

We demonstrate that the key target of increased PIP3 on cell polarity and migration is the excessive Akt signaling. In Dictyostelium, the defects in cells lacking PTEN are suppressed by the secondary deletions of PkbA, or its activator, TorC2. Multiple novel PkbA substrates are identified, and one of them, PakA, links PIP3 to downstream signaling.
Abstract
By limiting phosphotidylinositol 3,4,5-triphosphate (PIP3) levels, tumor suppressor PTEN not only controls cell growth but also maintains cell polarity required for cytokinesis and chemotaxis. To identify the critical targets of PIP3 that link it to the cytoskeleton, we deleted secondary genes to reverse the deficiencies of pten- cells in Dictyostelium. The polarity defects in pten- cells correlate with elevated phosphorylations of PKB substrates. Deletion of AKT orthologue, PkbA, or a subunit of its activator TORC2, reduced the phosphorylations and suppressed the cytokinesis and chemotaxis defects in pten- cells. In these double mutants, the excessive PIP3 levels and, presumably, activation of other PIP3-binding proteins had little or no effect on the cytoskeleton. In bands with increased phosphorylation in pten- cells, we found PKB substrates, PI5K, GefS, GacG, and PakA. Disruption of PakA in pten- cells restored a large fraction of the cells to normal behavior. Consistently, expression of phosphomimetic PakA in pten- cells exacerbated the defects but nonphosphorylatable PakA had no effect. Thus, among many putative PTEN- and PIP3-dependent events, phosphorylation of PKB substrates is the key downstream regulator of cell polarity.
INTRODUCTION
Motile cells are able to detect chemical gradients and move toward or away from the sources. This process, referred to as chemotaxis or directed cell migration, plays an important role in free living cells and in the physiology of organisms. In developing embryos, chemoattractants guide cells to correct locations during the formation of organs and wiring of the nervous system (Engelhardt, 2008; Mortimer et al., 2008). In adults, chemotaxis mediates the trafficking of immune cells, the closure of wounds, and the homing of stem cells to niches (Jones, 2000; Williams et al., 2008). Furthermore, chemotaxis may play a key role in pathological states, such as excessive inflammation and cancer metastasis (Moore, 2001; Savarin-Vuaillat and Ransohoff, 2007; Simon et al., 2009).
Chemoattractants typically bias the basal motile behavior of polarized cells toward the high side of the gradient (Van Haastert and Devreotes, 2004). In many cells, chemoattractants are sensed by G-protein-coupled receptors (GPCRs) and associated G-proteins, which, surprisingly, are uniformly distributed around the perimeter of the cell. In contrast, downstream signaling events are sharply localized. In amoeboid cells such as Dictyostelium and neutrophils, for example, phosphotidylinositol 3,4,5-triphosphate (PIP3) accumulation and new actin polymerization are localized at the tips of pseudopodia that extend rhythmically and propel the cells forward. The signaling pathways appear to be integrated into complex networks that control motility, polarity, and directional sensing (Swaney et al., 2010).
The role of localized PIP3 in these signaling networks has been explored by perturbations that overproduce or eliminate it. In Dictyostelium cells lacking a PI3-phosphatase, PTEN, or expressing constitutively active PI3Ks, PIP3 levels are elevated, actin polymerization responses are prolonged and exaggerated, and chemotaxis is impaired (Iijima and Devreotes, 2002; Huang et al., 2003). In neutrophils, disruption of PTEN or PI5-phosphatase, SHIP, produces a similar phenotype (Liu et al., 1999; Sarraj et al., 2009). Furthermore, synthetic activation of PI3K in neutrophils, which bypasses receptor stimulation and produces intracellular PIP3, triggers pseudopodia extension (Weiner et al., 2002; Inoue and Meyer, 2008). These phenotypes, which are partially suppressed by PI3K inhibitors, show that PIP3 is sufficient to cause the cellular projections. However, the chemotaxis of cells treated with PI3K inhibitors or lacking various PI3Ks is condition-dependent and often only slightly impaired, indicating that additional signals act in parallel to PIP3 (Chen et al., 2007; Hoeller and Kay, 2007; Bosgraaf et al., 2008; Yoo et al., 2010).
In Dictyostelium, two parallel pathways have been delineated. The first involves PLA2 as simultaneous inhibition of PLA2 and PI3K leads to a stronger defect than inhibition of either enzyme alone (Chen et al., 2007; van Haastert et al., 2007). The second parallel pathway results from the presence of both a conventional AKT homologue, PkbA, which is recruited to the membrane by PIP3, and a second PKB isoform, PkbR1, which is tethered to the membrane by myristoylation and can phosphorylate PKB substrates in the absence of PIP3. In cells lacking PI3K activity or PkbA, chemoattractants trigger nearly wild-type patterns of PKB substrate phosphorylation. We have previously shown that chemoattractants trigger activation of TORC2 and several PDKs, which are sufficient to regulate PKBR1 activity in the absence of PIP3. PKB substrate phosphorylation is significantly reduced only by simultaneous removal of both PKBs, the PDKs, or by deletion of PiaA, a key subunit of TORC2 (Kamimura et al., 2008; Kamimura and Devreotes, 2010; Liao et al., 2010). In these cases, the loss of PKB substrate phosphorylation is correlated with defects in chemotaxis (Kamimura et al., 2008).
Although the presence of parallel pathways has stirred considerable debate over the importance of PIP3, few studies have focused on how PIP3 exerts its effects on the cytoskeleton and cell morphology. It has been speculated that PIP3-sensitive Rac GEFs, such as Tiam, Dock 180, and Prex, might be recruited to the membrane and regulate the cytoskeleton (Cote et al., 2005; Fine et al., 2009; Nishikimi et al., 2009; Rivard, 2009). It has also been suggested that several PIP3-sensitive PH-domain-containing proteins of unknown function, such as Crac and PhdA in Dictyostelium, play a role in chemotaxis (Funamoto et al., 2001; Comer et al., 2005). However, individual disruptions of most of these candidates have not produced dramatic chemotaxis phenotypes. The mechanism of action of PIP3 is an important issue as elevated levels not only impair chemotaxis but also interfere with cytokinesis, phagocytosis, macropinocytosis, as well as apical–basalateral polarity of epithelial cells (Zhou et al., 1998; Larsen et al., 2003; Janetopoulos and Devreotes, 2006; Martin-Belmonte et al., 2007). Indeed, many of these altered processes are likely to contribute to the role of PTEN as a tumor suppressor.
We reasoned that because redundant pathways appear to regulate the cytoskeleton and cell shape changes, suppression strategies, rather than simple loss of function studies, would be a more effective means of determining the downstream elements that mediate the role of individual components such as PIP3. Therefore, we disrupted potential mediators in cells with elevated levels of PIP3 due to disruption of PTEN. We started with PkbA and PiaA and found that their removal significantly abrogated the effects of elevated PIP3. This observation prompted us to compare patterns of PKB substrate phosphorylation in the mutant cells lines. We identified specific substrates that were strongly correlated with cellular phenotype. Pursuing one of these, PakA, we found that PakA mediates some of the effects of elevated PIP3.
RESULTS
Effects of excessive PIP3 are mediated by excessive phosphorylation of PKB substrates
To determine whether the effects of elevated PIP3 resulting from loss of Pten are mediated through phosphorylation of PKB substrates, we examined the substrates as well as activation of PkbR1 and PkbA in wild-type and pten- cells. As shown in Figure 1A, consistent with our previous report, chemoattractant stimulation triggered transient phosphorylation of a series of bands, detected by the phospho-PKB substrate antibody, ranging from 66 to 300 kDa in wild-type cells. The levels of phosphorylation generally increased to a peak at 1 min and declined toward the prestimulus values by 2 min. The activation loops of PkbR1 and PkbA were transiently phosphorylated with similar time courses. In pten- cells, some of the bands showed elevated and more persistent phosphorylation. For example, the bands at pp90, pp110, and pp140 remained highly phosphorylated at the 2-min time point (Figure 1A). Interestingly, we previously had observed that bands in this region were slightly decreased pi3k1–5- cells, further suggesting that phosphorylation of these bands is regulated by PIP3. In pten- cells, the activation loop of PkbA showed increased and prolonged phosphorylation, whereas PKBR1 activation was slightly reduced and had a similar time course as in wild-type cells.
FIGURE 1:

Disruption of PkbA, but not PkbR1, reverses defects in pten- cells. (A) Developed cells were stimulated with 1 μM cAMP and collected at the indicated time points. Upper panels show Western blots using anti–phosphospecific PKB substrate antibodies that recognize R-X-R-X-X-S/T-X-X in a phosphorylation-dependent manner. Lower panels show Western blots using anti–phospho PKC (pan) antibodies that recognize phosphorylation of the activation loop in PkbA and PkbR1. Bands that show increased phosphorylation levels compared with wild-type cells are indicated by red dots. (B) Representative images of cells growing on glass substrates. Bar, 50 μm. (C) Chemotaxis of cells moving toward a micropipette filled with 10 μM cAMP. Cells were observed for 30 min at 30-s intervals. Upper panels show images at 30 min after chemotaxis was initiated. In lower panels, trajectories of the entire recording field are shown. The black diamond indicates the location of the micropipette tip. Bar, 50 μm. (D) Quantification of the chemotaxis movies. Average and standard deviation of at least three movies for each cell line are calculated. See Materials and Methods for definitions of these parameters.
To determine the contribution of each of the PKBs to these phosphorylations events, we disrupted PkbR1 or PkbA in the pten- background and examined the time courses of the modifications in each of the double mutants (Figure 1 and Supplemental Figure S1). In the pten-/pkbR1- cells, the activation of PkbA and the patterns of phosphorylation of PKB substrates were similar to those in pten- cells, with elevated and prolonged phosphorylation of pp90, pp110, and pp140. However, in the pten-/pkbA- cells, the elevated and prolonged phosphorylation seen in pten- cells was reversed and the pattern resembled that of wild-type cells. The activation of PKBR1 was unchanged, and, of course, PKBA was absent in the pten-/pkbA- cells.
As shown in Figure 1B, these patterns of phosphorylation were predictive of the morphology of the cells during steady-state growth on surfaces. As previously noted, most of the pten- cells displayed a flattened profile, and there were a number of very large cells. Further examination showed that many of the large cells were multinucleated (Janetopoulos et al., 2005; see below). The pten-/pkbR1- cells were essentially indistinguishable from the pten- cells. In contrast, the morphology of the pten-/pkbA- cells was similar to wild type, and few multinucleated cells were apparent (Figure 1B).
Because disruption of PkbA suppressed the effects of loss of PTEN on both PKB substrate phosphorylation and cell morphology, we further examined whether chemotaxis was improved in the pten-/pkbA- cells. After developing cells to chemotactic competence, we recorded migration toward a gradient formed by a cAMP-filled micropipette. As shown in Figure 1C and Supplemental Video 1, wild-type cells were polarized and migrated smoothly toward chemoattractant, with many reaching the micropipette tip within 30 min. The trajectories and tracings of the movement of five representative cells showed that most wild-type cells are polarized, migrated many cell lengths during 30 min, and some formed end-to-end contacts, called streams (Figure 1C and Supplemental Figure S1B). The average chemotactic speed and chemotactic index for the wild-type cells were 3.28 ± 1.46 μm/min and 0.55 ± 0.09, respectively. As previously reported, pkbA- cells were marginally impaired with average chemotactic speed and chemotactic index of 1.94 ± 1.44 μm/min and 0.30 ± 0.17, respectively (Supplemental Video 2). Consistent with previous reports, the pten- cells are not only less polarized, but also move toward cAMP very slowly on circuitous routes (Supplemental Video 3). Only a few nearby cells reached the micropipette tip by the end of the observation. The pten- cells had chemotactic speed and index of 0.10 ± 0.08 μm/min and 0.03 ± 0.04, respectively. Remarkably, the disruption of PkbA in the pten- cells suppressed these defects in polarity as well as chemotaxis. The pten-/pkbA- cells were elongated and migrated toward the tip as efficiently as the pkbA- cells with chemotactic speed and index of 1.02 ± 0.65 μm/min and 0.24 ± 0.14, respectively (Supplemental Video 4). The chemotaxis of pten-/pkbR1- cells was very defective, essentially the same as that of pten- cells (data not shown).
We next examined whether deletion of PiaA would suppress the defects in pten- cells. Activation of the PKBs involves phosphorylation of their hydrophobic motifs by TORC2, and deletion of the key TORC2 subunit PiaA reduces PKB activity and impairs chemotaxis (Kamimura et al., 2008). As shown in Figure 2A, the morphology of the pten-/piaA- cells growing on surfaces indeed was reverted toward that of wild-type cells. The pten-/piaA- cells were not as flattened as pten- cells, and neither tight foci nor large multinuclear cells were observed within the population. Importantly, when we exogenously expressed PiaA in the pten-/piaA- cells, the characteristic morphology of the pten- cells returned. Furthermore, when we expressed PTEN in the pten-/piaA- cells, the morphology resembled that of piaA- cells.
FIGURE 2:

Disruption of PiaA rescues defects in pten- cells. (A) Upper panels show representative images of wild-type, piaA-, pten-, and pten-/piaA- cells growing on glass substrates. Lower panels show pten-/piaA- cells carrying PTEN-GFP, PiaA-Flag, or empty vector. Bar, 50 μm. (B) Phosphorylation of PKB substrates and of activation loops in PKBR1 and PKBA. Cells were stimulated with 1 μM cAMP and analyzed with Western blot using antibodies against phosphospecific PKB substrates (upper panels) and phospho PKC (lower panels). (C) Chemotaxis assays. Cells were observed for 45 min at 30-s intervals. Upper panels show images at 45 min after chemotaxis was initiated. In lower panels, trajectories of the entire chemotaxis field are shown. The black diamond indicates the location of micropipette tip. Bar, 50 μm. (D) Quantification of the chemotaxis movies. Average and standard deviation of at least three movies for each cell line are calculated. See Materials and Methods for the definitions of these parameters.
We next examined PKB activation and PKB substrate phosphorylation in these cell lines (Figure 2B). As described above, the pten- cells showed elevated and prolonged phosphorylation of PKB substrates as well as the activation loop of PkbA, whereas the activation of PkbR1 was slightly reduced. Deletion of PiaA eliminated PkbR1 phosphorylation and greatly reduced PkbA phosphorylation. Accordingly, phosphorylation of most of PKB substrates was reduced or absent (Figure 2B). In this piaA- cell line, several possibly new bands between 100 and 150 kDa were more apparent than in piaA- cells created in other wild-type backgrounds (Kamimura et al., 2008). These bands were also apparent in the pten-/piaA- cells. Nevertheless, the pten-/piaA- cells had nearly identical patterns of phosphorylation of the PKBs and PKB substrates compared with piaA- cells.
We next compared the chemotaxis of the wild-type, pten-, piaA-, and pten-/piaA- cells in the micropipette assay (Figure 2C and Supplemental Figure S2). As previously reported, chemotaxis of the piaA- cells is impaired. The piaA- cells are less polarized, and both the chemotaxis speed and index were reduced to 0.69 ± 0.42 μm/min and 0.18 ± 0.09, respectively (Supplemental Video 5). However, these values were higher than those of pten- cells, which, in this experiment, did not move significantly and displayed chemotaxis speed and index of 0.06 ± 0.09 μm/min and 0.07 ± 0.13, respectively. Remarkably, these chemotaxis defects do not synergize; rather the pten-/piaA- cells closely resembled the piaA- cells. Most cells migrated toward the micropipette tip, and some reached it by the end of the observation. The chemotaxis speed and index for pten-/piaA- were 0.47 ± 0.19 μm/min and 0.16 ± 0.03, respectively. Interestingly, many of the pten-/piaA- cells appeared elongated, but closer examination showed that their direction of movement was perpendicular to cell length (Supplemental Video 6).
Blocking PKB-mediated phosphorylation events does not alter PIP3 levels or distribution
To rule out that the reversal of the pten- phenotype was due to reducing PIP3 levels, we expressed PHcrac-GFP in pten-/piaA- and pten-/pkbA- cells to monitor the levels and distribution of PIP3 (Figure 3 and Supplemental Videos 7–10). In contrast to wild-type cells, where PIP3 is localized only at the leading edge, PIP3 was localized over the entire plasma membrane in the pten- cells, as previously reported. We observed a similar localization of PIP3 in the pten-/piaA- and pten-/pkbA -cells. That is, although chemotaxis and cytokinesis were restored in these cells, PIP3 was distributed uniformly along the membrane as in pten- cells.
FIGURE 3:

Localization of PHcrac-GFP and LimEΔcoil-RFP in wild-type, pten-, pten-/pkbA-, and pten-/piaA- cells. Cells expressing PHcrac-GFP and LimEΔcoil-RFP were developed to chemotactic competent stages and observed by confocal microscopy. Images from GFP or RFP channel are shown on the top or middle panels, respectively. Bottom panels are the merged images. Bar, 5 μm.
Coexpression of LimEΔcoil-RFP allowed us to monitor actin polymerization in parallel with PIP3 (Figure 3 and Supplemental Videos 7–10). In wild-type cells, LimEΔcoil-RFP localizes to the leading edge, which corresponds closely to the localization of PIP3. In the pten- cells, LimEΔcoil was more concentrated in the pseudopods compared with the more uniform localization of PIP3. However, pseudopods extend and retract around the peripheral of the pten- cells and LimEΔcoil localization switches dynamically. This is not the case in either pten-/pkbA- or pten-/piaA- cells. In pten-/pkbA- cells, LimEΔcoil is sharply localized at the leading edge as in wild-type cells, even though PIP3 is evenly distributed along the membrane. In pten-/piaA- cells, localization of LimEΔcoil to the leading edge is less obvious because, as noted earlier, the cells tend to migrate in the direction perpendicular to cell length. Nevertheless, LimEΔcoil is localized in the direction of cell migration, while PIP3 is uniformly distributed.
Identification of excessively phosphorylated substrates in the pten- cells
Because elevated and prolonged phosphorylation of pp90, pp110, and pp140 correlated strongly with the phenotypes of the mutants, we hypothesized that one or all of these substrates might provide a link between the high level of PIP3 and hyperactive cytoskeleton observed in the pten- cells. We therefore sought to identify pp90, pp110, and pp140. Previously, we were able to identify two proteins, PI5K and GefS, in pp110 by directly immunoprecipitating the phosphorylated substrates using the phospho-PKB substrate antibody followed by mass spectrometry (Kamimura et al., 2008). However, pp140 and pp90 were not identified, probably due to the inefficiency of immunoprecipitation from whole cell lysates. Therefore, we decided to purify and concentrate the bands from cell lysates by ion-exchange chromatography prior to immunoprecipitation. To maintain the phosphorylation status of substrates during purification, we adjusted the cell lysates to 8 M urea during the process. As indicated in Figure 4, a Fast SP column effectively separated the bands. The pp140 band appeared in the flow through, the majority of pp90 eluted with 0.1 M NaCl, and pp110 eluted with 0.5 M NaCl. We next subjected these three fractions from the SP column to individual Fast Q columns and then removed the urea with a desalting column. We carried out immunoprecipitation of the desalted fractions with the phospho-PKB substrate antibody. In pp140, we identified GacG and PakA by mass spectrometry as proteins with the PKB substrate motif, RXRXXS/T (Supplemental Table 1). Consistent with our previous report, we also identified PI5K and GefS in pp110. The proteins identified in pp90 did not meet our validation tests as described below.
FIGURE 4:

Identification of PKB substrates that are hyperphosphorylated in pten- cells. (A) Separation of pp140, pp110, and pp90 by a Fast SP column. The whole cell lysate was loaded to a 2-ml Fast SP column. After washes, bound proteins were eluted using elution buffer containing 0.1 and 0.5 M NaCl. Proteins were analyzed by Western blot using anti–phosphopecific PKB substrate antibodies. pp140 did not bind to the column. pp90 and pp110 were eluted with 0.1 M and 0.5 M NaCl, respectively. (B) GFP fused to GacG, PakA, Pi5K, or GefS were expressed in wild-type cells. After developed to chemotactic competence, cells were stimulated with 1 μM cAMP and subjected to immunoprecipitation by anti-GFP antibodies and analyzed by Western blot with anti–phosphospecific PKB substrate antibodies. (C) PakA-GFP and GefS-GFP were expressed in wild-type or pten- cells. Cell extracts were prepared at different time points after cAMP stimulation, subjected to immunoprecipiation with anti-GFP antibodies, and analyzed with Western blotting using anti–phosphospecific PKB substrate antibodies.
To validate the mass spectroscopic results, we expressed each of the proteins as GFP fusions in wild-type cells. After developing the cells to chemotactic competence, we stimulated them with cAMP and immunoprecipitated the GFP-tagged proteins with the anti-GFP antibody. We then carried out immunoblots with the phospho-PKB substrate antibody. As shown in Figure 4B, the newly identified proteins, GacG and PakA, as well as the previously found proteins were selectively phosphorylated after cAMP stimulation, confirming that all are bona fide PKB substrates.
We also compared the time courses of phosphorylation of these GFP-tagged substrates in wild-type and pten- cells. As shown in Figure 4C, PakA-GFP remained highly phosphorylated in pten- cells at 5 min, whereas its phosphorylation had almost returned to the prestimulus level in wild-type cells by this time. This observation correlates well with the prolonged phosphorylation we observed in pp140. GefS-GFP also showed prolonged phosphorylation in pten- cells. However, differences in the phosphorylation patterns of GacG or PI5K between wild-type and pten- cells were less obvious (data not shown).
PakA mediates the effects of loss of PTEN
Because the phosphorylation kinetics of PakA-GFP and GefS-GFP correlate with the kinetics of pp140 and pp110 in wild-type and pten- cells, we decided to disrupt each of them in pten- cells to determine whether eliminating these substrates would reverse the phenotypes. As controls, we also disrupted PakA or GefS in wild-type cells. We did not observe obvious differences in the phenotypes of gefS- versus wild-type cells or pten-/gefS- versus pten- cells. Both pakA- and pten-/pakA- cells differed from wild-type and pten- cells, respectively.
In pakA- cells, fruiting body formation on non-nutrition agar was delayed (Supplemental Figure S3B). Whereas wild-type cells formed fruiting bodies after 18 h of starvation, at this point the pakA- cells were still at the “slugs” stage (equivalent to 15 h of wild-type cells) and formed fruiting bodies after another 8 h. Expression of PakA-GFP in pakA- reversed this phenotype. Western blot analysis showed that this delayed fruiting body formation was not likely due to delayed development, because the pakA- cells had the same amount of cAMP receptor (cAR1) expression as in wild-type cells (Supplemental Figure S3B). While growing on surfaces, individual pakA- cell resembled wild-type cells. Disruption of PakA in pten- cells partially rescued the cytokinesis defects caused by loss of PTEN. There were fewer large, multinuclear cells in the pten-/pakA- versus the pten- population. The pten- phenotype was restored by expressing PakA-GFP in pten-/pakA- cells (Figure 5, A and B). In random fields of pten-, pten-/pakA-, and PakA expressing pten-/pakA- cells, the area covered by large cells was 16.8% (n = 41), 3.9% (n = 35), and 10.3% (n = 64), respectively. DAPI staining showed that the large cells were multinucleated cells. The fraction of cells containing 6 to 10 and greater than 10 nuclei per cell, respectively, was 2.42% and 1.16% in pten- cells, compared with 0.58% and 0.07% in pten-/pakA- cells. The fraction of cells containing 3 to 5 nuclei per cell was also reduced to 4.89% in pten-/pakA- cells, compared with 6.51% in pten- cells (Figure 5B).
FIGURE 5:

Disruption of PakA partially rescues defects in pten- cells. (A) Representative images of indicated cell lines growing on glass substrates. Bar, 50 μm. Relative area occupied by large cells are shown (n > 35). Values are mean ± SE. (B) Representative DAPI images of pten- and pten-/pakA- cells are shown on the top panel. Corresponding phase images are shown on the bottom panel. Bar, 50 μm. Number of nuclei per cell were counted for each cell line (n = 12). Values are mean ± SE. (C) PakA-GFP localization in wild-type or pten- cells. Bar, 5 μm. (D) Chemotaxis assays. Cells were observed 30 min for wild-type and pakA- and 45 min for pten- and pten-/pakA-. Upper panels show the last frame of the area surrounding micropipette. Lower panels show the trajectories of the entire recording field. Bar, 50 μm. (E) Quantification of the chemotaxis movies. Average and standard deviation of at least three movies for each cell line are calculated. See Materials and Methods for the definitions of these parameters.
The fact that the cytokinesis defects of the pten- cells are mitigated in the pten-/pakA- cells suggests that PakA mediates the effects of excessive PIP3. To further test this, we exogenously expressed PakA-GFP in pten- cells in an attempt to exaggerate the pten-phenotype. Indeed, we observed more multinuclear cells growing on surfaces (Figure 5A). Interestingly, whereas PakA-GFP was tightly localized to the trailing edge of wild-type cells, as previously reported (Chung and Firtel, 1999), in pten- cells, Pak-GFP was uniformly distributed along the membrane except at sites of projections (Figure 5C).
We next compared the chemotaxis of wild-type, pakA-, pten-, and pten-/pakA- cells. The pakA- cells showed motility speed similar to that of the wild-type cells, 5.66 ± 0.44 μm/min compared with 5.24 ± 1.26 μm/min of wild type. However, movements of the pakA- cells were less directed toward the chemoattractant (Figure 5D and Supplemental Video 11). Chemotaxis speed and index were impaired compared with those of wild-type cells. The pten-/pakA- cells had improved chemotaxis speed of 0.58 ± 0.26 μm/min compared with 0.06 ± 0.10 μm/min of pten- cells. It is interesting to note that the pten-/pakA-, unlike pten-/pkbA- or pten-/piaA-, comprised two populations of cells. One population was polarized and moved toward chemoattractant as efficiently as wild-type cells from far distances. The other populations were much less motile and less polarized and did not migrate significantly even when they were closer to the cAMP source (Figure 5D and Supplemental Video 12).
To determine whether the phenotypic effects of excess PIP3 are mediated by phosphorylation of PakA at its PKB substrate motif site, we expressed either alanine (T585A) or glutamate (T585E) substituted version of PakA in pten-/pakA- cells. As shown in Figure 6A, expression of PakAT585A did not significantly alter the phenotype of pten-/pakA- cells. Quantitation showed that, in this cell’s line, large cells occupied only about 2.6% (n = 58) of the total cell area. In chemotaxis assays, the pten-/pakA- cells expressing PakAT585A behaved similarly to pten-/pakA- (compare Figure 6B with Figure 5D). About half of the cells were polarized, showed long chemotaxis tracks, and overall chemotaxis speed and index are 0.40 ± 0.25 μm/min and 0.16 ± 0.04, respectively (Figure 6B and Supplemental Video 13). In contrast, expression of PakAT585E caused a large fraction of the cells to be multinucleated: About 18% (n = 55) of the area is occupied by large cells (Figure 6A). These cells were flat and phase dark, resembling pten- cells. Chemotaxis of these cells was impaired compared with that of pten-/pakA-, with chemotaxis speed and index of 0.04 ± 0.03 μm/min and 0.03 ± 0.04, respectively (Figure 6B and Supplemental Video 14).
FIGURE 6:

Phosphorylation of PakA at its PKB substrate motif site is required for its function. PakAT585A-GFP or PakAT585E-GFP was expressed in pten-/pakA- cells. (A) Representative images of cells on glass substrates. Relative area occupied by multinucleated cells are shown (n > 55). Gray bars are replotted from Figure 5A. (B) Chemotaxis assays. Cells were observed for 45 min at 30-s intervals. Upper panels show images at 45 min after chemotaxis was initiated. Lower panels show trajectories of the entire recording field. Bar, 50 μm. (C) Quantification of the chemotaxis movies. Average and standard deviation of at least three movies for each cell line are calculated. See Material and Methods for the definitions of these parameters.
DISCUSSION
We have used genetic suppression to identify mediators that link PIP3 signaling to cytokinesis and chemotaxis. To identify the relevant targets of PIP3, we created specific gene disruptions that reversed the phenotypic defects of pten- cells. This approach circumvented complexities caused by the presence of pathways that act in parallel with PIP3 signaling. In pten- cells, several PKB substrates displayed excessive and prolonged phosphorylation. Deletion of PkbA in pten- cells restored a wild-type pattern of PKB substrate phosphorylation and nearly normal cytokinesis and chemotaxis, whereas deletion of PiaA, an essential subunit of TORC2, largely eliminated PKB substrate phosphorylation, suppressed the physiological defects, and led to a phenotype different than that of the pten- cells. Even though PKBR1 and PKBA overlap in substrate specificity, deletion of PkbR1 in pten- cells did not reverse the exaggerated substrate phosphorylation or abnormal phenotypes caused by elevated PIP3 levels. Because PKBA activation and/or expression is strongly up-regulated in the pten-/pkbR1- cells, the loss of PKBR1 is not significant at the level of substrate phosphorylation. Figure 7 illustrates a hypothetical scheme for a portion of the network linking chemoattractant to cytoskeletal activity. Elevated PIP3, together with TORC2, leads to the activation of the AKT orthologue, PKBA, which phosphorylates a series of PKB substrates, including PakA. PakA is partially responsible for transducing the PIP3 signal into cytoskeletal activity. The fact that not all pten-/pakA- cells display normal cell morphology and chemotaxis, whereas most pten-/pkbA- cells behave nearly as wild type, suggests that phosphorylation of some other PKBA substrates are also critical in regulating cell polarity and migration. Studies are currently underway to identify other important PKB substrates.
FIGURE 7:
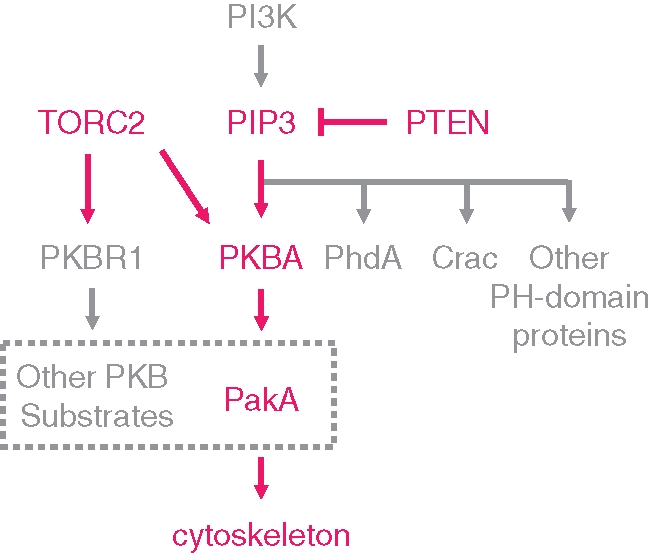
Network that links PIP3 to cytoskeleton in regulating chemotaxis. PIP3 levels are regulated by a balance of PI3K and PTEN activities. Disruption of PTEN leads to high levels of PIP3. Among many possible effectors, PKBA is the critical target of elevated PIP3 in regulating cell polarity and migration. Although there are many substrates of PKBA, phosphorylation of PakA appears to be one of the major players in transmitting the signal to the cytoskeleton.
These suppressors act downstream, not in parallel, with PTEN because proper regulation of PIP3 is not restored when these genes are disrupted in the pten- background. In wild-type cells, PIP3 and new actin polymerization colocalize at the poles of dividing cells and at leading edges of migrating cells. In pten- cells, the levels of PIP3 and new actin polymerization are elevated on many projections that extend nearly randomly around the cell perimeter, which greatly impairs cytokinesis, motility, and directional sensing. Whereas the cytokinesis and motility defects were suppressed in pten-/pkbA- and pten-/piaA- cells, PIP3 levels remained elevated and dispersed along the cell perimeter in pten- cells. This phenomenon was most striking in the pten-/pkbA- cells, in which new actin polymerization was confined to the leading edges as in wild-type cells, whereas PIP3, and presumably activation of other PIP3 binding proteins was dispersed uniformly along the length of the plasma membrane. This suggests that, among many PIP3-dependent events, PKBA signaling is a critical downstream target of elevated PIP3 in regulating cytoskeleton. In pten-/piaA- cells, new actin polymerization occurred on a very broad leading edge—curiously, the cells move with a fan-like morphology reminiscent of keratocytes—whereas PIP3 was again uniformly distributed around the cell border.
Positive feedback loops linking actin polymerization to activation of PI3K have been proposed (Charest and Firtel, 2006; Inoue and Meyer, 2008). In the pten-/pkbA- and pten-/piaA- cells, elevated levels of PIP3 no longer lead directly to actin polymerization because the downstream mediators have been removed. Nevertheless, based on the positive feedback models, we should expect the localized actin polymerization to enhance PIP3 synthesis locally. However, in pten-/pkbA- and pten-/piaA- cells, we could not detect enhancement of PIP3 accumulation in the positions where new actin polymerization was occurring. This may mean that the putative positive feedback from actin to PI3K is weak, and, because PTEN is absent, the dispersion length of the locally produced PIP3 is increased so that local accumulation is not observable above the uniform background. We are currently testing whether PI3K itself localizes to the sites of new actin polymerization. Nevertheless, we did notice that membrane localization of PHcrac-GFP is reduced in pten-/pkbA- cells compared with that of pten- cells. The membrane/cytoplasmic ratios of PHcrac-GFP for wild-type, pten-, and pten-/pkbA- were 0.00, 0.47, and 0.19, respectively. This may suggest that, in most regions of the pten- cell, a portion of the elevated PIP3 levels is due to positive feedback. Deletion of PkbA and restoration of polarity may restrict the effects of feedback to a narrow region at the leading edge and therefore reduce the overall amount of PIP3 on the membrane.
We noted that some PKB substrates are more affected than others by the elevated levels of PIP3 in the pten- cells. This suggests that there is a certain level of specificity displayed by PKBA and PKBR1. As we reported, the pp90, pp110, and pp140 bands were increased in intensity and duration of phosphorylation. We previously noticed that elimination of PIP3 or disruption of PKBA reduced the bands in this region of the gel more than others. The specificity is not strong because, in the pten-/pkbA- cells, the bands are still phosphorylated by PKBR1 but not to elevated levels and prolonged times. It is not clear whether the low level of specificity is related to the respective kinase domains of PKBA and PKBR1. It is also possible that the broader distribution of activated PKBA that occurs in the pten- background allows increased access to certain substrates such as PakA. Consistently, PakA is found toward the back of wild-type cells but is more broadly distributed in less polarized pten- cells.
Previous reports have shown that PakA localizes toward the back of the cell and is a substrate for PKBA, although there were discrepancies concerning the role of PakA (Chung and Firtel, 1999; Chung et al., 2001; Muller-Taubenberger et al., 2002). Chung et al. argued that pakA- cells phenocopied the loss of myosin II. That is, deletion of PakA led to a lateral pseudopod formation, loss of polarity, and cytokinesis defects (Chung and Firtel, 1999). In contrast, Muller-Taubenberger et al. did not observe cytokinesis or motility defects (Muller-Taubenberger et al., 2002). Our fresh disruption of PakA is more consistent with the results of Muller-Taubenberger. We see no evidence of increased multicellularity. As a matter of fact, deletion of PakA suppressed the cytokinesis defects seen in the pten- cells. Our data suggest that the broader distribution of PIP3 and PakA in pten- cells allows more overlap in their localization and leads to increased phosphorylation of PakA, which is evidenced on our gel profile. Phosphorylated PakA, in turn, alters cell morphology by an as yet unknown mechanism. We found that phosphorylation of PakA plays a role in its function but not its localization. Expression of PakAT585A or PakAT585E in wild-type cells does not alter PakA localization (data not shown). But replacement of PakA with PakAT585A, a version that cannot be phosphorylated, largely suppresses PakA function, even though PIP3 overlaps extensively with PakAT585A.
Mutations in the tumor suppressor gene PTEN are often found in melanomans, glioblastomas, and prostate cancer (Simpson and Parsons, 2001; Chalhoub and Baker, 2009). Our studies suggest that the inhibitory effects of loss of PTEN on cell polarity, cytokinesis, and migration should be considered along with the enhancement of cell growth. In breast and prostate epithelial cells, PIP3 and PTEN are found on the basal–lateral and apical membranes, respectively, in analogy to the polarized distributions in migrating cells (Martin-Belmonte et al., 2007; Martin-Belmonte and Mostov, 2008; Hill et al., 2010). In the cerebellum, loss of PTEN leads to aberrant migration of neurons, and this phenotype is suppressed by disruption of PDK1, which prevents activation of Akt, in strong analogy to the studies we present here (Chalhoub et al., 2009). It is widely accepted that elevations of PIP3, as occur with loss of PTEN or mutational activation of PI3K, contribute to tumor promotion due to enhancement of growth. It is reasoned that inhibition of downstream components of the pathways, such as AKT or TORC2, would be beneficial in reversing the defects. However, our studies, and those in the brain, suggest that high PIP3 levels are inhibitory to migration and that the same interventions that inhibit growth may enhance migration and therefore possibly invasion and metastasis. On the other hand, our studies suggest that inhibition of downstream signaling in cells with elevated PIP3 would tend to restore or maintain polarity in epithelial cells which may tend to ameliorate invasion. In any case, these complexities brought about by the many roles encompassed by PIP3 signaling need to be taken into account in designing therapeutic strategies.
MATERIALS AND METHODS
Cell growth and development
Cells were cultured in HL5 medium at 22ºC, except that pten-/pia-cells were grown in HL5 that contains heat-killed bacteria. Transformants carrying PHcrac-GFP, LimEΔcoil-RFP, or GFP-tagged PKB substrates were grown in HL5 containing 20 μg/ml G418. To develop cells to chemotactic competent stage, cells growing exponentially in HL5 were washed twice in development buffer (DB) and starved in DB for 1 h at 2 × 107 cells/ml and then pulsed with 100 nM cAMP every 6 min for 4.5 h.
Construction of deletion strains
To construct the PTEN disruption vector, DNA fragments containing the 5′ untranslated region (UTR) of PTEN were amplified from genomic DNA using primers Ppten1 and Ppten2 and digested with NotI and SmaI. DNA fragments containing the 3′UTR region of PTEN were amplified using primers Ppten3 and Ppten4 and digested with SmaI and SalI. Blasticidin-S-resistance (Bsr) cassette was excised from pLPBLPv2 using SmaI. These three DNA fragments were cloned into pBluescript (Stratagene, La Jolla, CA), forming the disruption vector pPTENdis. After digestion with SalI and NotI, linearized pPTENdis (5 μg) was introduced into growth phase of AX2 cells by electroporation. Transformants were selected in HL5 containing 10 μg/ml of Blasticidin S sulfate. After 10 days, cells were harvested and cloned to SM-agar plates with K. aerogenes. Colonies were picked and checked for positive disruption clones by PCR and Southern blot analysis.
Similar strategies were used to disrupt PkbA, PkbR1, and PiaA. Primers Ppkba1/Ppkba2 and Ppkba3/Ppkba4 were used to amplify the 5′ and 3′ region of PkbA, respectively. Primers Ppkbr1/Ppkbr2 and Ppkbr3/Ppkbr4 were used to amplify the 5′ and 3′ region of PkbR1, respectively. Primers YK118/YK119 and YK120/YK121 were used to amplify the 5′ and 3′ region of PiaA.
To make disruption construct of PakA, the 5′ region of the gene was cloned by MT60 and MT61, and the 3′ region of the gene was cloned by MT62 and MT63. These two fragments were linked together by bridge PCR and put into a TOPO vector. The Bsr gene was then inserted at the SmaI site that is between the 5′ and 3′ fragments. This disruption construct was then digested with PstI and NotI and electroporated into Ax2 or pten- cells. Selection of transformants and positive clones were carried out similarly as PTEN disruption described earlier.
Plasmid construction
The full-length PakA was obtained by bridge PCR of N-terminal and C-terminal halves of the PakA. The N-terminal half of the PakA was obtained by amplifying the Dicty cDNA library using MT64, which introduced a BamHI site at the 5′ end, and MT47. The C-terminal half of the PakA was obtained by amplifying genomic DNA using MT43 and MT41, which creates a XhoI site at the 3′ end. The full-length PakA with BamHI and XhoI site at each end was then ligated into a Dicty expression vector pJK1 that has a GFP sequence at the 3′ end of the insertion site.
GacG was amplified from genomic DNA by PCR using MT65 and MT53, which introduce BamHI and XhoI sites at the 5′ and 3′ end, respectively. The full-length GacG was inserted to the same vector as PakA.
PakAT585A was obtained by bridge PCR: the N-terminal half of the gene was amplified by MT64 and MT73, and the C-terminal half was amplified by MT72 and MT41. Alanine mutation was introduced on primers MT72 and MT73. For cloning of PakAT585E, primers MT74 and MT75 were used, instead of MT72 and MT73.
Immunoblotting
Chemotaxis-competent cells were treated with 5 mM caffeine and shaken at 200 rpm for 20 min. Cells were then washed with cold DB twice, resuspended in DB at 2 × 107 cells/ml, and kept on ice. We then stimulated cells with 1 μM cAMP and start shaking at 200 rpm. Samples were taken at various time points, lysed in SDS buffer, and boiled for 5 min. After running samples on 4–15% SDS–PAGE gel, proteins were transferred to a PVDF membrane. The membrane was then blocked with 5% milk and incubated with either α-phosphospecific PKB substrate antibody (Cell Signaling #9614; Beverly, MA) or α-phospho PKC (pan) antibody (Cell Signaling #2060) overnight.
Identification of PKB substrates
cAMP-stimulated 6 × 108 cells were filter lysed by Nuclepore Polycarbonate membrane (Whatman #110603; Clifton, NJ) in the presence of protease and phosphatase inhibitors (12.5 mM Na4P2O7, 25 mM NaF, 0.5× protease inhibitor complete EDTA free [Roche #11873580001; Nutley, NJ], 0.5% protease inhibitor cocktail [Sigma Aldrich #P-8215; St. Louis, MO], 1 mM Na3VO4, 50 mM Hepes [pH 7.2]). A total of 4 ml cell lysates were immediately mixed with 16 ml of 8 M Urea and 50 mM Hepes (pH 7.2) and then centrifuged for 5 min at 14,000 rpm to remove debris. The supernatants were loaded to a 2-ml Fast SP column that was equilibrated with 8 M urea and 50 mM Hepes (pH 7.2). Proteins were eluted by buffers containing 0, 0.1 M, and 0.5 M NaCl. Each fraction was then passed through a 0.5-ml Fast Q column. The final elution from Q column was subjected to Zeba Desalt Spin column (Thermo Scientific #89889; Rockford, IL) to remove the urea and change to immunoprecipitation buffer. Immunoprecipitation by α-phosphospecific PKB substrate antibody was carried out as described (Kamimura et al., 2008). For mass spectrometry, LC/MS/MS analysis was carried out on Thermo Finnigan LTQ (Proteomics Core Facility, JHMI).
Chemotaxis assay
Chemotactic-competent cells were plated on a one-well Lab-Tek chamber. A micropipette filled with 10 μM cAMP was placed in the center of the view, and the response of the cells were recorded by a time-lapse video every 30 s for 30 or 45 min. Chemotactic parameters were analyzed by a software package provided by Y. Xiong and P. A. Iglesias. Motility speed was defined as the total length of the track divided by the elapsed time. Chemotactic speed was calculated as the distance (d1 − d2) divided by the elapsed time, where d1 and d2 are the start and the end point of the migration path to the micropipette. The chemotactic index was calculated by an instantaneous, weighted chemotaxis index. For each frame, the cosine of the angle between the direction of movement and the direction to the micropipette was determined. These values were weighted according to the length of the movement step and averaged. Persistence was calculated as the shortest linear distance between the start point and end point of the migration path divided by the total distance traveled by the cell.
Supplementary Material
Acknowledgments
We thank Yuan Xiong and Pablo A. Iglesias for the help with video analysis. This work was supported by NIH GM28007 and GM34933 to P.N.D. and by NIH GM084015 and AHA 0765345U to M.I.
Abbreviations used:
- PakA
p21-activated kinase A
- PDK
phosphoinositide-dependent protein kinase
- PIP3
phosphotidylinositol 3,4,5-triphosphate
- PI3K
phosphoinositide 3-kinase
- PKB
protein kinase B
- PLA2
phospholipase A2
- PTEN
phosphatase and tensin homologue
- TORC2
target of rapamycin complex 2
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E10-06-0522) on December 17, 2010.
REFERENCES
- Bosgraaf L, Keizer-Gunnink I, Van Haastert PJ. PI3-kinase signaling contributes to orientation in shallow gradients and enhances speed in steep chemoattractant gradients. J Cell Sci. 2008;121:3589–3597. doi: 10.1242/jcs.031781. [DOI] [PubMed] [Google Scholar]
- Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, Zhu G, Zhu X, Baker SJ. Cell type specificity of PI3K signaling in Pdk1- and Pten-deficient brains. Genes Dev. 2009;23:1619–1624. doi: 10.1101/gad.1799609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charest PG, Firtel RA. Feedback signaling controls leading-edge formation during chemotaxis. Curr Opin Genet Dev. 2006;16:339–347. doi: 10.1016/j.gde.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Chen L, Iijima M, Tang M, Landree MA, Huang YE, Xiong Y, Iglesias PA, Devreotes PN. PLA2 and PI3K/PTEN pathways act in parallel to mediate chemotaxis. Dev Cell. 2007;12:603–614. doi: 10.1016/j.devcel.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Firtel RA. PAKa, a putative PAK family member, is required for cytokinesis and the regulation of the cytoskeleton in Dictyostelium discoideum cells during chemotaxis. J Cell Biol. 1999;147:559–576. doi: 10.1083/jcb.147.3.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CY, Potikyan G, Firtel RA. Control of cell polarity and chemotaxis by Akt/PKB and PI3 kinase through the regulation of PAKa. Mol Cell 7. 2001:937–947. doi: 10.1016/s1097-2765(01)00247-7. [DOI] [PubMed] [Google Scholar]
- Comer FI, Lippincott CK, Masbad JJ, Parent CA. The PI3K-mediated activation of CRAC independently regulates adenylyl cyclase activation and chemotaxis. Curr Biol. 2005;15:134–139. doi: 10.1016/j.cub.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Cote JF, Motoyama AB, Bush JA, Vuori K. A novel and evolutionarily conserved PtdIns(3,4,5)P3-binding domain is necessary for DOCK180 signalling. Nat Cell Biol. 2005;7:797–807. doi: 10.1038/ncb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274:23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Fine B, et al. Activation of the PI3K pathway in cancer through inhibition of PTEN by exchange factor P-REX2a. Science. 2009;325:1261–1265. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funamoto S, Milan K, Meili R, Firtel RA. Role of phosphatidylinositol 3’ kinase and a downstream pleckstrin homology domain-containing protein in controlling chemotaxis in dictyostelium. J Cell Biol. 2001;153:795–810. doi: 10.1083/jcb.153.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KM, Kalifa S, Das JR, Bhatti T, Gay M, Williams D, Taliferro-Smith L, De Marzo AM. The role of PI 3-kinase p110beta in AKT signally, cell survival, and proliferation in human prostate cancer cells. Prostate. 2010;70:755–764. doi: 10.1002/pros.21108. [DOI] [PubMed] [Google Scholar]
- Hoeller O, Kay RR. Chemotaxis in the absence of PIP3 gradients. Curr Biol. 2007;17:813–817. doi: 10.1016/j.cub.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Huang YE, Iijima M, Parent CA, Funamoto S, Firtel RA, Devreotes P. Receptor-mediated regulation of PI3Ks confines PI(3,4,5)P3 to the leading edge of chemotaxing cells. Mol Biol Cell. 2003;14:1913–1922. doi: 10.1091/mbc.E02-10-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima M, Devreotes P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell. 2002;109:599–610. doi: 10.1016/s0092-8674(02)00745-6. [DOI] [PubMed] [Google Scholar]
- Inoue T, Meyer T. Synthetic activation of endogenous PI3K and Rac identifies an AND-gate switch for cell polarization and migration. PLoS One. 2008;3:e3068. doi: 10.1371/journal.pone.0003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janetopoulos C, Borleis J, Vazquez F, Iijima M, Devreotes P. Temporal and spatial regulation of phosphoinositide signaling mediates cytokinesis. Dev Cell. 2005;8:467–477. doi: 10.1016/j.devcel.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Janetopoulos C, Devreotes P. Phosphoinositide signaling plays a key role in cytokinesis. J Cell Biol. 2006;174:485–490. doi: 10.1083/jcb.200603156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GE. Cellular signaling in macrophage migration and chemotaxis. J Leukoc Biol. 2000;68:593–602. [PubMed] [Google Scholar]
- Kamimura Y, Devreotes PN. Phosphoinositide-dependent protein kinase (PDK) activity regulates phosphatidylinositol 3,4,5-trisphosphate-dependent and -independent protein kinase B activation and chemotaxis. J Biol Chem. 2010;285:7938–7946. doi: 10.1074/jbc.M109.089235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura Y, Xiong Y, Iglesias PA, Hoeller O, Bolourani P, Devreotes PN. PIP3-independent activation of TorC2 and PKB at the cell’s leading edge mediates chemotaxis. Curr Biol. 2008;18:1034–1043. doi: 10.1016/j.cub.2008.06.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen M, Hoffman MP, Sakai T, Neibaur JC, Mitchell JM, Yamada KM. Role of PI 3-kinase and PIP3 in submandibular gland branching morphogenesis. Dev Biol. 2003;255:178–191. doi: 10.1016/S0012-1606(02)00047-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao XH, Buggey J, Kimmel AR. Chemotactic activation of Dictyostelium AGC-family kinases AKT and PKBR1 requires separate but coordinated functions of PDK1 and TORC2. J Cell Sci. 2010;123:983–992. doi: 10.1242/jcs.064022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sasaki T, Kozieradzki I, Wakeham A, Itie A, Dumont DJ, Penninger JM. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13:786–791. doi: 10.1101/gad.13.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Belmonte F, Mostov K. Regulation of cell polarity during epithelial morphogenesis. Curr Opin Cell Biol. 2008;20:227–234. doi: 10.1016/j.ceb.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Moore MA. The role of chemoattraction in cancer metastases. Bioessays. 2001;23:674–676. doi: 10.1002/bies.1095. [DOI] [PubMed] [Google Scholar]
- Mortimer D, Fothergill T, Pujic Z, Richards LJ, Goodhill GJ. Growth cone chemotaxis. Trends Neurosci. 2008;31:90–98. doi: 10.1016/j.tins.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Muller-Taubenberger A, Bretschneider T, Faix J, Konzok A, Simmeth E, Weber I. Differential localization of the Dictyostelium kinase DPAKa during cytokinesis and cell migration. J Muscle Res Cell Motil. 2002;23:751–763. doi: 10.1023/a:1024475628061. [DOI] [PubMed] [Google Scholar]
- Nishikimi A, et al. Sequential regulation of DOCK2 dynamics by two phospholipids during neutrophil chemotaxis. Sciences. 2009;324:384–387. doi: 10.1126/science.1170179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivard N. Phosphatidylinositol 3-kinase: a key regulator in adherens junction formation and function. Front Biosci. 2009;14:510–522. doi: 10.2741/3259. [DOI] [PubMed] [Google Scholar]
- Sarraj B, Massberg S, Li Y, Kasorn A, Subramanian K, Loison F, Silberstein LE, von Andrian U, Luo HR. Myeloid-specific deletion of tumor suppressor PTEN augments neutrophil transendothelial migration during inflammation. J Immunol. 2009;182:7190–7200. doi: 10.4049/jimmunol.0802562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savarin-Vuaillat C, Ransohoff RM. Chemokines and chemokine receptors in neurological disease: raise, retain, or reduce? Neurotherapeutics. 2007;4:590–601. doi: 10.1016/j.nurt.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SI, Sarantos MR, Green CE, Schaff UY. Leucocyte recruitment under fluid shear: mechanical and molecular regulation within the inflammatory synapse. Clin Exp Pharmacol Physiol. 2009;36:217–224. doi: 10.1111/j.1440-1681.2008.05083.x. [DOI] [PubMed] [Google Scholar]
- Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- Swaney KF, Huang CH, Devreotes PN. Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu Rev Biophys. 2010;39:265–289. doi: 10.1146/annurev.biophys.093008.131228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nat Rev Mol Cell Biol. 2004;5:626–634. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- van Haastert PJ, Keizer-Gunnink I, Kortholt A. Essential role of PI3-kinase and phospholipase A2 in Dictyostelium discoideum chemotaxis. J Cell Biol. 2007;177:809–816. doi: 10.1083/jcb.200701134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner OD, Neilsen PO, Prestwich GD, Kirschner MW, Cantley LC, Bourne HR. A PtdInsP(3)- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat Cell Biol. 2002;4:509–513. doi: 10.1038/ncb811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DA, Zheng Y, Cancelas JA. Rho GTPases and regulation of hematopoietic stem cell localization. Methods Enzymol. 2008;439:365–393. doi: 10.1016/S0076-6879(07)00427-2. [DOI] [PubMed] [Google Scholar]
- Yoo SK, Deng Q, Cavnar PJ, Wu YI, Hahn KM, Huttenlocher A. Differential regulation of protrusion and polarity by PI3K during neutrophil motility in live zebrafish. Dev Cell. 2010;18:226–236. doi: 10.1016/j.devcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Pandol S, Bokoch G, Traynor-Kaplan AE. Disruption of Dictyostelium PI3K genes reduces [32P]phosphatidylinositol 3,4 bisphosphate and [32P]phosphatidylinositol trisphosphate levels, alters F-actin distribution and impairs pinocytosis. J Cell Sci. 1998;111:283–294. doi: 10.1242/jcs.111.2.283. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.