndromes in relation to anti-ganglioside antibodies. J Neurol Sci. 1999;168:78–84. doi: 10.1016/s0022-510x(99)00173-2. [DOI] [PubMed] [Google Scholar]
- Cross AH, Trotter JL, Lyons J. B cells and antibodies in CNS demyelinating disease. J Neuroimmunol. 2001;112:1–14. doi: 10.1016/s0165-5728(00)00409-4. [DOI] [PubMed] [Google Scholar]
- Dell A, Reason AJ, Khoo KH, Panico M, McDowell RA, Morris HR. Mass spectrometry of carbohydrate-containing biopolymers. Methods Enzymol. 1994;230:108–132. doi: 10.1016/0076-6879(94)30010-0. [DOI] [PubMed] [Google Scholar]
- Enders U, Karch H, Toyka KV, Michels M, Zielasek J, Pette M, Heesemann J, Hartung HP. The spectrum of immune responses to Campylobacter jejuni and glycoconjugates in Guillain-Barre syndrome and in other neuroimmunological disorders. Ann Neurol. 1993;34:136–144. doi: 10.1002/ana.410340208. [DOI] [PubMed] [Google Scholar]
- Feasby TE, Gilbert JJ, Brown WF, Bolton CF, Hahn AF, Koopman WF, Zochodne DW. An acute axonal form of Guillain-Barré polyneuropathy. Brain. 1986;109:1115–1126. doi: 10.1093/brain/109.6.1115. [DOI] [PubMed] [Google Scholar]
- Gong Y, Tagawa Y, Lunn MP, Laroy W, Heffer-Lauc M, Li CY, Griffin JW, Schnaar RL, Sheikh KA. Localization of major gangliosides in the PNS: implications for immune neuropathies. Brain. 2002;125:2491–2506. doi: 10.1093/brain/awf258. [DOI] [PubMed] [Google Scholar]
- Goodfellow JA, Bowes T, Sheikh K, Odaka M, Halstead SK, Humphreys PD, Wagner ER, Yuki N, Furukawa K, Furukawa K, Plomp JJ, Willison HJ. Overexpression of GD1a ganglioside sensitizes motor nerve terminals to anti-GD1a antibody-mediated injury in a model of acute motor axonal neuropathy. J Neurosci. 2005;25:1620–1628. doi: 10.1523/JNEUROSCI.4279-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenshields KN, Halstead SK, Zitman FM, Rinaldi S, Brennan KM, O'Leary C, Chamberlain LH, Easton A, Roxburgh J, Pediani J, Furukawa K, Furukawa K, Goodyear CS, Plomp JJ, Willison HJ. The neuropathic potential of anti-GM1 autoantibodies is regulated by the local glycolipid environment in mice. J Clin Invest. 2009;119:595–610. doi: 10.1172/JCI37338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregson NA, Koblar S, Hughes RA. Antibodies to gangliosides in Guillain-Barre syndrome: specificity and relationship to clinical features. Q J Med. 1993;86:111–117. [PubMed] [Google Scholar]
- Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, Swan AV. Electrophysiological classification of Guillain-Barre syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barre Syndrome Trial Group. Ann Neurol. 1998;44:780–788. doi: 10.1002/ana.410440512. [DOI] [PubMed] [Google Scholar]
- Hadden RD, Karch H, Hartung HP, Zielasek J, Weissbrich B, Schubert J, Weishaupt A, Cornblath DR, Swan AV, Hughes RA, Toyka KV. Preceding infections, immune factors, and outcome in Guillain-Barre syndrome. Neurology. 2001;56:758–765. doi: 10.1212/wnl.56.6.758. [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y, Suzuki T, Suzuki Y, Taki T, Matsumoto M, Higashi H, Kato S. A new method for purification of anti-glycosphingolipid antibody-avian anti-hematoside (Neugc) antibody. J Biochem. 1983;94:327–330. doi: 10.1093/oxfordjournals.jbchem.a134350. [DOI] [PubMed] [Google Scholar]
- Ho TW, Mishu B, Li CY, Gao CY, Cornblath DR, Griffin JW, Asbury AK, Blaser MJ, McKhann GM. Guillain-Barre syndrome in northern China: relationship to Campylobacter jejuni infection and anti-glycolipid antibodies. Brain. 1995;118:597–605. doi: 10.1093/brain/118.3.597. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366:1653–1666. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- Ilyas AA, Mithen FA, Dalakas MC, Chen ZW, Cook SD. Antibodies to acidic glycolipids in Guillain-Barré syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 1992;107:111–121. doi: 10.1016/0022-510x(92)90217-9. [DOI] [PubMed] [Google Scholar]
- Jacobs BC, van Doorn PA, Schmitz PI, Tio-Gillen AP, Herbrink P, Visser LH, Hooijkass H, van der Meché FG. Campylobacter jejuni infections and anti-GM1 antibodies in Guillain-Barré syndrome. Ann Neurol. 1996;40:181–187. doi: 10.1002/ana.410400209. [DOI] [PubMed] [Google Scholar]
- Jacobs BC, Hazenberg MP, Van Doorn PA, Endtz HP, van der Meché FGA. Cross-reactive antibodies against gangliosides and Campylobacter jejuni lipopolysaccharides in patients with Guillain-Barré or Miller Fisher syndrome. J Infect Dis. 1997;175:729–733. doi: 10.1093/infdis/175.3.729. [DOI] [PubMed] [Google Scholar]
- Kanter JL, Narayana S, Ho PP, Catz I, Warren KG, Sobel RA, Steinman L, Robinson WH. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- Koga M, Yuki N, Hirata K, Morimatsu M, Mori M, Kuwabara S. Anti-GM1 antibody IgG subclass: a clinical recovery predictor in Guillain-Barre syndrome. Neurology. 2003;60:1514–1518. doi: 10.1212/01.wnl.0000061615.77865.83. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, Noda M. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barre syndrome. Ann Neurol. 1998a;44:202–208. doi: 10.1002/ana.410440210. [DOI] [PubMed] [Google Scholar]
- Kuwabara S, Asahina M, Koga M, Mori M, Yuki N, Hattori T. Two patterns of clinical recovery in Guillain-Barre syndrome with IgG anti-GM1 antibody. Neurology. 1998b;51:1656–1660. doi: 10.1212/wnl.51.6.1656. [DOI] [PubMed] [Google Scholar]
- Lehmann HC, Lopez PH, Zhang G, Ngyuen T, Zhang J, Kieseier BC, Mori S, Sheikh KA. Passive immunization with anti-ganglioside antibodies directly inhibits axon regeneration in an animal model. J Neurosci. 2007;27:27–34. doi: 10.1523/JNEUROSCI.4017-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez PH, Irazoqui FJ, Nores GA. Normal human plasma contains antibodies that specifically block neuropathy-associated human anti-GM1 IgG-antibodies. J Neuroimmunol. 2000;105:179–183. doi: 10.1016/s0165-5728(99)00276-3. [DOI] [PubMed] [Google Scholar]
- Lopez PH, Villa AM, Sica RE, Nores GA. High affinity as a disease determinant factor in anti-GM(1) antibodies: comparative characterization of experimentally induced vs disease-associated antibodies. J Neuroimmunol. 2002;128:69–76. doi: 10.1016/s0165-5728(02)00139-x. [DOI] [PubMed] [Google Scholar]
- Lopez PH, Zhang G, Bianchet MA, Schnaar RL, Sheikh KA. Structural requirements of anti-GD1a antibodies determine their target specificity. Brain. 2008;131:1926–1939. doi: 10.1093/brain/awn074. [DOI] [PubMed] [Google Scholar]
- MacKenzie CR, Hirama T, Lee KK, Altman E, Young NM. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J Biol Chem. 1997;272:5533–5538. doi: 10.1074/jbc.272.9.5533. [DOI] [PubMed] [Google Scholar]
- Nores GA, Hanai N, Levery SB, Eaton HL, Salyan EK, Hakomori S. Synthesis and characterization of lyso-GM3 (II3Neu5Ac Lactosyl sphingosine), de-N-acetyl-GM3 (II3NeuNH2 lactosyl Cer), and related compounds. Carbohydr Res. 1988;179:393–410. doi: 10.1016/0008-6215(88)84135-1. [DOI] [PubMed] [Google Scholar]
- Ogawara K, Kuwabara S, Mori M, Hattori T, Koga M, Yuki N. Axonal Guillain-Barre syndrome: relation to anti-ganglioside antibodies and Campylobacter jejuni infection in Japan. Ann Neurol. 2000;48:624–631. [PubMed] [Google Scholar]
- O'Hanlon GM, Paterson GJ, Wilson G, Doyle D, McHardie P, Willison HJ. Anti-GM1 ganglioside antibodies cloned from autoimmune neuropathy patients show diverse binding patterns in the rodent nervous system. J Neuropathol Exp Neurol. 1996;55:184–195. doi: 10.1097/00005072-199602000-00007. [DOI] [PubMed] [Google Scholar]
- Press R, Matá S, Lolli F, Zhu J, Andersson T, Link H. Temporal profile of anti-ganglioside antibodies and their relation to clinical parameters and treatment in Guillain-Barre syndrome. J Neurol Sci. 2001;190:41–47. doi: 10.1016/s0022-510x(01)00580-9. [DOI] [PubMed] [Google Scholar]
- Quattrini A, Lorenzetti I, Sciorati C, Corbo M, Previtali SC, Feltri ML, Canal N, Wrabetz L, Nemni R, Clementi E. Human IgM anti-GM1 autoantibodies modulate intracellular calcium homeostasis in neuroblastoma cells. J Neuroimmunol. 2001;114:213–219. doi: 10.1016/s0165-5728(00)00452-5. [DOI] [PubMed] [Google Scholar]
- Sántha P, Jancsó G. Transganglionic transport of choleragenoid by capsaicin-sensitive C-fibre afferents to the substantia gelatinosa of the spinal dorsal horn after peripheral nerve section. Neuroscience. 2003;116:621–627. doi: 10.1016/s0306-4522(02)00701-7. [DOI] [PubMed] [Google Scholar]
- Schnaar RL, Fromholt SE, Gong Y, Vyas AA, Laroy W, Wayman DM, Heffer-Lauc M, Ito H, Ishida H, Kiso M, Griffin JW, Shiekh KA. IgG-class mouse monoclonal antibodies to major brain gangliosides. Anal Biochem. 2002;302:276–284. doi: 10.1006/abio.2001.5540. [DOI] [PubMed] [Google Scholar]
- Schön A, Freire E. Thermodynamics of intersubunit interactions in cholera toxin upon binding to the oligosaccharide portion of its cell surface receptor, ganglioside GM1. Biochemistry. 1989;28:5019–5024. doi: 10.1021/bi00438a017. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Spirman N. Sprouting from chicken-embryo dorsal root-ganglia induced by nerve growth-factor is specifically inhibited by affinity-purified antiganglioside antibodies. Proc Natl Acad Sci U S A. 1982;79:6080–6083. doi: 10.1073/pnas.79.19.6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G. An update on pathobiologic roles of anti-glycan antibodies in Guillain-Barré syndrome. F1000 Biology Reports. 2010;2:21. doi: 10.3410/B2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh KA, Nachamkin I, Ho TW, Willison HJ, Veitch J, Ung H, Nicholson M, Li CY, Wu HS, Shen BQ, Cornblath DR, Asbury AK, McKhann GM, Griffin JW. Campylobacter jejuni lipopolysaccharides in Guillain-Barre syndrome: molecular mimicry and host susceptibility. Neurology. 1998;51:371–378. doi: 10.1212/wnl.51.2.371. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Deerinck TJ, Ellisman MH, Griffin JW. The distribution of ganglioside-like moieties in peripheral nerves. Brain. 1999a;122:449–460. doi: 10.1093/brain/122.3.449. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Sun J, Liu Y, Kawai H, Crawford TO, Proia RL, Griffin JW, Schnaar RL. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc Natl Acad Sci U S A. 1999b;96:7532–7537. doi: 10.1073/pnas.96.13.7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G, Gong Y, Schnaar RL, Griffin JW. An anti-ganglioside antibody-secreting hybridoma induces neuropathy in mice. Ann Neurol. 2004;56:228–239. doi: 10.1002/ana.20173. [DOI] [PubMed] [Google Scholar]
- Sheikh KA, Zhang G, Lehmann HC. Fc gamma receptors on injured Schwann cells facilitate antibody-mediated modulation of axon regeneration. J Peripher Nerv Syst; Presented at the Peripheral Nerve Society Meeting; July 4–8, 2009; Würzburg, Germany. 2009. p. 136. [Google Scholar]
- Simone IL, Annunziata P, Maimone D, Liguori M, Leante R, Livrea P. Serum and CSF anti-GM1 antibodies in patients with Guillain-Barre syndrome and chronic inflammatory demyelinating polyneuropathy. J Neurol Sci. 1993;114:49–55. doi: 10.1016/0022-510x(93)90048-4. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, McGuinness C, Schwartz M, Grafstein B. Antibodies to gangliosides inhibit goldfish optic-nerve regeneration in vivo. J Neurosci Res. 1984;12:233–243. doi: 10.1002/jnr.490120211. [DOI] [PubMed] [Google Scholar]
- Spirman N, Sela BA, Schwartz M. Antiganglioside antibodies inhibit neuritic outgrowth from regenerating goldfish retinal explants. J Neurochem. 1982;39:874–877. doi: 10.1111/j.1471-4159.1982.tb07974.x. [DOI] [PubMed] [Google Scholar]
- Spoerri PE, Rapport MM, Mahadik SP, Roisen FJ. Inhibition of conditioned media-mediated neuritogenesis of sensory ganglia by monoclonal antibodies to GM1 ganglioside. Brain Res. 1988;469:71–77. doi: 10.1016/0165-3806(88)90170-8. [DOI] [PubMed] [Google Scholar]
- Stefansson K, Marton LS, Dieperink ME, Molnar GK, Schlaepfer WW, Helgason CM. Circulating autoantibodies to the 200,000-dalton protein of neurofilaments in the serum of healthy individuals. Science. 1985;228:1117–1119. doi: 10.1126/science.4039466. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Boström K, Fredman P, Jungbjer B, Månsson JE, Rynmark BM. Membrane lipids of human peripheral nerve and spinal cord. Biochim Biophys Acta. 1992;1128:1–7. doi: 10.1016/0005-2760(92)90250-y. [DOI] [PubMed] [Google Scholar]
- Tong YG, Wang HF, Ju G, Grant G, Hökfelt T, Zhang X. Increased uptake and transport of cholera toxin B-subunit in dorsal root ganglion neurons after peripheral axotomy: possible implications for sensory sprouting. J Comp Neurol. 1999;404:143–158. [PubMed] [Google Scholar]
- Toyka KV, Drachman DB, Griffin DE, Pestronk A, Winkelstein JA, Fishbeck KH, Kao I. Myasthenia gravis: study of humoral immune mechanisms by transfer to mice. N Engl J Med. 1977;296:125–131. doi: 10.1056/NEJM197701202960301. [DOI] [PubMed] [Google Scholar]
- Vriesendorp FJ, Triggs WJ, Mayer RF, Koski CL. Electrophysiological studies in Guillain-Barré syndrome: correlation with antibodies to GM1, GD1B and Campylobacter jejuni. J Neurol. 1995;242:460–465. doi: 10.1007/BF00873550. [DOI] [PubMed] [Google Scholar]
- Wang J, Lu ZH, Gabius HJ, Rohowsky-Kochan C, Ledeen RW, Wu G. Cross-linking of GM1 ganglioside by galectin-1 mediates regulatory T cell activity involving TRPC5 channel activation: possible role in suppressing experimental autoimmune encephalomyelitis. J Immunol. 2009;182:4036–4045. doi: 10.4049/jimmunol.0802981. [DOI] [PubMed] [Google Scholar]
- Willison HJ, Yuki N. Peripheral neuropathies and anti-glycolipid antibodies. Brain. 2002;125:2591–2625. doi: 10.1093/brain/awf272. [DOI] [PubMed] [Google Scholar]
- Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990;40:1900–1902. doi: 10.1212/wnl.40.12.1900. [DOI] [PubMed] [Google Scholar]
- Yuki N, Handa S, Taki T, Kasama T, Takahashi M, Saito K. Cross-reactive antigen between nervous tissue and a bacterium elicits Guillain-Barre syndrome: molecular mimicry between gangliocide GM1 and lipopolysaccharide from Penner's serotype 19 of Campylobacter jejuni. Biomed Res. 1992;13:451–453. [Google Scholar]
- Zhang J, Zhang G, Morrison B, Mori S, Sheikh KA. Magnetic resonance imaging of mouse skeletal muscle to measure denervation atrophy. Exp Neurol. 2008;212:448–457. doi: 10.1016/j.expneurol.2008.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]