

Abstract
The large GTPase atlastin belongs to the dynamin superfamily that has been widely implicated in facilitating membrane tubulation, fission, and in select cases, fusion. Mutations spread across atlastin isoform 1 (atlastin-1) have been identified in patients suffering from hereditary spastic paraplegia (HSP), a neurodegenerative disorder affecting motor neuron function in the lower extremities. On a molecular level, atlastin-1 associates with high membrane curvature and fusion events at the endoplasmic reticulum and cis-Golgi. Here we report crystal structures of atlastin-1 comprising the G and middle domains in two different conformations. Although the orientation of the middle domain relative to the G domain is different in the two structures, both reveal dimeric assemblies with a common, GDP-bound G domain dimer. In contrast, dimer formation in solution is observed only in the presence of GTP and transition state analogs, similar to other G proteins that are activated by nucleotide-dependent dimerization. Analyses of solution scattering data suggest that upon nucleotide binding, the protein adopts a somewhat extended, dimeric conformation that is reminiscent of one of the two crystal structures. These structural studies suggest a model for nucleotide-dependent regulation of atlastin with implications for membrane fusion. This mechanism is affected in several mutants associated with HSP, providing insights into disease pathogenesis.
Keywords: protein structure, neuropathogenic mechanism, tubular membrane network
The superfamily of dynamin GTPases comprises a group of proteins that are involved in diverse cellular functions and are often closely associated with biological membranes (1). They function in a wide range of cellular scenarios including vesicle scission, fusion and fission of organelles, cytokinesis, and antiviral activity (1). Although they contain G domains that are structurally related to the small G proteins of the Ras superfamily, the corresponding domains in the dynamin superfamily are significantly larger (300 vs. 180 residues) and have a low intrinsic affinity for nucleotide. In addition, GTPases of this family often depend on nucleotide-dependent homodimerization to facilitate GTP hydrolysis rather than heterodimerization with a GTPase activating protein (2). Such a regulatory mechanism has been established for guanylate-binding protein (GBP) and dynamin (3, 4).
Dynamin and most dynamin-related proteins have a common domain architecture comprising a middle domain and a GTPase effector domain (GED) in addition to the N-terminal G domain (1). To date, the molecular mechanism responsible for nucleotide-dependent functional and structural transitions is best understood for the prototypical member dynamin, a protein that catalyzes membrane fission (5). In low-resolution models of dynamin, the middle domain and GED form a stalk-like structure that is involved in self-assembly (6, 7). Furthermore, the GED and the C terminus of the G domain form a three-helix bundle, called the bundle signaling element (BSE), which modulates dynamin function (4, 8). A recent crystal structure of a minimal G domain-GED fusion protein revealed dimeric G domains in a catalytically competent transition state that was proposed to play a role in the disassembly of dynamin coats from membrane tubes proceeding the fission event (4, 9). Membrane binding and the GTPase of dynamin are further regulated by the pleckstrin homology (PH) domain, which is located at the tip of the stalk distal to the G domain and serves an autoregulatory function (7). Membrane-mediated, higher-order oligomerization of dynamin stimulates its basal GTPase activity further (10, 11). A similar model has been proposed for MxA, a dynamin-like protein involved in innate immunity (12).
Atlastin, a large G protein most closely related to the GBP subfamily, has been identified as a crucial protein in maintaining endoplasmic reticulum (ER) morphology and vesicle trafficking (13–17). Atlastin lacks a GED but instead contains two transmembrane helices and a C-terminal domain in addition to its large G and middle domains, both of which face the cytoplasm (18). Three isoforms of atlastin are found in humans, differing in their N and C termini as well as their expression patterns. Atlastin-2 and -3 are found ubiquitously and localize to the ER. In contrast, atlastin-1 is mainly found in the central nervous system, especially in neurons of the corticospinal tract as well as the cerebral cortex and hippocampus and is localized to the cis-Golgi and to a lesser extent in the ER (18, 19). On a microscopic level, they are found to associate with three-way junctions in the ER, and their absence results in a loss of this morphology in exchange for long, unbranched tubules (14, 20). At the ER, atlastin interacts with proteins from the reticulon and DP1 families (14), spastin/SPG4 (21, 22), and REEP1/SPG31 (20). Notably, members of these families have been found to be among the most frequently mutated loci in the neurodegenerative disorder hereditary spastic paraplegia (HSP; also known as familial spastic paraplegia or Strumpell–Lorrain disease) (23).
HSP is a group of inherited disorders that cause progressive spasticity and weakness in the lower extremities. Mutations in the atlastin gene account for ∼10% of autosomal dominant HSP cases (20). Nearly 40 atlastin-1 mutations have been identified in HSP patients, which affect upper motor neurons of the cortical spinal tract (23). The vast majority of disease-associated alleles encode missense mutations, with one deletion and several nonsense mutations also identified (24, 25). Although atlastin accounts for only a tenth of the total HSP cases, it is the most common form in instances where the disorder occurs in infancy or childhood. In addition, many forms of atlastin show very slow progression or no apparent progression when the disease occurs in childhood, which led to the presumption that atlastin may have its effect in development rather than maintenance of affected neurons (26).
Understanding the molecular basis of atlastin function and of HSP-associated alleles will be important for diagnostic and prognostic applications as well as for a basic understanding of HSP pathogenesis. Furthermore, a structural analysis may reveal basic mechanisms controlling the cellular function of atlastin and related proteins. In this study, we determined the crystal structure of the cytoplasmic domain of human atlastin-1 in two different crystal forms, both bound to GDP. This portion of the protein comprises the G domain followed by the middle domain, a common modular architecture found in other members of the large G-protein family. Additionally, we determined the apo- and nucleotide-bound oligomerization states and low-resolution structures of the cytoplasmic domain in solution by using static light scattering and small-angle X-ray scattering (SAXS). Several mutations associated with HSP affect GTPase activity and/or nucleotide-dependent dimerization suggesting a basis of disease pathogenesis.
Results and Discussion
Crystal Structures of Atlastin-1.
The purified, cytoplasmic domain of human atlastin-1 (residues 1–446; Fig. 1A) crystallized in two distinct crystal forms (form 1: space group p6522; form 2: p212121; Table S1). The form 1 crystals grown from selenomethionine-derivatized protein diffracted X-rays to a maximal resolution of 2.7 Å with one molecule in the asymmetric unit, and the structure was solved with single anomalous dispersion phasing (Fig. 1B). The form 2 crystals yielded a native dataset at 3.0 Å (four molecules/asymmetric unit), and the structure was solved by molecular replacement using the separated GTPase and middle domains as the search models (Fig. 1C). Although crystallization for both forms was carried out in the presence of GTPγS, only density for the GDP moiety and a magnesium ion was observed (Fig. 1B, Inset and Fig. S1A). In the present structures, the side chain of F76 within the P loop is positioned in such a way that the γ-phosphate moiety cannot be accommodated (Fig. S1A). Conformational changes typically accompanying GTP binding to G proteins would allow F76 to flip out to accommodate the terminal phosphate. Both the slow hydrolysis of GTPγS as well as the presence of GDP as a major contaminant in the nucleotide solution may have contributed to this result (Fig. S2).
Fig. 1.

Structure of atlastin-1. (A) Domain organization of atlastin-1. The large G domain (orange) is connected to the middle domain (blue) by a short flexible linker (green) and is followed by two transmembrane helices (black) that span the membrane and a C-terminal domain. A topological model is shown (Right). The fragment used in this study consists of residues 1–446 of human atlastin-1. (B) Protomer structure of atlastin-1 in crystal form 1. (Inset) GDP/Mg2+-coordinating residues as sticks. (C) Protomer structure of atlastin-1 in crystal form 2. The G domain is shown in a similar orientation as shown in B. GDP and Mg2+ are shown as sticks and spheres, respectively.
In both crystal forms, the N terminus of atlastin forms a globular GTPase domain fold similar to that of GBP1 (rmsd of 4.5 Å) (27). The middle domain that connects to the transmembrane helices folds into a three-helix bundle (Fig. 1 B and C). Most strikingly, the structures in the two crystal forms differ in their relative orientation of the G and middle domains with minimal variance of the individual domains (rmsd of 0.7 Å for both the G and middle domains; Fig. 1 and Fig. S1D). A rigid body rotation by ∼90° of the middle domain relative to the G domain separates the two conformations, with the difference being realized by an alternative conformation of the connecting linker segment (residues 337–346). In contrast to the extended, random-coil conformation of the linker in form 1, it adopts a U-shaped turn in the form 2 crystals. In the latter case, it contributes an additional turn to the first helix of the middle domain, consistent with secondary structure predictions, and one of the two proline residues (P344) in the linker segment serves as a helix cap (Fig. S1D). Mutation of this residue in the cytoplasmic domain of atlastin-1 (P344G) renders the protein insoluble. The second proline (P342) is located at the center of the linker. Similarly, proline residues are present at comparable positions in dynamin and other G proteins, where they serve as pivot points during nucleotide-dependent transitions (4).
Although atlastin lacks a GED and its sequence conservation to other dynamin-like proteins predominantly resides in the G domain, there is a remarkable conservation with regard to domain arrangements, in particular, when the form 2 crystal structure is considered. In this conformation, atlastin’s middle domain folds back onto the G domain and protrudes at a similar position as the BSE (including the GED) and middle domain in dynamin and GBP1, respectively (3, 4, 27, 28) (Fig. 2 A and B). Although the contact region in GBP1 and atlastin is preserved, containing in part hydrophobic contacts between corresponding secondary structure motifs (Fig. S3A), the available dynamin structures lack the middle domain impeding a more detailed comparison. Based on low-resolution structural reconstructions of near-full-length dynamin, the BSE and middle domain form a structural unit (7), and the BSE position in the crystal structure serves as an approximate projection of the middle domain (4). The conservation of the overall architecture suggests a common molecular mechanism underlying the regulation and function of these distantly related proteins.
Fig. 2.

Crystallographic dimers of atlastin-1. (A) GBP1. A model for GTP-bound, full-length GBP1 was constructed according to ref. 27. (B) Dynamin. A dynamin G domain-GED fusion dimer bound to 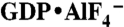 is shown (PDB ID code 2X2E) (4). (C) Atlastin-1. Dimers observed in crystal lattices with P212121 symmetry (form 2, Upper) and with P6522 symmetry (form 1, Lower) are shown. GDP and Mg2+ positions are indicated and are shown as sticks and spheres, respectively. Hypothetical membrane positions based on the location of the C terminus of the cytosolic domain are indicated. Positions of missense mutation associated with HSP are shown as residues displayed as red spheres in both forms. (D) Nucleotide arrangement and G domain dimer interface. The G domain dimer interaces of atlastin-1 (form 1; Left) and GBP1 (Right; PDB ID code 2BC9) (3) are shown. The protomers are colored in light and dark gray, with the nucleotide-binding regions (G1–G4) shown in shades of blue. Nucleotide and Mg2+ are shown as sticks and spheres, respectively. The dashed, red line separates the dimer halves, marking the interfacial region.
is shown (PDB ID code 2X2E) (4). (C) Atlastin-1. Dimers observed in crystal lattices with P212121 symmetry (form 2, Upper) and with P6522 symmetry (form 1, Lower) are shown. GDP and Mg2+ positions are indicated and are shown as sticks and spheres, respectively. Hypothetical membrane positions based on the location of the C terminus of the cytosolic domain are indicated. Positions of missense mutation associated with HSP are shown as residues displayed as red spheres in both forms. (D) Nucleotide arrangement and G domain dimer interface. The G domain dimer interaces of atlastin-1 (form 1; Left) and GBP1 (Right; PDB ID code 2BC9) (3) are shown. The protomers are colored in light and dark gray, with the nucleotide-binding regions (G1–G4) shown in shades of blue. Nucleotide and Mg2+ are shown as sticks and spheres, respectively. The dashed, red line separates the dimer halves, marking the interfacial region.
GBP1 and dynamin form G domain-mediated homooligomers in the presence of GTP and transition state analogs, but not in their apo- and GDP-bound states (Fig. 2 A and B) (4, 27). Considering the symmetry mates in each crystal lattice, the atlastin structures provide insights into potential modes for dimerization, revealing a common interface observed in both structures formed between adjacent G domains (Fig. 2C). The two atlastin-1 G domain dimers align with an rmsd of 2 Å, with the main difference observed in the angle the G domains take with respect to each other (Fig. S1C). As a result, the interfacial area differs by 470 Å2 considering only interactions between the G domains (1226 Å2 and 756 Å2 for form 1 and form 2 G domain dimers, respectively; Fig. S3 C and D). The dimerization involves residues close to the nucleotide-binding pocket, especially within the P loop (G1), Walker B (G3) motif, and the guanosine-binding loop (G4) (Fig. S1 A and B).
The relative position and orientation of the G domains and nucleotide are reminiscent of those seen in the crystal structures of GppNHp-bound GBP1 and dynamin when bound to 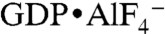 (Fig. 2 and Fig. S3B) (4, 27). Specifically, there is a remarkable structural conservation with regard to the involvement of nucleotide-binding motifs between GBP1 and atlastin-1 (regions G1–G4) in dimerization, despite the difference in bound nucleotide species (Fig. 2D). Dynamin and GBP1 dimers bound to
(Fig. 2 and Fig. S3B) (4, 27). Specifically, there is a remarkable structural conservation with regard to the involvement of nucleotide-binding motifs between GBP1 and atlastin-1 (regions G1–G4) in dimerization, despite the difference in bound nucleotide species (Fig. 2D). Dynamin and GBP1 dimers bound to 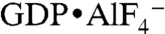 or GppNHp, respectively, involve more extensive contributions of the P loop (G1) and the Walker A (G2) motif compared to the GDP-bound dimers of atlastin-1 (Fig. 2D) (3, 4).
or GppNHp, respectively, involve more extensive contributions of the P loop (G1) and the Walker A (G2) motif compared to the GDP-bound dimers of atlastin-1 (Fig. 2D) (3, 4).
Most notably, the two crystallographic atlastin-1 dimer forms differ in their relative position of the middle domains with respect to the G domain dimers (Fig. 2C). Although the form 2 dimer resembles GBP1 or dynamin assemblies with the middle domains pointing in opposite directions (3, 4, 27), the form 1 dimer of atlastin is distinct from those because the middle domains run in parallel (Fig. 2). In this conformation, the linker segments and middle domains cross over, contributing an additional 1571 Å2 to the overall interfacial area (Fig. 2C and Fig. S3C). Although the middle domains in the form 1 structure protrude in the same direction and would locate to proximal membrane regions via the successive transmembrane domains (Fig. 2C), the functional relevance for this conformation remains elusive.
Nucleotide-Dependent Dimerization of Atlastin.
GTP-dependent dimerization of dynamin and GBP1 establish the catalysis-competent states of these enzymes (4, 27). Several lines of evidence support the presence of a conserved dimerization-based GTPase mechanism for atlastin-1, in addition to the crystallographic observation that GDP-bound atlastin dimers are reminiscent of dimeric states of dynamin and GBP1. Similar to the latter cases, nucleotide-free (apo) and GDP-bound atlastin-1 (residues 1–446) elute in single peaks from a size-exclusion column with molecular weights determined by in-line, multiangle light scattering close to that calculated for monomers based on the primary sequence (49.2 or 49.3 kDa, respectively; theoretical molecular weight: 51.1 kDa) (Fig. 3 and Table S2). Incubation of the protein with the GTP analog GppNHp or the transition state analog GDP•AlFx promotes dimerization, indicated by an earlier elution volume and an average molecular weight of 99.2 kDa or 98.2 kDa, respectively.
Fig. 3.

Atlastin-1 oligomerization in solution. SEC-MALS data are shown for wild-type atlastin-1 (residues 1–446) in its apo-state (red) or bound to GppNHp (green), GDP (purple), or GDP•AlFx (orange). The signal from the 90°-scattering detector and refractive index detector are shown as solid, colored lines and black dashed lines, respectively (Left, Y axis). Average molecular weight as calculated every second across the protein elution peak is shown as black circles (Right, Y axis). Theoretical molecular weights based on primary sequence for the monomer and dimer are indicated as horizontal, dashed lines. Proteins (20 μM) were incubated with nucleotides (2 mM) prior to SEC-MALS analysis.
A nonconservative mutation in residue R77 (R77E), a residue located at the G domain dimer interface in the crystal structures of atlastin (Fig. S3C), renders the protein monomeric in the presence of GDP•AlFx, and only partially dimeric when bound to GppNHp, indicating that the crystallographic interface is also involved in the nucleotide-dependent dimerization of the protein in solution (Fig. S3E and Table S2). The mutant protein is also devoid of GTPase activity (Table S2), indicating that dimerization can have an impact on enzymatic activity.
Next, we turned to SAXS to gain further insight into the solution structure of dimeric atlastin. By using a monodisperse protein sample in diffraction experiments, accurate shape information in the form of the radius of gyration (Rg), the maximal diameter of the particle (Dmax), and the distance distribution function (P(r)) can be obtained (29, 30). The experimental scattering curves and P(r) functions for the cytoplasmic domain of atlastin-1 differ in the nucleotide-free, GDP-, GppNHp- and GDP•AlFx-bound states (Fig. 4A), indicating that the structures of these intermediates feature differences (a detailed analysis of the monomeric, apo-, and GDP-bound states is provided as SI Text). The dimeric, GppNHp-, and GDP•AlFx-bound states have overall similar P(r) profiles but vary slightly in their Dmax and Rg with the GDP•AlFx-bound specimen being more extended (Dmax/Rg: 120/36 Å vs. 125/37 Å for the GppNHp- and GDP•AlFx-bound states, respectively; Fig. 4A and Table S3).
Fig. 4.

Small-angle X-ray scattering. (A) Solution scattering data for atlastin-1. Intensity plots and P(r) functions for apo (red), GDP-bound (purple), GppNHp-bound (green), and GDP•AlFx-bound (orange) atlastin-1 (residues 1–446) are shown. (Left, Inset) Corresponding Guinier plots. (B) SAXS-based shape reconstructions for nucleotide-bound solution states of atlastin-1. The dimeric form 2 crystal structure was docked in the models as a reference and is shown as a cartoon presentation.
We compared the solution scattering data with the crystallographic dimer models (Fig. S4). Both solution states are more extended than the crystallographic form 1 dimer but shorter than the form 2 dimer (Fig. S4A and Table S3). The position and shape of the main peak in the theoretical P(r) functions calculated from the two crystallographic, GDP-bound dimers are very similar to the corresponding region of the experimental curves, consistent with common folds of the domains and similar G domain dimerization. Yet, in general the crystalline conformations represent inaccurate models for GTP-bound atlastin based on the overall fit (Table S3 and Fig. S4A).
Modeling of the SAXS data can yield low-resolution reconstructions representing the shape of a protein. A similar approach has been described recently for near-full-length dynamin revealing the overall organization of the protein in solution (7). Unbiased models for the solution states of GTP-bound and the transition state atlastin-1 were obtained by using a free-atom, simulated annealing approach with the respective P(r) functions as the target (31). For each state, 20 models were computed, averaged, and filtered to yield a consensus envelope. The shape reconstructions for atlastin-1 bound to GppNHp or GDP•AlFx are consistent with a central G domain dimer carrying lateral middle domains. Despite the inaccurate fits between the SAXS data and crystallographic models (Table S3), the overall conformation of the dimeric solution states has some resemblance with the extended conformation of the dimeric, form 2 crystal structure, in which the middle domains fold back onto their respective G domains and point in opposite directions (Fig. 4B). The G domain dimer fits the mass observed at the center of both models, placing the middle domains roughly into the skinnier density at the distal tips. Subtle differences in the SAXS-based models for atlastin-1 bound to GppNHp and GDP•AlFx concern these distal regions, extending radially from the center of the G domain dimer, but a more exact prediction of the structural changes is likely beyond the accuracy of the method.
In summary, the nucleotide-dependent oligomerization via central G domain dimers indicates related mechanisms between atlastin, GBP, dynamin, and other large G proteins that are activated by nucleotide-dependent dimerization (2). As described in the next section, further corroboration comes from several atlastin-1 mutants associated with the neurodegenerative disorder HSP, many of which display defects in GTP-dependent dimerization when introduced into the cytosolic domain, suggesting that disruption of this mechanism contributes to the pathogenesis in HSP.
Structure-Based Mapping and Characterization of HSP-Associated Mutations of Atlastin-1.
To date, 28 atlastin-1 missense mutants associated with cases of HSP have been identified within the construct studied here (24, 25). Within the primary sequence, the mutations are located in the second half of the G domain and the middle domain (Fig. S5A). By mapping the mutations onto the three-dimensional structures determined here, HSP-associated mutants cluster in the regions of the protein that vary most between the two crystal forms, including the putative dimer interface, the nucleotide-binding pocket, and the region of the intramolecular rigid body rotation (Fig. 2 and Fig. S5).
For a functional characterization, we introduced the HSP-associated, single-point mutations into the soluble, cytoplasmic domain of atlastin-1 used in this study. The majority of the mutants (16 out of 28) expressed and purified indistinguishably compared to the wild-type protein and elute from the gel filtration column as a single, monodisperse peak (Table S2). Those mutants that did not express in our system likely introduce significant folding defects (black spheres, Fig. S5). In contrast, the soluble mutants exhibit similar thermal melting temperatures that are comparable to that of the wild-type protein (Table S2 and Fig. S6C), indicating that folding defects, if any, are local with no apparent effect on protein stability.
Half of the soluble mutants exhibited dimerization behavior indistinguishable from the wild-type protein (Fig. S6A and Table S2). In contrast, several mutant proteins displayed defects in dimerization (Fig. 5 and Figs. S6A and S7). These include L157W, Q191R, Y196C, R217Q, H247R, Y336H, and M408V/T (orange and red residues, Fig S5). R217Q, the only mutation located in the nucleotide-binding site, displays the most drastic effect. R217 is the first residue in the RD motif and interacts with the nucleotide’s guanine base (Fig. 1B; red residue, Fig. S5) (32). The protein remains monomeric irrespective of the presence of nucleotide and is defective with regard to nucleotide binding and GTPase activity (Fig. 5 and Figs. S2 and S7), consistent with its direct role in nucleotide coordination. The oligomerization propensities of the mutants T157W, Q191R, Y196C, H247R, Y336H, and M408V/T are peculiar in that they exhibit dimer formation similar to wild-type protein upon binding of the transition state analog, but show a significant fraction of monomeric species upon incubation with GppNHp (Fig. 5 and Fig. S7). Although they show variable degrees of GTPase activity and no direct correlation between GppNHp-dependent dimer formation and activity, they coalesce at or close to a surface on the G domain that is part of the interdomain interfaces in both crystal forms (Fig. 5 and Fig. S5). Although the location of the mutations in the crystal structures and type of amino acid change do not allow for any obvious predictions regarding their effect on oligomerization or enzymatic activity, the defective dimerization upon GppNHp incubation and the spatial proximity suggest a common mode of action.
Fig. 5.

Characterization of HSP-associated mutations in atlastin-1. (A) Mapping of HSP mutant residues that affect dimerization (red and orange) onto the crystal structures of atlastin-1. Protomers are colored in light and dark gray. R217 (red) is located in the nucleotide-binding pocket. Dimer interface mutant R77 (blue) is not associated with HSP but was chosen based on the crystallographic dimer interface. (B) Nucleotide-dependent dimerization of HSP-associated atlastin-1 mutants. Molecular weight distributions for GppNHp- or AlFx-bound atlastin-1 variants (residues 1–446) were determined by static multiangle light scattering. Errors correspond to calculated errors in the fitting function parameters. (C) GTPase activity. GTPase activity was determined by measuring the production of inorganic phosphate upon GTP hydrolysis.
Taken together, the subset of atlastin-1 mutations with reduced dimerization propensity further support a dimer-dependent molecular mechanism for atlastin function and reveal a prominent mode of action for genetic alterations that contribute to HSP pathogenesis. Despite the measurable reduction in dimer formation upon incubation with GppNHp, the transition state dimers appear to be less affected by the mutations (with the exception of the nucleotide-binding-deficient mutant R217Q), which may explain why changes in GTPase activity are less pronounced. Alternatively, nucleotide binding and dimerization may not be necessarily interdependent and could potentially constitute distinct mechanisms underlying HSP. In addition, other mutants showed only modestly decreased GTPase activity and near wild-type dimerization (green spheres, Fig. S5; Fig. S6; Table S2), indicating that they confer defects in some other, yet unknown aspect.
Conclusions
In summary, the structural and functional analyses revealed a mechanistic conservation between atlastin and other dynamin superfamily proteins with regard to overall architecture, in particular the position of the respective stalk-like protrusions relative to their G domains, and nucleotide-dependent homodimerization. The apparent differences in the solution structures in different nucleotide-bound states, the distinct conformations observed in the two different crystal forms, and the unique behavior of several HSP-associated mutants lead one to speculate that the overall structure and function of atlastin is coupled to its GTP hydrolysis cycle. The indication that GppNHp and GDP•AlFx induce distinct conformational and functional states is analogous to both Gα subunits and small GTPases where these differences are attributed to the GTP-bound ground state and transition state for GTP hydrolysis (33, 34).
The antiparallel orientation of the middle domains in one of the crystal structures (form 2) and the elongated, dimeric solution states suggests that atlastin monomers from opposite membranes may engage in this oligomerization. GTP hydrolysis and/or the transition state may trigger a conformational change in atlastin, facilitating the formation of three-way junctions and membrane fusion (14, 15). The association with highly curved membranes and interactions with other proteins will further contribute to these processes (14, 20). Such interactions could trigger allosteric changes across the middle domain modulating atlastin’s GTPase activity, similar to the autoregulatory role of the PH domain in dynamin (7). Although we have not observed the second crystalline conformation (form 1) in solution, we speculate it may be relevant in the native environment of the protein and could represent an end state (and/or inhibited, GDP-bound state).
Many mutations occurring in HSP patients impair oligomerization of atlastin, nucleotide hydrolysis, or both. Given the minor impact of several mutants on protein stability, it is likely that they retain the ability to interact with some (if not all) binding partners in the cell. Mixed dimer formation and engagement in dysfunctional complexes may affect the wild-type alleles, providing a rationale for the dominant phenotype of the mutations. Although many mutations coalesce in the region between the nucleotide-binding site and the intramolecular domain interface, their exact pathological mechanism remains elusive. One hypothesis awaiting further analysis is that these alterations may perturb communication between the G domain and middle domain during the nucleotide-dependent, functional cycle.
Other mutations associated with HSP that occur in regions outside of the crystallized fragment would be predicted to affect atlastin’s stability in the membrane as well as its interactions with the reticulon and DP1 families (e.g., mutations near or in the transmembrane domains; Y459C, G469A, G482V, R495W) (14, 15). Furthermore, there are several mutations within the C-terminal domain of atlastin, including missense or nonsense mutations, that truncate the C terminus (A492fsX522, E502fsX522, I507fsX522, S519N), which would be expected to affect atlastin’s interactions with other binding partners, such as spastin, as has been shown previously for I507fsX522 (21).
Material and Methods
Protein Expression, Purification and Characterization.
The cytosolic domain of human atlastin-1 (residues 1–446) was produced following standard molecular biology and liquid chromatography techniques. Detailed descriptions of the protein stability and nucleotide-binding measurements are provided in SI Text.
Crystallization, X-Ray Data Collection, and Structure Solution.
Crystallization conditions and crystal morphologies are described in detail in SI Text. Datasets were collected using synchrotron radiation at the Cornell High Energy Synchrotron Source (CHESS, beamline A1). Data reduction, heavy atom searches, phasing, and refinement were carried out with the software packages HKL2000 (35), PHENIX (36), and COOT (37).
Small-Angle X-Ray Scattering.
SAXS data were collected at the CHESS (beamline G1) at an electron energy of 8 KeV. Data were collected at 20 °C on homogeneous, monodisperse samples. Data reduction, analysis, and free-atom modeling of the SAXS data were carried out by using the program package ATSAS (38). A more detailed description is provided in SI Text.
Size-Exclusion Chromatography-Coupled Multiangle Light Scattering (SEC-MALS).
Purified protein (∼1 μg/μL or 20 μM, injected concentration) was subjected to SEC using a WTC-030S5 column (Wyatt Technology) equilibrated in MALS buffer (25 mM Tris-HCl pH 7.5, 100 mM NaCl, 4 mM MgCl2, and 2 mM EGTA). Where specified, wild-type or mutant atlastin was incubated with GDP, GppNHp, or GDP•AlFx (2 mM) for 30 min at room temperature prior to injection. The SEC was coupled to a static 18-angle light scattering detector (DAWN HELEOS-II) and a refractive index detector (Optilab T-rEX) (Wyatt Technology). Data were collected every second at a flow rate of 1 mL/ min. Data analysis was carried out using the program ASTRA, yielding the molar mass and mass distribution (polydispersity) of the sample. Molecular weight distributions were determined by using the Multipeak Fitting Package in Igor Pro (WaveMetrics). For normalization of the light scattering detectors and data quality control, monomeric BSA (Sigma) was used.
GTPase Activity Assay.
GTPase activity was measured using the Enzchek Phosphate Assay kit (Molecular Probes) following the manufacturer’s instructions. Recombinant wild-type or mutant atlastin-1 was incubated in the reaction mixture at room temperature for 10 min, after which reactions were started by addition of 400 μM GTP and the production of inorganic phosphate was monitored by measuring the absorbance at 360 nm in a microplate reader. A detailed description of the assay is provided in SI Text.
Supplementary Material
Acknowledgments.
We are grateful to Richard Gillilan, Soeren Nielson, and Qi Wang for SAXS data collection and analysis and to Marcos Navarro for help with structure refinement. This work is based upon research conducted at the CHESS. The facility is supported by Grant DMR-0225180 from the National Science Foundation and Grant RR-01646 from the National Institutes of Health (NIH) (to CHESS). This work was supported by the NIH under Grant T32 GM008267 (to L.J.B.) and a PEW scholar award in Biomedical Sciences (to H.S.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3Q5D and 3Q5E).
See Commentary on page 2175.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1012792108/-/DCSupplemental.
References
- 1.Praefcke GJK, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 2.Gasper R, Meyer S, Gotthardt K, Sirajuddin M, Wittinghofer A. It takes two to tango: Regulation of G proteins by dimerization. Nat Rev Mol Cell Biol. 2009;10:423–429. doi: 10.1038/nrm2689. [DOI] [PubMed] [Google Scholar]
- 3.Prakash B, Praefcke GJ, Renault L, Wittinghofer A, Herrmann C. Structure of human guanylate-binding protein 1 representing a unique class of GTP-binding proteins. Nature. 2000;403:567–571. doi: 10.1038/35000617. [DOI] [PubMed] [Google Scholar]
- 4.Chappie JS, Acharya S, Leonard M, Schmid SL, Dyda F. G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature. 2010;465:435–440. doi: 10.1038/nature09032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mettlen M, Pucadyil T, Ramachandran R, Schmid SL. Dissecting dynamin’s role in clathrin-mediated endocytosis. Biochem Soc Trans. 2009;37:1022–1026. doi: 10.1042/BST0371022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang P, Hinshaw JE. Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol. 2001;3:922–926. doi: 10.1038/ncb1001-922. [DOI] [PubMed] [Google Scholar]
- 7.Kenniston JA, Lemmon MA. Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J. 2010;29:3054–3067. doi: 10.1038/emboj.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chappie JS, et al. An intramolecular signaling element that modulates dynamin function in vitro and in vivo. Mol Biol Cell. 2009;20:3561–3571. doi: 10.1091/mbc.E09-04-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bashkirov PV, et al. GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell. 2008;135:1276–1286. doi: 10.1016/j.cell.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tuma PL, Collins CA. Activation of dynamin GTPase is a result of positive cooperativity. J Biol Chem. 1994;269:30842–30847. [PubMed] [Google Scholar]
- 11.Warnock DE, Hinshaw JE, Schmid SL. Dynamin self-assembly stimulates its GTPase activity. J Biol Chem. 1996;271:22310–22314. doi: 10.1074/jbc.271.37.22310. [DOI] [PubMed] [Google Scholar]
- 12.Gao S, et al. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature. 2010;465:502–506. doi: 10.1038/nature08972. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 14.Hu J, et al. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orso G, et al. Homotypic fusion of ER membranes requires the dynamin-like GTPase Atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 16.Namekawa M, et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis. Mol Cell Neurosci. 2007;35:1–13. doi: 10.1016/j.mcn.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Muriel M, et al. Atlastin-1, the dynamin-like GTPase responsible for spastic paraplegia SPG3A, remodels lipid membranes and may form tubules and vesicles in the endoplasmic reticulum. J Neurochem. 2009;110:1607–1616. doi: 10.1111/j.1471-4159.2009.06258.x. [DOI] [PubMed] [Google Scholar]
- 18.Rismanchi N, Soderblom C, Stadler J, Zhu P-P, Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu P-P, et al. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 20.Park SH, Zhu P-P, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evans K, et al. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc Natl Acad Sci USA. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanderson CM, et al. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum Mol Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 24.Smith BN, et al. Four novel SPG3A/atlastin mutations identified in autosomal dominant hereditary spastic paraplegia kindreds with intra-familial variability in age of onset and complex phenotype. Clin Genet. 2009;75:485–489. doi: 10.1111/j.1399-0004.2009.01184.x. [DOI] [PubMed] [Google Scholar]
- 25.McCorquodale D, III, et al. Mutation screening of spastin, atlastin, and REEP1 in hereditary spastic paraplegia. Clin Genet. 2010 doi: 10.1111/j.1399-0004.2010.01501.x. 10.1111/j.1399-0004.2010.01501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu P-P, Soderblom C, Tao-Cheng J-H, Stadler J, Blackstone C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum Mol Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh A, Praefcke GJK, Renault L, Wittinghofer A, Herrmann C. How guanylate-binding proteins achieve assembly-stimulated processive cleavage of GTP to GMP. Nature. 2006;440:101–104. doi: 10.1038/nature04510. [DOI] [PubMed] [Google Scholar]
- 28.Low HH, Lowe J. A bacterial dynamin-like protein. Nature. 2006;444:766–769. doi: 10.1038/nature05312. [DOI] [PubMed] [Google Scholar]
- 29.Putnam CD, Hammel M, Hura GL, Tainer JA. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q Rev Biophys. 2007;40:191–285. doi: 10.1017/S0033583507004635. [DOI] [PubMed] [Google Scholar]
- 30.Koch MH, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Q Rev Biophys. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- 31.Svergun DI, Petoukhov MV, Koch MHJ. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Praefcke GJK, et al. Identification of residues in the human guanylate-binding protein 1 critical for nucleotide binding and cooperative GTP hydrolysis. J Mol Biol. 2004;344:257–269. doi: 10.1016/j.jmb.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 33.Sprang SR. Conformational display: A role for switch polymorphism in the superfamily of regulatory GTPases. Sci STKE. 2000;2000((50)) doi: 10.1126/stke.2000.50.pe1. pe1. [DOI] [PubMed] [Google Scholar]
- 34.Oldham WM, Hamm HE. Structural basis of function in heterotrimeric G proteins. Q Rev Biophys. 2006;39:117–166. doi: 10.1017/S0033583506004306. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Petoukhov MV, Konarev PV, Kikhney AG, Svergun DI. ATSAS 2.1—Towards automated and web-supported small-angle scattering data analysis. J Appl Crystallogr. 2007;40 doi: 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.