

Abstract
Background
Plasma levels of γ-glutamyl transpeptidase (GGT) are associated with risk factors for nonalcoholic fatty liver disease (NAFLD) such as dyslipidemia, insulin resistance (IR), and hypertension. Limited data exist on whether there is genetic covariance between plasma levels of GGT and NAFLD risk factors. Variants of β2-adrenergic receptor gene (ADRB2) have been associated with dyslipidemia, IR, and hypertension, but its affect on GGT secretion is not known. We estimated the heritability of GGT using a twin-study design and examined the genetic co-variance between GGT levels, IR, hypertension, levels of low-density lipoproteins and triglycerides, and ADRB2 variants.
Methods
We studied phenotypes of 362 twins; the heritability of increased GGT activity and genetic covariance with NAFLD risk factors were estimated by variance-component methodology. ADRB2 genotype associations with plasma GGT activity were examined using generalized estimating equations (GEE) to account for intra-twinship correlations.
Results
Increased GGT activity was heritable in 49%±8% of the twin cohort and had significant covariance with IR; insulin, triglyceride, and uric acid levels; and diastolic blood pressure. In multivariate GEE models, the most common haplotypes of ADRB2 were significantly associated with plasma GGT activity; 5 single nucleotide polymorphisms (SNPs) within ADRB2 were associated with increased GGT activity (28.6 IU/L for 1 or 2 copies of the ADRB2 haplotype vs. 23.0 IU/L for 0 copies; P=0.02). Five SNPs in ADRB2 were associated with levels of GGT; ADRB2 haplotypes were pleiotropic for GGT and triglyceride levels.
Conclusions
In a twin study, GGT shared genetic co-determination with traits of metabolic syndrome. The ADRB2 gene had pleiotropic effects on plasma levels of GGT and triglycerides, indicating linked pathways (e.g. adrenergic) between genetic susceptibility to NAFLD and metabolic syndrome.
Keywords: GGT, NAFLD, ADRB2, metabolic syndrome, LDL, TG
INTRODUCTION
Gamma-glutamyl transpeptidase (GGT) is a hepatic and biliary enzyme synthesized by hepatocytes as well as epithelial cells of intra-hepatic bile ducts (1–4). Elevated plasma GGT enzymatic activity is a marker of fatty liver disease resulting from either obesity or alcohol in the general population (5, 6). Recent studies suggest that plasma GGT is a significant predictor of the metabolic syndrome, independent of alcohol intake (7, 8), as well as its hepatic manifestation, non-alcoholic fatty liver disease (NAFLD) (9, 10). Several studies have shown that plasma GGT is cross-sectionally and longitudinally associated with metabolic syndrome risk factors such as diabetes, insulin resistance, dyslipidemia, elevated uric acid, hypertension and NAFLD (7, 8, 11–13). It is unclear whether the association between plasma GGT, as a marker of NAFLD, and metabolic syndrome traits is genetic and/or environmental in origin. Therefore, we propose to examine whether plasma GGT shares genetic or environmental covariance with metabolic syndrome risk factors in a well-characterized, US twin cohort (14, 15).
Wessel et al. showed that C-reactive protein (an acute phase protein made in hepatocytes), which correlates with plasma GGT, is associated with multiple features of the metabolic syndrome and is linked to genes that regulate adrenergic activity and metabolism (15). Several studies have shown that genetic variation at ADRB2 is associated with insulin resistance, regulation of glucose metabolism in liver, hypertriglyceridemia, and hypertension (16–20). Such metabolic traits are significant correlates of GGT and NAFLD (21). Therefore, we also propose to examine whether common variants at ADRB2 in twins is associated with plasma GGT. Our study is based upon the hypotheses that: 1) Plasma GGT shares genetic determination (co-variance) with metabolic risk factors such as insulin resistance (measured by homeostasis model of insulin resistance, HOMA-IR), hypertension, obesity, and elevated plasma triglycerides. 2) Genetic variants within the ADRB2 gene predict plasma GGT. To test these hypotheses, we conducted a cross-sectional twin-study design in a previously well-defined and characterized twin cohort. STROBE (22) guidelines for reporting cross-sectional studies were followed.
METHODS
Participants and study design
The University of California at San Diego twin cohort recruitment has been previously described (14, 15, 23, 24). In brief, the cohort was recruited by access to a twin-birth registry and newspaper advertisements. This study included 380 Caucasian twins with 128 mono-zygotic twins (24 male pairs and 104 female pairs) and 62 di-zygotic twins (14 male pairs, 36 female pairs and 12 male-female pairs). None of the twins had diabetes, and the prevalence of obesity (BMI ≥30 kg/m2) was 13.9%. Zygosity was confirmed by use of either >100 microsatellites (chromosomes 1 and 2) for self-identified DZ twins, or single nucleotide polymorphism (SNP) data (11–177 SNPs) as well as the TH (TCAT)n microsatellite (24) for self-reported MZ and DZ pairs. To qualify for analysis, twins of white (European) ancestry identified not only themselves but also both parents and all four grandparents as being of that biogeographic ancestry group. Twins were between 18–81 years of age. None of the twins had a history of renal failure, and plasma creatinine concentrations were ≤1.5 mg/dl. None of the twins provided a history of known liver disease. Definitions of subject characteristics have been previously published in prior manuscripts from our laboratory (15). Study participants were twin volunteers from urban southern California (principally the San Diego area). Written informed consent was obtained from each participant, and the research protocol was approved by the institutional review board of UCSD.
Blood pressure and heart rate phenotyping
Brachial cuff BP (mmHg) and heart rate (beats/min) were obtained in seated subjects with a DynaPulse oscillometric device (PulseMetric, San Diego, California, USA), as described previously (25), to obtain blood pressure (in mmHg) and heart rate (beats/min) in the seated position. The recordings were validated by triplicate determinations of BP and heart rate, until each value was within 10% of the mean value.
Genomics
Details regarding genotyping methodology used in our laboratory have been previously published (15, 23, 24). SNP selection across the ADRB2 locus proceeded as we have previously described for studies of hypertension (16). The same SNPs were also described by Drysdale et al (26) and McGraw et al (27). Genomic DNA was isolated from blood leukocytes. SNP polymorphisms were scored in a two-stage assay (28). In stage one, PCR primers flanking the polymorphism were used to amplify the target region from 5 ng of genomic DNA. In stage two, an oligonucleotide primer flanking the variant was annealed to the amplified template, and extended across the variant base. The mass of the extension product (wild-type versus variant) was scored by MALDI-TOF mass spectrometry (low mass allele versus high mass allele).
Biochemical phenotyping
Plasma GGT activity
GGT enzymatic activity in plasma was measured by an enzymatic rate method (29) in the UCSD Medical Center clinical chemistry laboratory. The lower limit of detection of the assay was 5 IU/L and the repeated sample correlation coefficient (r) was 0.9985. The precision of the assay was robust with CV <3.5%. Elevated GGT was defined as >40 IU/L in men or >30 IU/L in women. The prevalence of elevated GGT was 19.3% in men and 15.8% in women.
Catecholamines, cytokines, and lipids
Samples of plasma for measurement of catecholamines, cytokines and lipids were quickly frozen at −70 C. Details of methods for the assays have been previously published (15).
Insulin resistance
Fasting plasma glucose and plasma insulin levels were obtained. Insulin resistance was measured by HOMA-IR, the homeostasis assessment model of insulin resistance, where HOMA-IR (mmol/L × µU/ml) = fasting glucose (mmol/L) × fasting insulin (µU/ml)/22.5).
Statistical analyses
Baseline descriptive statistics were obtained in all the twins. Twins were then classified into two groups by dividing about the median GGT value into lower versus upper quantiles, which were compared using generalized estimating equation (GEE: PROC GENMOD) using SAS version 9.1, SAS Institute , Cary, NC; to account for within twin-pair correlations.
Heritability (h2) was estimated based upon following equation: h2 = VG/VP, where VG indicates additive genetic variance and VP is total phenotypic variance, by variance-component methodology implemented in the Sequential Oligogenic Linkage Analysis Routines (SOLAR) package (available at http://www.sfbr.org.solar/). This method maximizes the likelihood assuming a multivariate normal distribution of phenotypes in twin pairs (monozygotic versus dizygotic) with a mean dependent on a particular set of explanatory variables. Heritability (h2 = VA/VP) is the proportion of trait variance (VP) that is explained by the genetic variation VG (relative to environmental variation VE) in a population. Heritability and environmentability are relative concepts regarding genetic or environmental trait determination in a population, and do not give any information regarding which genes influence the trait. The null hypothesis (H0) of no heritability is tested by comparing the full model, which assumes genetic variation (VG), and a reduced model, which assumes no genetic variation, using a likelihood ratio test. Heritability estimates were adjusted for age and sex, because of the effects of these covariates on several traits. The primary study outcome for gene effects on plasma GGT was based upon haplotype analysis in age/sex-adjusted GEE models in Caucasian twins, and p-values ≤0.05 were considered significant. We examined the association between ADRB2 haplotype-1 (AGGCCGGGC, the most common haplotype) and plasma GGT concentrations. Haplotypes were inferred from diploid genotypic data using the HAP algorithm (30) (http://research.calit2.net/hap); haplotypes at a locus were numbered in order of frequency (1: most frequent). Patterns of linkage disequilibrium (LD) between/among SNPs across the ADRB2 locus were visualized with Haploview (31).
SOLAR was also used to examine the contribution of allelic variation at the locus to the heritability (i.e., locus-specific VG) heritability, by comparing models including or excluding the genotype as a covariate. During allele, haplotype and diplotype (haplotype pair) associations, multivariable analyses were carried out to control for confounding by age and sex.
Pleiotropy (shared genetic determination; genetic covariance for two correlated, heritable traits) was estimated as RhoG (ρG) and the environmental covariance (shared environmental determination) as parameter RhoE (ρE) using SOLAR.
RESULTS
Heritability (h2) of GGT in twins
Heritability was estimated from twin pairs based upon correlations among monozygotic and dizygotic twins as shown in Figure 1. Heritability was significant for plasma GGT, at 49±9% of trait variance (p <0.001). Figure 1 displays the heritability estimates of other associated metabolic syndrome traits in this twin cohort; such heritability estimates were similar to other reports.
Figure 1. Heritability (h2) estimates of GGT and fatty liver risk factors shown in the present twin cohort.
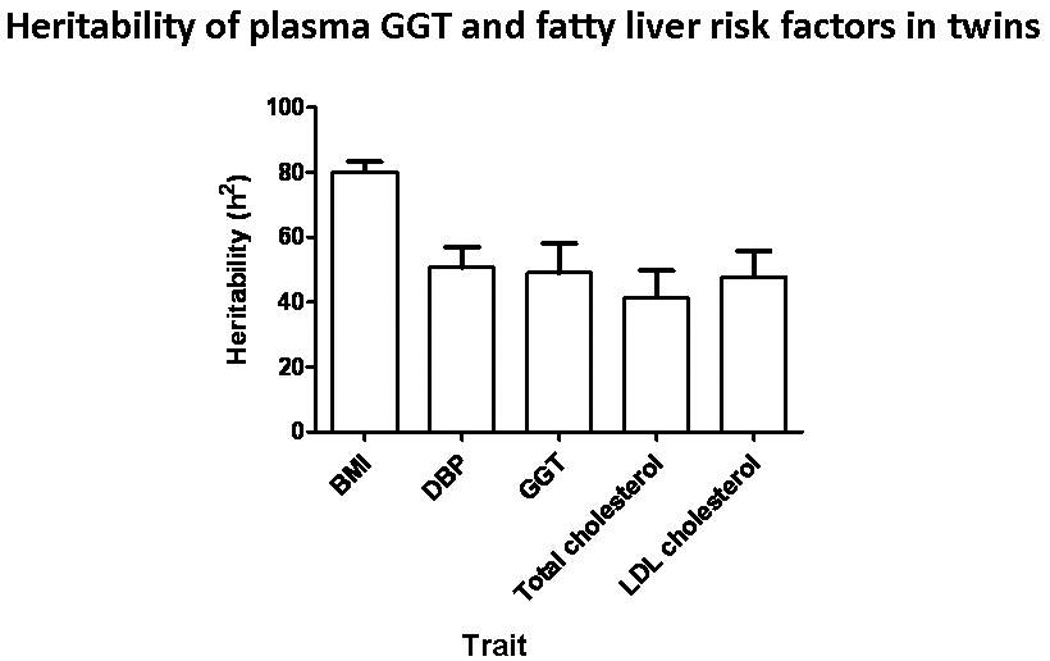
These h2 estimates (± SEM) are similar to previously published studies, suggesting generalizability of findings from our cohort. GGT has substantial heritability, at 49±8%. Abbreviations: BMI, body mass index; DBP, diastolic blood pressure; GGT, gamma-glutamyl transpeptidase; LDL, low-density lipoprotein..
The average (± SEM) plasma GGT in the twin cohort was 23.7 ± 1.2 IU/L. The prevalence of elevated plasma GGT (≥40 IU/L in men or ≥30 U/L in women) was 16.6% in this cohort.
GGT associations with other traits
Table 1 provides a description of this twin cohort by dividing the cohort into two groups (quantiles) around the GGT median. As expected, plasma GGT increased with age and was higher in men than women. Participants with higher GGT showed significant (p<0.05) trait differences with markers of inflammation, adrenergic activity, and the metabolic syndrome: body mass index, epinephrine, IL-6, P-selectin, triglycerides, HDL, glucose, insulin and HOMA-IR, homocysteine, uric acid, butyrylcholinesterase (BChE), and blood pressure. Sex-specific data is also provided in the appendix supplementary table.
Table 1.
Gamma-glutamyl transpeptidase (GGT) stratification into upper and lower quantiles: Association with demographic, biochemical and physiological traits in twins.
| Trait | GGT lower quantile, ≤16.6 IU/L (n=182) |
GGT upper quantile, >16.6 IU/L (n=180) |
|||
|---|---|---|---|---|---|
| Mean | SEM | Mean | SEM | p-value | |
| Demographic | |||||
| Age (years) | 37.7 | 1.2 | 43.7 | 1.2 | 0.001 |
| Sex (M/F) | 30/152 | 50/130 | 0.01 | ||
| Ethnicity | All Caucasian | All Caucasian | -- | ||
| State (CA/non CA) | 178/4 | 176/4 | 0.99 | ||
| Obesity (Obese: BMI≥30 kg/m2) Non-obese/Obese |
164/14 | 147/33 | 0.003 | ||
| Physical | |||||
| BMI (kg/m2) | 24.3 | 1.3 | 25.6 | 0.4 | 0.016 |
| Physiological | |||||
| Systolic blood pressure (mmHg) | 129 | 1.1 | 132 | 1.3 | 0.08 |
| Diastolic blood pressure (mmHg) | 70 | 0.7 | 72.3 | 0.8 | 0.03 |
| Biochemical (plasma) | |||||
| C-reactive protein (ng/ml) | 2214 | 235 | 2610 | 318 | 0.3 |
| Uric acid (mg/dl) | 3.9 | 0.1 | 4.5 | 0.1 | <0.0001 |
| Homocysteine (µmol/L) | 6.2 | 0.2 | 7.0 | 0.3 | 0.02 |
| Butyryl-cholinesterase (IU/L) | 9.4 | 0.15 | 10.0 | 0.2 | 0.03 |
| IL-6 (pg/ml) | 1.6 | 0.1 | 2.2 | 0.2 | 0.019 |
| vWF (%) | 53.2 | 3.8 | 60.2 | 4.3 | 0.2 |
| P-selectin (ng/ml) | 101 | 4.5 | 125.9 | 6.4 | 0.001 |
| Metabolic (plasma) | |||||
| Glucose (mg/dl) | 79 | 0.6 | 84.8 | 1.9 | 0.006 |
| Insulin (U/L) | 11.5 | 0.6 | 15.7 | 1.2 | 0.001 |
| HOMA-IR (score) | 2.3 | 0.13 | 3.5 | 0.3 | 0.0003 |
| Gamma-glutamyl transpeptidase (IU/L) | 10.5 | 0.3 | 37.1 | 2.1 | <0.0001 |
| Total cholesterol (mg/dl) | 174.6 | 3 | 180.1 | 2.8 | 0.19 |
| Triglycerides (mg/dl) | 97.3 | 3.8 | 124.7 | 7.3 | 0.001 |
| HDL-cholesterol (mg/dl) | 54.4 | 1.3 | 50.6 | 1.2 | 0.037 |
| LDL-cholesterol (mg/dl) | 100.7 | 2.5 | 105 | 2.5 | 0.222 |
| Adrenergic (plasma) | |||||
| Epinephrine (pg/ml) | 24.4 | 1.3 | 30.0 | 1.9 | 0.013 |
| Norepinephrine (pg/ml) | 312.3 | 12.2 | 348.6 | 14.1 | 0.052 |
| Autonomic | |||||
| Baroreflex coupling at 0.05–0.15 Hz, msec/mmHg |
17.0 | 1.5 | 13.0 | 0.8 | 0.018 |
Descriptive statistics for twin study population are provided above. Values are means ± one SEM (standard error of the mean) or n and (percentage) derived from generalized estimating equations (GEEs; PROC GENMOD) in SAS software version 9.1, to account for intra-pair correlations within twinships. GGT (gamma-glutamyl transpeptidase, dichotomized around the median concentration of 16.6 IU/L) is listed for all individuals. The mean or N (%) gives the mean for continuous variables, or percentage of the trait observed for dichotomous variables (listed in parentheses). A two-tailed p-value ≤ 0.05 is considered statistically significant (in bold type). Abbreviations: BMI, body mass index; CRP; C-reactive protein; BChE, butylcholinesterase; IL-6, interleukin-6; vWF, Von-Willebrand factor; HOMA-IR, homeostasis assessment model of insulin resistance (HOMA-IR (mmol/L × µU/ml) = fasting glucose (mmol/L) × fasting insulin (µU/ml)/22.5); HDL, high-density lipoprotein; LDL, low-density lipoprotein.
Shared heritability in twins: Genetic covariance between GGT and metabolic risk factors
Metabolic syndrome traits that correlated with plasma GGT were tested for shared genetic determination (pleiotropy) with the GGT trait (Table 2). The following traits showed significant (p<0.05) genetic covariance with plasma GGT: diastolic blood pressure (ρG 27%, p<0.05), uric acid (ρG 46%, p<0.005), insulin (ρG 50%, p<0.005), HOMA-IR (ρG 39.6%, p<0.02), and triglycerides (ρG 53%, p<0.005). The only trait to share significant environmental covariance with GGT was BChE (ρE-24%, p<0.025).
Table 2.
Shared genetic and environmental determination for metabolic and adrenergic traits with gamma-glutamyl transpeptidase in twins.
| Shared genetic determination (pleiotropy): ρG |
Shared environmental determination: ρE |
|||||
|---|---|---|---|---|---|---|
| Trait | ρG estimate | SEM | P value | ρE estimate | SEM | P value |
| Metabolic pathway | ||||||
| BMI | 0.03 | 0.10 | 0.8 | −0.08 | 0.09 | 0.8 |
| Blood pressure* Systolic Diastolic |
0.06 0.27 |
0.14 0.13 |
0.7 0.04 |
−0.02 −0.07 |
0.09 0.09 |
0.8 0.4 |
| Total cholesterol | 0.23 | 0.18 | 0.2 | −0.14 | 0.10 | 0.07 |
| LDL-cholesterol | 0.40 | 0.28 | 0.12 | −0.19 | 0.10 | 0.5 |
| HDL-cholesterol | −0.21 | 0.13 | 0.09 | 0.11 | 0.10 | 0.2 |
| Triglycerides | 0.53 | 0.19 | 0.004 | 0.21 | 0.08 | 0.6 |
| Homocysteine | 0.31 | 0.21 | 0.2 | 0.02 | 0.12 | 0.9 |
| Uric acid | 0.46 | 0.14 | 0.002 | −0.023 | 0.10 | 0.8 |
| Glucose | 0.12 | 0.16 | 0.4 | 0.06 | 0.09 | 0.5 |
| Insulin | 0.50 | 0.16 | 0.003 | −0.07 | 0.11 | 0.5 |
| HOMA-IR | 0.40 | 0.15 | 0.011 | −0.02 | 0.11 | 0.9 |
| IL-6 | −0.01 | 0.28 | 1 | 0.06 | 0.09 | 0.5 |
| P-selectin | 0.15 | 0.12 | 0.2 | 0.14 | 0.10 | 0.2 |
| Butyryl-cholinesterase | 0.21 | 0.12 | 0.07 | −0.24 | 0.10 | 0.02 |
| Adrenergic pathway | ||||||
| Epinephrine | 0.02 | 0.16 | 0.9 | 0.04 | 0.10 | 0.7 |
| Norepinephrine | −0.03 | 0.12 | 0.8 | 0.01 | 0.10 | 0.9 |
| Baroreflex coupling at 0.05–0.15 Hz |
−0.05 | 0.20 | 0.8 | −0.1 | 0.10 | 0.4 |
Also involved in the adrenergic pathway.
Significant (p<0.05) coefficients are designated in bold type. ρG indicates genetic co-variance that detects the shared genetic determination of two traits. ρE indicates environmental co-variance that suggests the shared environmental determination of two traits. Abbreviations and units: see Table 1.
β2-adrenergic receptor gene effect on GGT in twins
We examined the effect of SNPs across the ADRB2 locus on plasma GGT. Graphical representation of linkage disequilibrium (LD) across the ADRB2 locus (Figure 2) suggests that the SNPs we evaluated lie within a single block of LD. Hardy-Weinberg Equilibrium was obtained for each SNP across ADRB2 except promoter T-47C, at p=0.02 (Table 3).
Figure 2. Linkage equilibrium (LD) across the ADRB2 locus using Haploview.
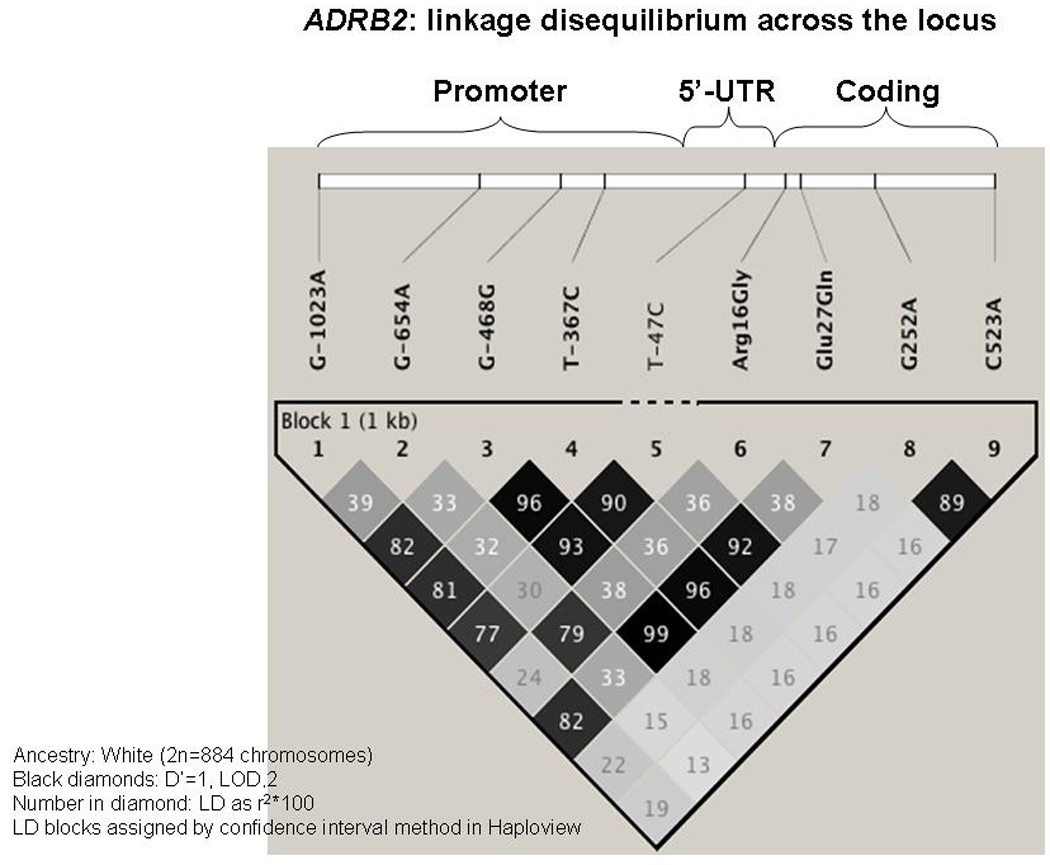
Darker block color denotes stronger pair-wise correlation among SNPs, within one major LD block.
Table 3.
ADRB2 allele frequency and Hardy-Weinberg Equilibrium (HWE) for single nucleotide polymorphisms across the ADRB2 locus.
| Domain | RefSNP | Major allele freq (p) |
Minor allele freq (q) |
HWE | ||
|---|---|---|---|---|---|---|
| N | χ2 | p | ||||
| Promoter G-1023A | rs2053044 | 161 | 0.55 | 0.45 | 2.90 | 0.09 |
| Promoter G-654A | none | 162 | 0.65 | 0.35 | 0.20 | 0.65 |
| Promoter C-468G | rs11168070 | 165 | 0.55 | 0.45 | 1.51 | 0.22 |
| Promoter T-367C | rs11959427 | 166 | 0.55 | 0.45 | 2.48 | 0.12 |
| Promoter T-47C | rs1042711 | 162 | 0.56 | 0.44 | 5.63 | 0.02 |
| Gly16Arg, G265A | rs1042713 | 159 | 0.66 | 0.34 | 1.13 | 0.29 |
| Glu27Gln, G298C | rs1042714 | 164 | 0.55 | 0.45 | 1.72 | 0.19 |
| Leu84Leu, G252A | rs1042717 | 164 | 0.80 | 0.20 | 1.88 | 0.17 |
| Arg175Arg, C523A | rs1042718 | 159 | 0.82 | 0.18 | 0.001 | 0.97 |
Footnote: Domains are numbered with respect to the translational start (AUG) codon. All ADRB2 SNPs except promoter T-47C fulfill the criteria of Hardy-Weinberg equilibrium. One member of each twinship was included in the analysis.
Five SNPs across ADRB2 were significantly associated with plasma GGT, independent of age and sex in GEE models, as shown in Table 3. Each of the five GGT-associated SNPs occurred in functional domains: four in the promoter region (G-1023A, C-468G, T-367C, and T-47C), and one non-synonymous coding variant (Glu27Gln). Since the SNPs across ADRB2 have strong pair-wise correlations (Figure 2), it is plausible that one or more functional variants in this region (e.g., T-47C (32)) may play a dominant role in plasma GGT regulation; however, previous functional studies of the ADRB2 promoter (32) indicate that the effects of individual SNPs on gene expression are interactive (i.e., are best explained by haplotypes rather than individual SNPs).
In this twin cohort, there was an association between β2-adrenergic receptor (ADRB2) haplotype-1 (the most frequent haplotype: AGGCCGGGC) and plasma GGT in age/sex-adjusted GEE models (p<0.024), in a dominant model (Figure 3A).
Figure 3. Figure 3A. ADRB2 haplotype-1 (AGGCCGGGC) effect on GGT in age-/sex-adjusted models.
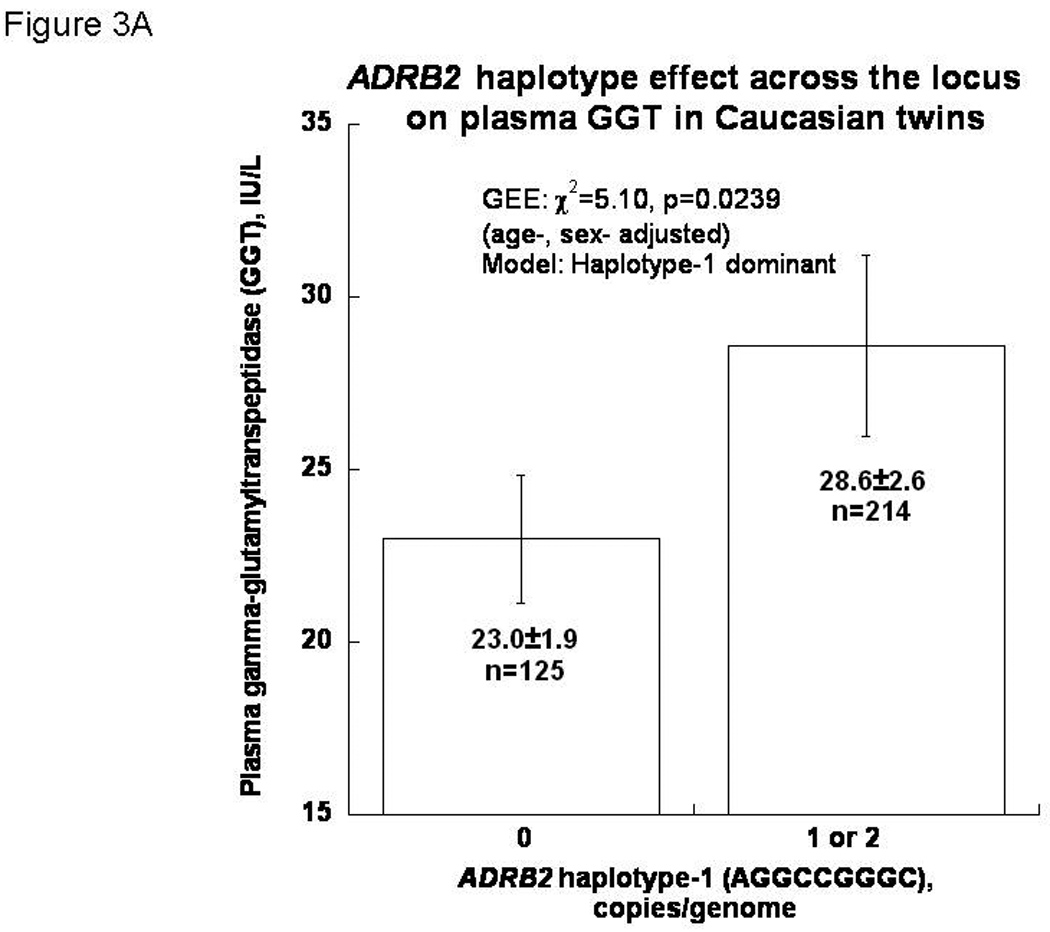
Results demonstrate a significant association (p <0.024), suggesting that ADRB2 plays a role in regulation of GGT secretion.
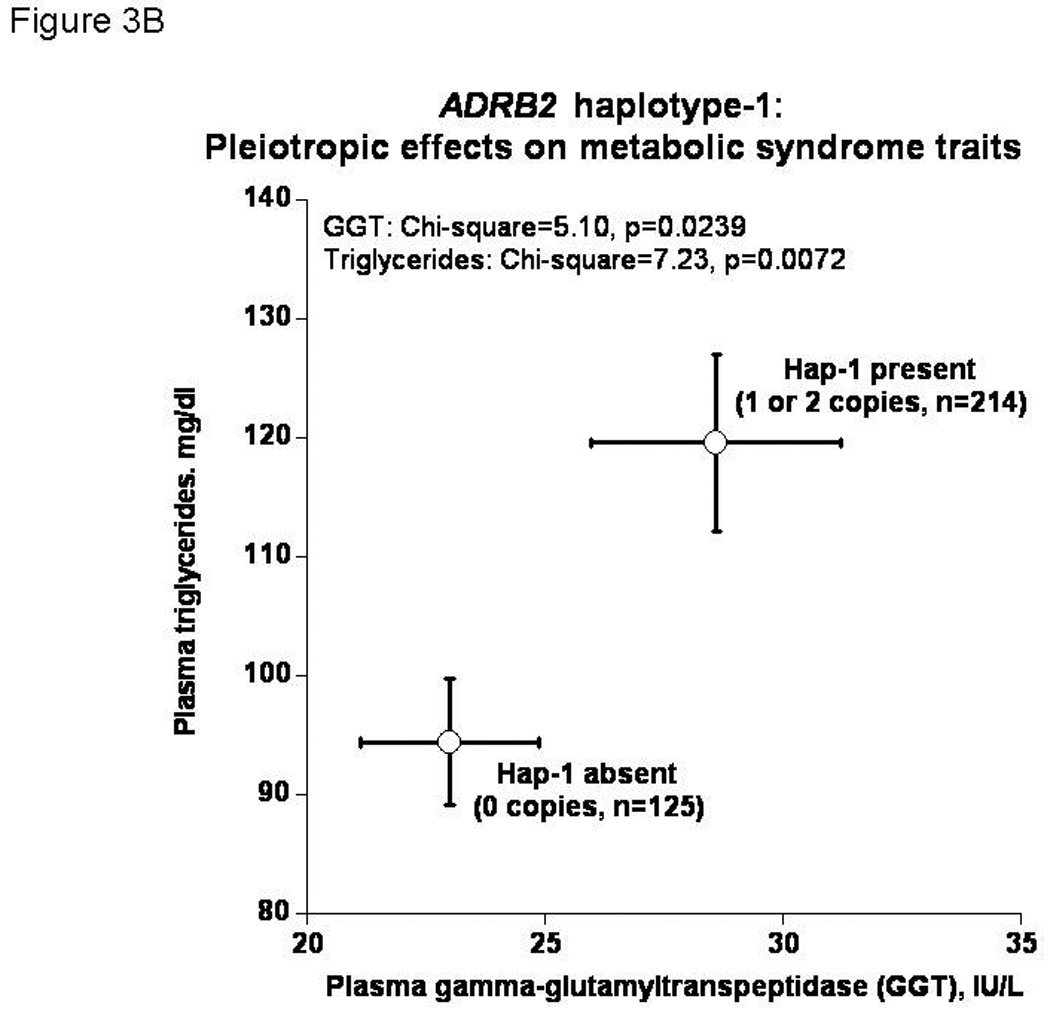
Figure 3B. ADRB2 haplotype-1 (AGGCCGGGC) increases both GGT and triglycerides in plasma. Results illustrate the pleiotropic effect of ADRB2 genetic variation on fatty liver risk factors.
Genetic co-determination by ADRB2 of metabolic traits
ADRB2 haplotype-1 (the most frequent haplotype: AGGCCGGGC) predicted not only GGT but also plasma triglycerides (p=0.0071). Figure 3B illustrates joint effect of ADRB2 haplotype-1 on both plasma GGT (p<0.024) and triglycerides (p=0.0071), suggesting pleiotropic effects of the gene on multiple metabolic traits.
We conducted sensitivity analyses after excluding the HWE-violating SNP (-47 T/C). We examined the association between ADRB2 haplotype-1 (excluding -47 T/C SNP) and plasma GGT as well as pleiotropic effects on plasma GGT and triglyceride. The results remained consistent and statistically significant (please see supplemental figure).
DISCUSSION
Main findings
Using a twin study design, we confirmed that plasma GGT is a heritable trait (at h2 = 49±8%, p<0.00001; Figure 1) and demonstrate genetic covariance (or shared heritable determination) between GGT, a marker of fatty liver disease, and metabolic syndrome traits such as insulin resistance, increased triglycerides, uric acid, and blood pressure (Table 2). This is the first study to report an association between plasma GGT and beta2-adrenergic receptor genetic variation (Figure 3A). The findings of phenotypic pleiotropy (Table 2) as well as the joint effect of ADRB2 variants on both GGT and lipids (Figure 3B) thus contribute to a genetic basis for the clinical observation that NAFLD patients display elevations of both GGT and triglycerides. The results provide a potential mechanistic link between adrenergic genetic variation and shared genetic susceptibility between NAFLD and the metabolic syndrome.
Our study suggests that GGT shares significant genetic covariance with uric acid, insulin, HOMA-IR, triglycerides, and BP (Table 2). These traits are common to the metabolic syndrome and frequently associated with NAFLD, and document the contribution of common genetic variation at one particular adrenergic locus: ADRB2. Our study thus suggests that the adrenergic system may be a key mediator linking metabolic risk factors with NAFLD.
Elevated GGT is a marker of future risk of diabetes, the metabolic syndrome and all-cause cardiovascular (CVD) and liver disease mortality (7). ADRB2 genotypes predict not only BP (16) but also survival after acute coronary syndrome (33) underscoring the prognostic significance of such variants. Therefore, the associations we described between adrenergic activity (Tables 1&2), ADRB2 genotype and plasma GGT (Figures 2&3) may ultimately have clinical implications, opening novel pathways to probe the emerging link between NAFLD and CVD.
Biological rationale and proposed mechanism of adrenergic involvement
The genetic polymorphisms within ADRB2 that predict plasma GGT and triglyceride concentrations (Table 4) confer functional variation upon the gene: the promoter variants (especially T-47C) alter reporter gene expression in transfected ADRB2 promoter/luciferase plasmids (32), while non-synonymous variant Gln27Glu enhances ADRB2 down-regulation in the face of repeated agonist exposure (34). Based upon our findings, we propose a model for the role of the adrenergic pathway in GGT regulation and its association with NAFLD. First of all, in vitro and in vivo studies demonstrate that hepatocytes and hepatic stellate cells (HSC), as well as intra-hepatic biliary epithelial cells, possess adrenergic receptors and respond to adrenergic stimulation (35–37). Catecholamines such as epinephrine stimulate hepatocytes to synthesize and release C-reactive protein (38), which correlates with elevations of plasma GGT and other features of the metabolic syndrome and NAFLD (Table 1) (15). Additionally, norepinephrine is required for activation of HSC and may be a key mediator of fibrogenesis (37). Both inflammatory and fibrogenic responses may lead to release of GGT by epithelial cells of intrahepatic bile duct and hepatocytes (21, 36, 39, 40). In addition, ADRB2 receptors are responsible for maintaining glucose homeostasis by hepatocytes, as well as release of free fatty acids (41), which play a role in the pathogenesis of NAFLD (42). Taken these findings together, it is plausible to suggest that increased adrenergic activity may predispose to both increased GGT and fibrogenic processes leading to NAFLD. The present study supports the viewpoint that the adrenergic system may be one common denominator between metabolic risk factors and NAFLD, and this nexus may be exploited, in future, to improve our understanding of the development of NAFLD in humans.
Table 4.
Genotype-phenotype association between ADRB2 (beta-2 adrenergic receptor) polymorphisms and gamma-glutamyl transpeptidase in twins.
| SNP domain | Diploid genotype code |
Gamma-glutamyl transpeptidase, IU/L |
Diploid genotype bases |
N | P value | |
|---|---|---|---|---|---|---|
| Mean | SEM | |||||
| Promoter G-1023A | 1 | 18.2 | 1.4 | G/G | 109 | 0.025 |
| (rs2053044) | 2 | 23.3 | 1.7 | G/A | 136 | |
| 3 | 24.9 | 3.3 | A/A | 80 | ||
| Promoter C-468G | 1 | 19.4 | 1.1 | C/C | 106 | 0.015 |
| (rs11168070) | 2, 3 | 24.7 | 1.9 | C/G or G/G | 221 | |
| Promoter T-367C | 1 | 19.6 | 1.1 | T/T | 112 | 0.021 |
| (rs11959427) | 2, 3 | 24.7 | 2.0 | T/C or C/C | 219 | |
| Promoter T-47C | 1 | 19.5 | 1.1 | T/T | 117 | 0.012 |
| (rs1042711) | 2, 3 | 24.8 | 1.9 | T/C or C/C | 213 | |
| Glu27Gln, G298C | 1 | 19.5 | 1.1 | C/C | 104 | 0.016 |
| (rs1042714) | 2, 3 | 24.8 | 1.9 | C/G or G/G | 224 | |
Footnote:
This table shows that five SNPs in the beta-2-adrenergic receptor gene (ADRB2) predict with plasma GGT, a marker of fatty liver. SNP statistics for twin study population are derived from generalized estimating equations (GEEs; PROC GENMOD) in SAS, version 9.1 (SAS Institute, Cary, NC) to account for correlations within twinships. Results are displayed for trait (plasma GGT) mean (IU/L, ± one SEM). All analyses are adjusted for age and sex. GGT was treated as a continuous outcome variable. A two-tailed p-value of ≤0.05 was considered statistically significant.
Strengths and limitations of the study
The strengths of this study are several: 1. Well-characterized cohort with comprehensive phenotyping of multiple metabolic risk factors (23). 2. Twin study design, which allowed us to examine both the heritability of the GGT trait, as well as genetic and environmental covariances associated with complex inheritance of GGT (and perhaps NAFLD). 3. Documentation of joint (pleiotropic) effects of ADRB2 genetic variation, both haplotypes and single SNPs, on both GGT and lipid traits. 4. The trait-associated ADRB2 SNPs are known to be functional, suggesting mechanistic links among the pathogenic metabolic traits.
Limitations of this study include that only one ethnicity (European ancestry) was examined. Therefore, these data may not be generalizable to individuals of other ethnicities. However, homogeneity of biogeographic ancestry in our study population may alleviate concerns regarding the internal validity of the results. The average BMI in our cohort was 25 kg/m2 that is lower than the reported average BMI of the US population (NHANES 1999–2002). Our twin population was exclusively white, predominantly female (72% of the population) and mainly derived from residents of Southern California, who may not be representative of entire US population, given demographic differences. Alcohol use, detailed socioeconomic and marital status data were not available. Effect modifications due to gender, alcohol use and age could not be adequately analyzed due to the lack of power. Additionally, only ADRB2 gene effects were examined. However, expanding the study to include a larger number of genes, utilizing a hypothesis-free, genomic approach, creates statistical problems in achieving the necessary modified alpha-threshold to avoid false-positive conclusions.
Results in context with other published studies
Our results suggest that GGT is heritable, and the heritability estimate (at h2=49±8%; Figure 1) is similar to previously published studies (43–46). The heritability estimate reported in our study is consistent with previous published twin study by Whitfield et al (GGT heritability is 49% in our study and 52% in Whitfield et al. study) (43). The mean age in Whitfield’s study was 23 versus 40 years in our study. Therefore, results remain consistent across at least these age groups of 23 through 40 among Caucasian twins. The average BMI was 25 kg/m2 in the Whitfield study, which is similar to our cohort. Therefore, we believe that our findings are consistent with prior published data.
Furthermore, Bathum et al have shown that the effect of heredity on plasma GGT is independent of BMI and alcohol among 580 Danish twins (47). Valle and colleagues have shown that butyryl-cholinesterase (BChE) has shared genetic association with the metabolic syndrome (41, 48), but here we find that GGT and BChE share environmental covariance but genetic covariance did not reach statistical significance. However, we only tested covariance models considering additive genetic variance for GGT and BChE, and cannot exclude other potentially shared effects such as dominance genetic variance or gene-by-environment interactions between these two traits. Additionally, a Japanese study of NAFLD showed that the same ADRB2 non-synonymous SNP that we associated with GGT (Gln27Glu) may be associated with both hypertriglyceridemia and NAFLD susceptibility (17).
A recent genome-wide association conducted in the Dallas Heart Study found that PNPLA3 (patatin-like phospholipase-3) genotype is associated with fatty liver in that cohort (49). PNPLA3 is a member of a newly recognized family of adipocyte triglyceride lipases (50–52). The previously described adipocyte triglyceride lipase, known as HSL (hormone sensitive lipase) or LIPE (lipase, hormone-sensitive), is positively regulated by catecholamines acting through ADRB2; the expression of LIPE seems to be decreased in obesity despite a rise in catecholamines (increased adrenergic activity), perhaps contributing to a state of catecholamine- and insulin-resistance (51–53). Both the newer family of lipases (including PNPLA3) and LIPE catalyze triglyceride hydrolysis in adipocytes (51, 52). Therefore, our study provides an additional mechanistic link between these parallel lipolytic processes and metabolic syndrome traits of increased adrenergic activity, triglycerides and GGT. Since NAFLD is a complex disease with substantial hereditary determination, there are likely several genes that may contribute to genetic susceptibility for the trait (54–56).
Utilizing the twin-study design, we demonstrate significant shared genetic and environmental covariance between metabolic risk factors and GGT (Table 2). In addition, we found that five functional (32) SNPs at ADRB2 are associated with plasma GGT levels in humans. In vitro and in vivo studies have shown that plasma epinephrine is involved in progression of NAFLD (35, 36), but human data to confirm these findings has been lacking. It is well-recognized that adrenergic activity plays a critical role in portal hypertension, and use of β-blockade in the setting of advanced portal hypertension (especially after variceal bleeding with elevated hepatic wedge pressure gradient) improves survival (57, 58), and ADRB2 gene polymorphisms influence hemodynamic response to β-blockade (59) . Our study suggests that increased epinephrine levels are associated with plasma GGT elevation (Table 1). It is thus plausible that increased adrenergic activity may be involved in the pathogenesis of NAFLD, as well as the progression of NAFLD to nonalcoholic steatohepatitis (NASH) and ulimately cirrhosis. Further studies are needed to examine these hypotheses.
Conclusions and perspectives
We conclude that GGT is heritable and shares significant genetic co-determination with multiple features of the metabolic syndrome as well as elevated adrenergic activity in Caucasians. ADRB2 genetic variation is predictive of plasma GGT concentration in humans, and the ADRB2 locus may thus participate in the complex heritability of GGT, and perhaps NAFLD in this population. Future longitudinal studies are needed to examine whether ADRB2 genetic variants increase susceptibility of NAFLD and/or progression from steatosis to steatohepatitis in humans.
Supplementary Material
ACKNOWLEDGEMENTS
Funding Support: This work was supported in part by the National Institutes of Health grants to Daniel T. O’Connor, MD, and an American Gastroenterological Association Foundation – Sucampo – ASP Designated Research Scholar Award in Geriatric Gastroenterology (Supported by a T. Franklin Williams Scholarship Award; Funding provided by: Atlantic Philanthropies, Inc, the John A. Hartford Foundation, the Association of Specialty Professors, and the American Gastroenterological Association) to Rohit Loomba, MD, MHSc.
Role of funding agencies: Funding agencies did not have any role in the design and conduct of the study, collection, management, analysis and interpretation of the data; preparation, review, or approval of the manuscript.
We appreciate the support of the Department of Veterans Affairs, the NIH/NCMHD-sponsored (MD000220) EXPORT minority health center, as well as the NIH/NCRR-sponsored (RR00827) General Clinical Research Center.
Abbreviations
- NAFLD
non-alcoholic fatty liver disease
- SNP
single nucleotide polymorphism
- NASH
non-alcoholic steatohepatitis
- GGT
gamma-glutamyl transpeptidase
- ADRB2
β2-adrenergic receptor
- IR
insulin resistance
- HOMA
homeostasis model of insulin resistance
- h2
heritability
- TG
triglycerides
- LDL
low density lipoprotein
- CRP
C-reactive protein
- LD
linkage disequilibrium
- ρG
Rho-G (genetic co-variance between traits)
- ρE
Rho-E (environmental co-variance between traits)
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- CVD
cardiovascular disease
- GEE
generalized estimating equation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: None to disclose.
Author contribution:
Study concept and design: Rohit Loomba and Daniel T O'Connor
Acquisition of data: Fangwen Rao, Srikrihna Khandrika, Michael Ziegler
Analysis and interpretation of data: Fangwen Rao, Rohit Loomba, Lian Zhang, and Daniel T O'Connor
Drafting of the manuscript: Rohit Loomba and Daniel T O'Connor
Critical revision of the manuscript for important intellectual content: Lian Zhang, Michael Ziegler and David Brenner
Statistical analysis: Fangwen Rao
Obtained funding: Daniel T O'Connor and Michael Ziegler
Technical or material support and study supervision: Daniel T O'Connor
REFERENCES
- 1.Ruttenburg AM, Goldbarg JA, Pineda EP. Serum Gamma-Glutamyl Transpeptidase Activity in Hepatobiliary Pancreatic Disease. Gastroenterology. 1963;45:43–48. [PubMed] [Google Scholar]
- 2.Nemesanszky E, Lott JA. Gamma-glutamyltransferase and its isoenzymes: progress and problems. Clin Chem. 1985;31:797–803. [PubMed] [Google Scholar]
- 3.Irie M, Suzuki N, Sohda T, Anan A, Iwata K, Takeyama Y, Watanabe H, et al. Hepatic expression of gamma-glutamyltranspeptidase in the human liver of patients with alcoholic liver disease. Hepatol Res. 2007;37:966–973. doi: 10.1111/j.1872-034X.2007.00151.x. [DOI] [PubMed] [Google Scholar]
- 4.Whitfield JB. Gamma glutamyl transferase. Crit Rev Clin Lab Sci. 2001;38:263–355. doi: 10.1080/20014091084227. [DOI] [PubMed] [Google Scholar]
- 5.Ikai E, Honda R, Yamada Y. Serum gamma-glutamyl transpeptidase level and blood pressure in nondrinkers: a possible pathogenetic role of fatty liver in obesity-related hypertension. J Hum Hypertens. 1994;8:95–100. [PubMed] [Google Scholar]
- 6.Ikai E, Noborizaka Y, Tsuritani I, Honda R, Ishizaki M, Yamada Y. Serum gamma-glutamyl transpeptidase levels and hypertension in non-drinkers: a possible role of fatty liver in the pathogenesis of obesity related hypertension. Obes Res. 1993;1:469–474. doi: 10.1002/j.1550-8528.1993.tb00029.x. [DOI] [PubMed] [Google Scholar]
- 7.Lee DS, Evans JC, Robins SJ, Wilson PW, Albano I, Fox CS, Wang TJ, et al. Gamma glutamyl transferase and metabolic syndrome, cardiovascular disease, and mortality risk: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2007;27:127–133. doi: 10.1161/01.ATV.0000251993.20372.40. [DOI] [PubMed] [Google Scholar]
- 8.Jo SK, Lee WY, Rhee EJ, Won JC, Jung CH, Park CY, Oh KW, et al. Serum gamma-glutamyl transferase activity predicts future development of metabolic syndrome defined by 2 different criteria. Clin Chim Acta. 2009;403:234–240. doi: 10.1016/j.cca.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 9.Jo SK, Lee WY, Rhee EJ, Won JC, Jung CH, Park CY, Oh KW, et al. Serum gamma-glutamyl transferase activity predicts future development of metabolic syndrome defined by 2 different criteria. Clin Chim Acta. 2009 doi: 10.1016/j.cca.2009.03.035. [DOI] [PubMed] [Google Scholar]
- 10.Dixon JB, Bhathal PS, O'Brien PE. Weight loss and non-alcoholic fatty liver disease: falls in gamma-glutamyl transferase concentrations are associated with histologic improvement. Obes Surg. 2006;16:1278–1286. doi: 10.1381/096089206778663805. [DOI] [PubMed] [Google Scholar]
- 11.Lee DH, Jacobs DR, Jr, Gross M, Kiefe CI, Roseman J, Lewis CE, Steffes M. Gamma-glutamyltransferase is a predictor of incident diabetes and hypertension: the Coronary Artery Risk Development in Young Adults (CARDIA) Study. Clin Chem. 2003;49:1358–1366. doi: 10.1373/49.8.1358. [DOI] [PubMed] [Google Scholar]
- 12.Saely CH, Vonbank A, Rein P, Woess M, Beer S, Aczel S, Jankovic V, et al. Alanine aminotransferase and gamma-glutamyl transferase are associated with the metabolic syndrome but not with angiographically determined coronary atherosclerosis. Clin Chim Acta. 2008;397:82–86. doi: 10.1016/j.cca.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 13.Yamada J, Tomiyama H, Yambe M, Koji Y, Motobe K, Shiina K, Yamamoto Y, et al. Elevated serum levels of alanine aminotransferase and gamma glutamyltransferase are markers of inflammation and oxidative stress independent of the metabolic syndrome. Atherosclerosis. 2006;189:198–205. doi: 10.1016/j.atherosclerosis.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 14.Rao F, Zhang L, Wessel J, Zhang K, Wen G, Kennedy BP, Rana BK, et al. Tyrosine hydroxylase, the rate-limiting enzyme in catecholamine biosynthesis: discovery of common human genetic variants governing transcription, autonomic activity, and blood pressure in vivo. Circulation. 2007;116:993–1006. doi: 10.1161/CIRCULATIONAHA.106.682302. [DOI] [PubMed] [Google Scholar]
- 15.Wessel J, Moratorio G, Rao F, Mahata M, Zhang L, Greene W, Rana BK, et al. C-reactive protein, an 'intermediate phenotype' for inflammation: human twin studies reveal heritability, association with blood pressure and the metabolic syndrome, and the influence of common polymorphism at catecholaminergic/beta-adrenergic pathway loci. J Hypertens. 2007;25:329–343. doi: 10.1097/HJH.0b013e328011753e. [DOI] [PubMed] [Google Scholar]
- 16.Bao X, Mills PJ, Rana BK, Dimsdale JE, Schork NJ, Smith DW, Rao F, et al. Interactive effects of common beta2-adrenoceptor haplotypes and age on susceptibility to hypertension and receptor function. Hypertension. 2005;46:301–307. doi: 10.1161/01.HYP.0000175842.19266.95. [DOI] [PubMed] [Google Scholar]
- 17.Iwamoto N, Ogawa Y, Kajihara S, Hisatomi A, Yasutake T, Yoshimura T, Mizuta T, et al. Gln27Glu beta2-adrenergic receptor variant is associated with hypertriglyceridemia and the development of fatty liver. Clin Chim Acta. 2001;314:85–91. doi: 10.1016/s0009-8981(01)00633-7. [DOI] [PubMed] [Google Scholar]
- 18.Vardeny O, Detry MA, Moran JJ, Johnson MR, Sweitzer NK. The beta2 adrenergic receptor Gln27Glu polymorphism affects insulin resistance in patients with heart failure: possible modulation by choice of beta blocker. J Cardiovasc Pharmacol. 2008;52:500–506. doi: 10.1097/FJC.0b013e31818f5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isaza CA, Henao J, Sanchez JC, Porras GL, Cardona J, Bedoya G. Beta-2-adrenergic receptor polymorphisms and changes in lipids induced by metoprolol. Pharmacology. 2007;80:279–285. doi: 10.1159/000106554. [DOI] [PubMed] [Google Scholar]
- 20.Svetkey LP, Timmons PZ, Emovon O, Anderson NB, Preis L, Chen YT. Association of hypertension with beta2- and alpha2c10-adrenergic receptor genotype. Hypertension. 1996;27:1210–1215. doi: 10.1161/01.hyp.27.6.1210. [DOI] [PubMed] [Google Scholar]
- 21.Patton HM, Lavine JE, Van Natta ML, Schwimmer JB, Kleiner D, Molleston J. Clinical correlates of histopathology in pediatric nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1961–1971. e1962. doi: 10.1053/j.gastro.2008.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Ann Intern Med. 2007;147:573–577. doi: 10.7326/0003-4819-147-8-200710160-00010. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Rao F, Zhang K, Khandrika S, Das M, Vaingankar SM, Bao X, et al. Discovery of common human genetic variants of GTP cyclohydrolase 1 (GCH1) governing nitric oxide, autonomic activity, and cardiovascular risk. J Clin Invest. 2007;117:2658–2671. doi: 10.1172/JCI31093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Rao F, Wessel J, Kennedy BP, Rana BK, Taupenot L, Lillie EO, et al. Functional allelic heterogeneity and pleiotropy of a repeat polymorphism in tyrosine hydroxylase: prediction of catecholamines and response to stress in twins. Physiol Genomics. 2004;19:277–291. doi: 10.1152/physiolgenomics.00151.2004. [DOI] [PubMed] [Google Scholar]
- 25.Brinton TJ, Kailasam MT, Wu RA, Cervenka JH, Chio SS, Parmer RJ, DeMaria AN, et al. Arterial compliance by cuff sphygmomanometer. Application to hypertension and early changes in subjects at genetic risk. Hypertension. 1996;28:599–603. doi: 10.1161/01.hyp.28.4.599. [DOI] [PubMed] [Google Scholar]
- 26.Drysdale CM, McGraw DW, Stack CB, Stephens JC, Judson RS, Nandabalan K, Arnold K, et al. Complex promoter and coding region beta 2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. Proc Natl Acad Sci U S A. 2000;97:10483–10488. doi: 10.1073/pnas.97.19.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGraw DW, Forbes SL, Kramer LA, Liggett SB. Polymorphisms of the 5' leader cistron of the human beta2-adrenergic receptor regulate receptor expression. J Clin Invest. 1998;102:1927–1932. doi: 10.1172/JCI4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buetow KH, Edmonson M, MacDonald R, Clifford R, Yip P, Kelley J, Little DP, et al. High-throughput development and characterization of a genomewide collection of gene-based single nucleotide polymorphism markers by chip-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc Natl Acad Sci U S A. 2001;98:581–584. doi: 10.1073/pnas.021506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goldbarg JA, Pineda EP, Smith EE, Friedman OM, Rutenburg AM. A method for the colorimetric determination of gamma-glutamyl transpeptidase in human serum; enzymatic activity in health and disease. Gastroenterology. 1963;44:127–133. [PubMed] [Google Scholar]
- 30.Halperin E, Eskin E. Haplotype reconstruction from genotype data using Imperfect Phylogeny. Bioinformatics. 2004;20:1842–1849. doi: 10.1093/bioinformatics/bth149. [DOI] [PubMed] [Google Scholar]
- 31.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 32.Johnatty SE, Abdellatif M, Shimmin L, Clark RB, Boerwinkle E. Beta 2 adrenergic receptor 5' haplotypes influence promoter activity. Br J Pharmacol. 2002;137:1213–1216. doi: 10.1038/sj.bjp.0704935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanfear DE, Jones PG, Marsh S, Cresci S, McLeod HL, Spertus JA. Beta2-adrenergic receptor genotype and survival among patients receiving beta-blocker therapy after an acute coronary syndrome. JAMA. 2005;294:1526–1533. doi: 10.1001/jama.294.12.1526. [DOI] [PubMed] [Google Scholar]
- 34.Small KM, McGraw DW, Liggett SB. Pharmacology and physiology of human adrenergic receptor polymorphisms. Annu Rev Pharmacol Toxicol. 2003;43:381–411. doi: 10.1146/annurev.pharmtox.43.100901.135823. [DOI] [PubMed] [Google Scholar]
- 35.Oben JA, Yang S, Lin H, Ono M, Diehl AM. Norepinephrine and neuropeptide Y promote proliferation and collagen gene expression of hepatic myofibroblastic stellate cells. Biochem Biophys Res Commun. 2003;302:685–690. doi: 10.1016/s0006-291x(03)00232-8. [DOI] [PubMed] [Google Scholar]
- 36.Oben JA, Roskams T, Yang S, Lin H, Sinelli N, Torbenson M, Smedh U, et al. Hepatic fibrogenesis requires sympathetic neurotransmitters. Gut. 2004;53:438–445. doi: 10.1136/gut.2003.026658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oben JA, Roskams T, Yang S, Lin H, Sinelli N, Li Z, Torbenson M, et al. Norepinephrine induces hepatic fibrogenesis in leptin deficient ob/ob mice. Biochem Biophys Res Commun. 2003;308:284–292. doi: 10.1016/s0006-291x(03)01360-3. [DOI] [PubMed] [Google Scholar]
- 38.O'Riordain MG, Ross JA, Fearon KC, Maingay J, Farouk M, Garden OJ, Carter DC. Insulin and counterregulatory hormones influence acute-phase protein production in human hepatocytes. Am J Physiol. 1995;269:E323–E330. doi: 10.1152/ajpendo.1995.269.2.E323. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Oben JA, Yang S, Lin H, Stafford EA, Soloski MJ, Thomas SA, et al. Norepinephrine regulates hepatic innate immune system in leptin-deficient mice with nonalcoholic steatohepatitis. Hepatology. 2004;40:434–441. doi: 10.1002/hep.20320. [DOI] [PubMed] [Google Scholar]
- 40.Oben JA, Roskams T, Yang S, Lin H, Sinelli N, Li Z, Torbenson M, et al. Sympathetic nervous system inhibition increases hepatic progenitors and reduces liver injury. Hepatology. 2003;38:664–673. doi: 10.1053/jhep.2003.50371. [DOI] [PubMed] [Google Scholar]
- 41.Van Heeswijk JC, Vianen GJ, van den Thillart GE. The adrenergic control of hepatic glucose and FFA metabolism in rainbow trout (Oncorhynchus mykiss): increased sensitivity to adrenergic stimulation with fasting. Gen Comp Endocrinol. 2006;145:51–61. doi: 10.1016/j.ygcen.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Hargrove DM, Bagby GJ, Lang CH, Spitzer JJ. Adrenergic blockade prevents endotoxin-induced increases in glucose metabolism. Am J Physiol. 1988;255:E629–E635. doi: 10.1152/ajpendo.1988.255.5.E629. [DOI] [PubMed] [Google Scholar]
- 43.Whitfield JB, Zhu G, Nestler JE, Heath AC, Martin NG. Genetic covariation between serum gamma-glutamyltransferase activity and cardiovascular risk factors. Clin Chem. 2002;48:1426–1431. [PubMed] [Google Scholar]
- 44.Rahmioglu N, Andrew T, Cherkas L, Surdulescu G, Swaminathan R, Spector T, Ahmadi KR. Epidemiology and genetic epidemiology of the liver function test proteins. PLoS ONE. 2009;4:e4435. doi: 10.1371/journal.pone.0004435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin JP, O'Donnell CJ, Fox CS, Cupples LA. Heritability of serum gamma-glutamyltransferase level: genetic analysis from the Framingham Offspring Study. Liver Int. 2009 doi: 10.1111/j.1478-3231.2008.01965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan X, Waterworth D, Perry JR, Lim N, Song K, Chambers JC, Zhang W, et al. Population-based genome-wide association studies reveal six loci influencing plasma levels of liver enzymes. Am J Hum Genet. 2008;83:520–528. doi: 10.1016/j.ajhg.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bathum L, Petersen HC, Rosholm JU, Hyltoft Petersen P, Vaupel J, Christensen K. Evidence for a substantial genetic influence on biochemical liver function tests: results from a population-based Danish twin study. Clin Chem. 2001;47:81–87. [PubMed] [Google Scholar]
- 48.Valle A, O'Connor DT, Taylor P, Zhu G, Montgomery GW, Slagboom PE, Martin NG, et al. Butyrylcholinesterase: association with the metabolic syndrome and identification of 2 gene loci affecting activity. Clin Chem. 2006;52:1014–1020. doi: 10.1373/clinchem.2005.065052. [DOI] [PubMed] [Google Scholar]
- 49.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase family. J Lipid Res. 2006;47:1940–1949. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Jocken JW, Langin D, Smit E, Saris WH, Valle C, Hul GB, Holm C, et al. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J Clin Endocrinol Metab. 2007;92:2292–2299. doi: 10.1210/jc.2006-1318. [DOI] [PubMed] [Google Scholar]
- 52.Ryden M, Jocken J, van Harmelen V, Dicker A, Hoffstedt J, Wiren M, Blomqvist L, et al. Comparative studies of the role of hormone-sensitive lipase and adipose triglyceride lipase in human fat cell lipolysis. Am J Physiol Endocrinol Metab. 2007;292:E1847–E1855. doi: 10.1152/ajpendo.00040.2007. [DOI] [PubMed] [Google Scholar]
- 53.Mulder H, Sorhede-Winzell M, Contreras JA, Fex M, Strom K, Ploug T, Galbo H, et al. Hormone-sensitive lipase null mice exhibit signs of impaired insulin sensitivity whereas insulin secretion is intact. J Biol Chem. 2003;278:36380–36388. doi: 10.1074/jbc.M213032200. [DOI] [PubMed] [Google Scholar]
- 54.Day CP. Genes or environment to determine alcoholic liver disease and non-alcoholic fatty liver disease. Liver Int. 2006;26:1021–1028. doi: 10.1111/j.1478-3231.2006.01323.x. [DOI] [PubMed] [Google Scholar]
- 55.Speliotes EK. Genetics of common obesity and nonalcoholic fatty liver disease. Gastroenterology. 2009;136:1492–1495. doi: 10.1053/j.gastro.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis. 2007;27:44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 57.Lebrec D, Poynard T, Hillon P, Benhamou JP. Propranolol for prevention of recurrent gastrointestinal bleeding in patients with cirrhosis: a controlled study. N Engl J Med. 1981;305:1371–1374. doi: 10.1056/NEJM198112033052302. [DOI] [PubMed] [Google Scholar]
- 58.Pascal JP, Cales P. Propranolol in the prevention of first upper gastrointestinal tract hemorrhage in patients with cirrhosis of the liver and esophageal varices. N Engl J Med. 1987;317:856–861. doi: 10.1056/NEJM198710013171403. [DOI] [PubMed] [Google Scholar]
- 59.Turnes J, Hernandez-Guerra M, Abraldes JG, Bellot P, Oliva R, Garcia-Pagan JC, Bosch J. Influence of beta-2 adrenergic receptor gene polymorphism on the hemodynamic response to propranolol in patients with cirrhosis. Hepatology. 2006;43:34–41. doi: 10.1002/hep.21000. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.