

Abstract
Field measurements of secondary organic aerosol (SOA) find significantly higher mass loads than predicted by models, sparking intense effort focused on finding additional SOA sources but leaving the fundamental assumptions used by models unchallenged. Current air-quality models use absorptive partitioning theory assuming SOA particles are liquid droplets, forming instantaneous reversible equilibrium with gas phase. Further, they ignore the effects of adsorption of spectator organic species during SOA formation on SOA properties and fate. Using accurate and highly sensitive experimental approach for studying evaporation kinetics of size-selected single SOA particles, we characterized room-temperature evaporation kinetics of laboratory-generated α-pinene SOA and ambient atmospheric SOA. We found that even when gas phase organics are removed, it takes ∼24 h for pure α-pinene SOA particles to evaporate 75% of their mass, which is in sharp contrast to the ∼10 min time scale predicted by current kinetic models. Adsorption of “spectator” organic vapors during SOA formation, and aging of these coated SOA particles, dramatically reduced the evaporation rate, and in some cases nearly stopped it. Ambient SOA was found to exhibit evaporation behavior very similar to that of laboratory-generated coated and aged SOA. For all cases studied in this work, SOA evaporation behavior is nearly size-independent and does not follow the evaporation kinetics of liquid droplets, in sharp contrast with model assumptions. The findings about SOA phase, evaporation rates, and the importance of spectator gases and aging all indicate that there is need to reformulate the way SOA formation and evaporation are treated by models.
Keywords: single-particle mass spectrometry, morphology
Atmospheric particles have a strong, yet poorly characterized effect on climate (1). Organic aerosols (OA) comprise 20–90% of atmospheric dry particles mass (2), the majority and least understood of which is secondary organic aerosol (SOA), formed from oxidation of gas phase organic vapors in the atmosphere (3–6). Despite an ongoing intense research effort aimed at understanding the formation and atmospheric evolution of OA, current models severely underestimate the formation of SOA in the atmosphere (5, 7). The effort to resolve the persistent discrepancy between field measurements and the amount of SOA predicted by atmospheric chemistry models has mostly focused on improving the understanding of SOA formation yields and finding new sources (8–11). In contrast, present models maintain the following fundamental assumptions: (i) Gas-particle partitioning is modeled assuming that all organics form a pseudoideal solution in the condensed particle phase, (ii) SOA particles remain liquid-like throughout their lifetime in the atmosphere, (iii) reversible thermodynamic equilibrium exists between gas and particle phases, and (iv) adsorption of other organic species and their effects on SOA properties and evaporation are ignored.
The assumption that particles are liquid is central to all modeling studies and affects calculations of condensation and evaporation of particles in the atmosphere. Properties and behavior of multicomponent liquids are clearly very different from multicomponent solids. Seven orders of magnitude lower diffusion rates within a solid phase limit most gas–particle interactions to surface layer for solid particles (12). As mixed solid particle evaporation proceeds, the solid surfaces change resulting in dynamic changes in evaporation rates.
As we show below, the evaporation kinetics of laboratory-generated and ambient SOA indicate that these particles are not liquid and do not behave as expected by the models. These findings are in accord with a recent report, in which SOA particle bouncing behavior was used to conclude that SOA particles are amorphous solids (13).
In their box modeling study of the Mexico City plume, Dzepina et al. (6) found that adding precursors and SOA formation mechanisms (9) increases modeled SOA mass, bringing model predictions closer to measurements. However, when evaporation is considered, their models predict that more than 3/4 of the SOA mass evaporates upon dilution by a factor of 10, in sharp contrast with measurements. The same study attempted to model measured SOA evaporation in thermodenuder (TD) and concluded that to get better agreement with field measurements requires SOA evaporation coefficients that are 1–3 orders of magnitude below unity (6), which leads to inconsistency with smog chamber SOA formation measurements.
Nearly all SOA evaporation studies rely on TD, which involve measurements on particles exposed for a short time to elevated temperatures. Using TD, Huffman et al. (14) observed that laboratory-generated SOA is more volatile than ambient SOA. Most recent analysis of field TD datasets concludes that a significant fraction of the atmospheric OA consist of nonvolatile components (15). Although TD produces valuable data, their interpretation is challenging. There are issues related to nonlinear coupling effects between various parameters such as initial aerosol compositions, enthalpies of vaporization, volatility distributions, and evaporation coefficients governing particle evaporation rates (16). Most importantly, if the SOA particles are not liquid droplets, increasing particle temperature may result in phase change and with it a change in evaporation behavior.
Grieshop et al. (17) studied the kinetics of dilution-induced evaporation of polydisperse SOA in a smog chamber at room temperature and concluded that the system returns to equilibrium by evaporation within ∼2.5 h. They attributed this “surprisingly slow” evaporation rate to the formation of low-volatility oligomers (17). However, comparison between the evaporation kinetics observed in Grieshop et al. (17) and in our laboratory indicates that 2.5 h is most likely the point at which the evaporation rate slows down and not where it ends. It has also been suggested that this dilution-induced evaporation type of experiment could be strongly affected by material deposited on smog chamber walls (18).
In this paper, we present results from a comprehensive investigation of SOA evaporation kinetics using a newly developed, highly sensitive experimental setup that allowed us to conduct a number of measurements that were not made before. We (i) characterized the evaporation of size-selected SOA particles at room temperature, (ii) monitored SOA evaporation for over 24 h, (iii) investigated the effect of hydrophobic “spectator” organic vapors on SOA evaporation, (iv) quantified the effect of aging on the evaporation kinetics of pure and coated SOA particles, and (v) studied the evaporation kinetics of size and chemically resolved ambient SOA particles at room temperature.
Results and Discussion
The schematic of the experimental setup is shown in Fig. 1. Laboratory SOA particles were generated in a Teflon bag by the reaction of α-pinene with ozone. Once reaction was complete, they were classified using a differential mobility analyzer (DMA), and the resulting monodisperse aerosol was transported through two room-temperature activated charcoal denuders and loaded into an evaporation chamber containing activated charcoal. The fact that particles were generated in a separate chamber and that gas phase organics in the evaporation chamber were continuously removed eliminates the issue of gas–wall repartitioning. Low particle number–concentrations in the chamber (∼10–200 particles cm-3) provide the means to follow kinetics of single-particle evaporation. SOA evaporation kinetics were monitored for over a ∼24-h period by periodically measuring changes in particle vacuum aerodynamic diameter, dva, distributions, and mass spectra using our single-particle mass spectrometer, SPLAT II (19). In experiments involving spectator gases, small amounts of dioctyl phthalate (DOP), dioctyl sebacate (DOS), and pyrene (PY), or a mixture of organics were placed at the bottom of the Teflon bag and allowed to equilibrate before reaction started. As evident by the particle mass spectra, SOA formed under these conditions acquire a coating of the spectator organic compound (20). Ambient particles were dried and then treated in the same manner as the laboratory particles.
Fig. 1.

Schematic of the experimental setup.
Evaporation Kinetics of Single-Component Test Particles.
We first present, in Fig. 2, results of evaporation studies for single-component particles composed of DOP. Fig. 2A shows how the dva of DOP particles with initial diameter d0 = 500 nm decreases as a function of time as the particles evaporate. Fig. 2B shows how particle diameters, d, decrease during evaporation in a plot of 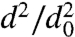 vs. time. The data agree with traditional models of single-component liquid droplet evaporation, which predict that the square of the particle diameter decreases nearly linearly with time (21) and that the slope of
vs. time. The data agree with traditional models of single-component liquid droplet evaporation, which predict that the square of the particle diameter decreases nearly linearly with time (21) and that the slope of 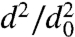 vs. time is size-dependent; that is, smaller particles evaporate faster. This size-dependent evaporation behavior also quantitatively explains the observed increase in the width of the dva distributions as the particles evaporate. DOP particles on the left edge of the size distribution (slightly smaller) evaporate faster than the larger particles on the right edge of the distribution. Applying the analysis of Zhang et al. (22) to these data, we obtain, from the slope of
vs. time is size-dependent; that is, smaller particles evaporate faster. This size-dependent evaporation behavior also quantitatively explains the observed increase in the width of the dva distributions as the particles evaporate. DOP particles on the left edge of the size distribution (slightly smaller) evaporate faster than the larger particles on the right edge of the distribution. Applying the analysis of Zhang et al. (22) to these data, we obtain, from the slope of 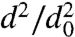 vs. time, a vapor pressure of 10-7 Torr, in excellent agreement with previously reported values (23). These measurements validate the experimental methodology and introduce the time scales expected for particle evaporation: 300-nm DOP particles, whose vapor pressure is 10-7 Torr, lose more than 70% of their volume by evaporation in 1 h, whereas 700-nm DOP particles lose less than 20% of their volume over the same time.
vs. time, a vapor pressure of 10-7 Torr, in excellent agreement with previously reported values (23). These measurements validate the experimental methodology and introduce the time scales expected for particle evaporation: 300-nm DOP particles, whose vapor pressure is 10-7 Torr, lose more than 70% of their volume by evaporation in 1 h, whereas 700-nm DOP particles lose less than 20% of their volume over the same time.
Fig. 2.

Evaporation kinetics of pure DOP particles. (A) dva distributions of 500-nm DOP particles at the marked evaporation times (in minutes), showing how particle size decreases and linewidths increase. (B) Evaporation kinetics (reported as 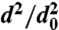 vs. time) for DOP particles with different initial diameters indicated in the legend.
vs. time) for DOP particles with different initial diameters indicated in the legend.
Evaporation Kinetics of Pure Laboratory SOA.
We proceed to investigate the evaporation kinetics of α-pinene SOA particles. The results of these measurements are summarized in Fig. 3. Fig. 3A shows how the dva of SOA particles with d0 = 160 nm, i.e., dva0 = 187 nm, decreases during evaporation. After comparison with Fig. 2, two observations are notable. First, the diameters of SOA particles decrease with time significantly slower, and second, the dva distributions remain narrow during SOA particles evaporation. Fig. 3B shows 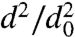 as a function of evaporation time for SOA particles with different initial diameters. Evidently, SOA particles undergo evaporation in two distinct stages. In the fast evaporation stage particles lose ∼50% of their volume in less than 100 min, whereas in the slow stage, it takes an additional 1,400 min to lose another ∼25% of their initial volume. Moreover, Fig. 3B shows that the evaporation kinetics of SOA particles is nearly size-independent.
as a function of evaporation time for SOA particles with different initial diameters. Evidently, SOA particles undergo evaporation in two distinct stages. In the fast evaporation stage particles lose ∼50% of their volume in less than 100 min, whereas in the slow stage, it takes an additional 1,400 min to lose another ∼25% of their initial volume. Moreover, Fig. 3B shows that the evaporation kinetics of SOA particles is nearly size-independent.
Fig. 3.

Evaporation kinetics of α-pinene SOA particles. (A) dva distributions of 160-nm SOA particles at the marked evaporation times (in minutes) during the evaporation process, showing how during evaporation particle sizes decrease and linewidths remain narrow. (B) Evaporation kinetics (reported as 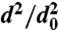 vs. time) for SOA particles with different initial diameters as indicated in the legend. (Inset) The fast evaporation stage in expanded scale. (C) Comparison between calculated and observed SOA evaporation behavior (reported as remaining volume fraction vs. time). See text for the details.
vs. time) for SOA particles with different initial diameters as indicated in the legend. (Inset) The fast evaporation stage in expanded scale. (C) Comparison between calculated and observed SOA evaporation behavior (reported as remaining volume fraction vs. time). See text for the details.
Fig. 3C presents a comparison between modeled and observed SOA evaporation behavior, plotted here and in the rest of the figures as 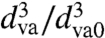 , i.e., the remaining volume fraction, as a function of time. Calculations for three particle sizes (100, 160, and 251 nm) are performed using a detailed kinetic model that employs the seven-product volatility basis sets developed to fit SOA formation data during α-pinene ozonolysis (24), and using two different mass accommodation coefficients (α) of 1.0 and 0.05 (16). Note that although α = 1 is consistent with observed SOA formation rates, α = 0.05 leads to SOA formation rates that are inconsistent with smog chamber measurements. Nevertheless, we use both values to illustrate that even unreasonably low α do not yield agreement with experiment. There are two important differences between modeled and observed evaporation kinetics. First, calculated evaporation rates are much faster than observed. For example, according to the model, 151-nm particles will lose 75% of their volume in 1 to 20 min of evaporation, depending on the mass accommodation coefficient, whereas the data show that in reality it takes over 24 h, independent of particle size. Second, calculated SOA evaporation behavior shows the expected size dependence, with larger particles evaporating significantly slower. In contrast, the observed data are nearly size-independent. It should be noted that all models based on fits to smog chamber data yield similar fast evaporation rates and size-dependent behavior. The simple fact that SOA evaporation does not follow the size-dependent evaporation behavior expected of liquid droplets indicates that these particles are not liquid-like.
, i.e., the remaining volume fraction, as a function of time. Calculations for three particle sizes (100, 160, and 251 nm) are performed using a detailed kinetic model that employs the seven-product volatility basis sets developed to fit SOA formation data during α-pinene ozonolysis (24), and using two different mass accommodation coefficients (α) of 1.0 and 0.05 (16). Note that although α = 1 is consistent with observed SOA formation rates, α = 0.05 leads to SOA formation rates that are inconsistent with smog chamber measurements. Nevertheless, we use both values to illustrate that even unreasonably low α do not yield agreement with experiment. There are two important differences between modeled and observed evaporation kinetics. First, calculated evaporation rates are much faster than observed. For example, according to the model, 151-nm particles will lose 75% of their volume in 1 to 20 min of evaporation, depending on the mass accommodation coefficient, whereas the data show that in reality it takes over 24 h, independent of particle size. Second, calculated SOA evaporation behavior shows the expected size dependence, with larger particles evaporating significantly slower. In contrast, the observed data are nearly size-independent. It should be noted that all models based on fits to smog chamber data yield similar fast evaporation rates and size-dependent behavior. The simple fact that SOA evaporation does not follow the size-dependent evaporation behavior expected of liquid droplets indicates that these particles are not liquid-like.
Evaporation Kinetics of Coated Laboratory SOA.
In the real atmosphere SOA particles form in the presence of a mixture of organic vapors, some of which are hydrophobic. We have previously demonstrated that 158-nm SOA particles nucleated in the presence of DOP vapor acquire by adsorption a ∼4-nm DOP coating (20). Because it has been shown that organic coatings on inorganic particles decrease/inhibit their evaporation rates (25, 26), we hypothesized that the evaporation kinetics of SOA particles coated by adsorbed hydrophobic organics could be similarly affected.
To investigate this hypothesis we studied the evaporation of SOA particles formed in the presence of the vapors of a number of hydrophobic organics: DOP and DOS, both of which are liquids; PY, a solid polyaromatic hydrocarbon (PAH); and the mixture of these three organics plus benzo[ghi]perylene, another solid PAH. Similar to our previous observations with DOP (20), in all four cases SOA particles that were formed in the presence of the vapors of hydrophobic organics adsorbed noticeable amounts of these organics, forming what we term here “coated” SOA particles.
The evaporation kinetics measurements of pure and coated SOA particles are presented in Fig. 4. In all cases, evaporation rates are nearly size-independent and evaporation appears to proceed in two stages: fast and slow. All lines in Fig. 4 represent fits to the evaporation data using biexponential functions (included in SI Text). DOP or DOS-coated particles lose only ∼30% of the particle volume during fast evaporation, and their slow evaporation parallels that of pure SOA particles. PY-coated SOA particles lose only ∼20% of the particle volume during the fast stage and show 10 times slower rate during slow evaporation stage. The evaporation of SOA particles formed in the presence of mixed organics (not shown here) is between that of DOP and PY-coated SOA. Importantly, the particles composed of pure coating materials (DOP, DOS, or PY) exhibit drastically higher evaporation rates.
Fig. 4.

Evaporation kinetics (reported as remaining volume fraction vs. time) for a number of the particle types indicated in the legend. All lines represent biexponential fits to the data. For each particle type, a single line is used to fit different sizes, and a single line is used to fit the DOP- and DOS-coated particles. Dashed lines represent evaporation of aged particles.
We next examine the effect of aging on the evaporation behavior, as SOA properties have been shown to evolve over time (27). Aged SOA particles, both coated and uncoated, were generated by allowing the particles to remain in the Teflon bag in the presence of ozone for ∼10–15 h before performing the evaporation experiments. As Fig. 4 shows in a set of dashed lines, for all cases aging decreases the fast evaporating fraction, while leaving the slow evaporation rate unchanged. These data also indicate that aging has the most pronounced effect on the evaporation of coated particles. After more than 24 h of evaporation, aged PY-coated SOA particles lose only ∼11% of their volume or ∼4% of their diameter. These results are interesting because the aging process involves only continued reaction with excess ozone that, in the gas phase, should affect the SOA material, but not the coatings. However, PY adsorbed to silica particles has been shown to undergo heterogeneous reaction with ozone (28), which may point to the importance of surface chemistry in determining the volatility of atmospheric particles. The observed reduction in SOA evaporation rate by the presence of hydrophobic organic vapors and aging may explain the reasons that laboratory-generated pure SOA is more volatile than SOA characterized in field studies (14).
Further insight may be obtained from the evolution of the particles’ mass spectra with evaporation. They indicate only minor changes for m/z smaller than 200 amu, disappearance of the peak at m/z = 201 after 2 min of evaporation, and, at later times, slow increase of intensity of peaks with m/z higher than 202, which could indicate oligomer formation (29). However, we note that aging alone produces no observable changes in particle mass spectra. The changes in mass-spectral intensities with evaporation time are illustrated in Fig. S1. The intensity of the DOP-characteristic peak (C6H4(CO)2OH+, m/z = 149) in DOP-coated SOA particles, rapidly drops with evaporation time, and then remains constant. The PY parent ion peak (m/z = 202), in PY-coated SOA, decreases slowly, such that ∼50% of it remains even after 25 h of evaporation. The comparison between PY and DOP suggests that there are differences in morphological distribution of solid PY and liquid DOP in the coated SOA.
Evaporation Kinetics of Atmospheric SOA.
To address the question of how closely the laboratory observations described above reflect reality in the atmosphere, we characterized the evaporation kinetics of atmospheric SOA particles sampled in situ during the recent Carbonaceous Aerosols and Radiative Effects Study (CARES) field campaign in Sacramento, CA, in June 2010. Because in the real atmosphere aerosol compositions change throughout the day, the first task was to identify the point at which SOA dominates the particle compositions. This was accomplished by following the diurnal evolution of particle number concentrations, size distributions, compositions, and densities. Early in the morning aerosol loadings and number concentrations were low, and most particles were composed of organics mixed with a significant fraction of sulfate, with larger particles containing a larger fraction of sulfate (30). By ∼9:00 AM, the number concentrations of very small particles (< 14 nm) started to increase, indicating the beginning of SOA nucleation events. As the day progressed and emissions of the volatile organic compounds were processed, SOA particles increased in size, making it possible to characterize their size, composition, and density with SPLAT II. By early afternoon aerosol composition was dominated by oxygenated organics mixed with a small amount of organic amines and sulfate (∼12% volume fraction), and the density of both 100-nm and 151-nm particles was measured to be 1.32 ± 0.02 g cm-3. At this point size-selected 100- and 151-nm particles were loaded into the evaporation chamber and their evaporation behavior was studied.
Fig. 5 shows the changes in the measured dva distributions of 100-nm ambient particles measured on June 26 over 208 min of evaporation. At t = 0 the particles average dva is 133 nm, which yields a density of 1.33 g cm-3 (see SI Text). With time in the chamber, the particles evaporate and after 208 min they shrink to 125 nm, i.e., lose 17% of their volume. Note that as the organic fraction of the particle evaporates, its sulfate fraction, and hence density, slightly increases, which when taken into account means that the actual evaporated volume of organics is 20%. Fig. 5B shows the remaining volume fraction for 100-nm and 151-nm particles as a function of time. Notably, the evaporation behavior of the atmospheric SOA is similar to that of laboratory-generated coated SOA. It is slow, size-independent, and can be fit with a biexponential curve that represents the fast and slow evaporation phases (see SI Text). Here, only a small fraction of the volume (0.135) is lost during the fast evaporation phase.
Fig. 5.

Evaporation kinetics of ambient particles. (A) dva distributions of 100-nm ambient particles at the marked evaporation times (in minutes). (B) Evaporation kinetics (reported as remaining volume fraction) for ambient 100-nm and 151-nm particles. The solid line is a biexponential fit to the data.
Although it is true that the vast majority of atmospheric particles were SOA, it is worth keeping in mind that the data in Fig. 5 are an average of all sampled particle types. It is possible to refine the analysis by classifying the data according to particle compositions and examine the evaporation of the individual classes. The mass spectra show that in addition to SOA-dominated particles, a small fraction of biomass burning and soot particles were also present, but their small numbers prevent us from analyzing their behavior in the evaporation chamber.
Two distinct types of SOA particles mixed with sulfate were observed on June 25, 26, and 27. Both types, labeled Type 44 and Type 43 to denote which of these two mass-spectral peaks has higher intensity prior to particle evaporation (Fig. S2), are dominated by oxygenated organics, have identical density, and are mixed with the same amount of sulfate (∼12%). Type 43 particles contain in addition to oxygenated organics and sulfate a small amount of organic amines.
Fig. 6 shows the evaporation data acquired during the four-day study. Comparison between the evaporation data of 100-nm and 151-nm particles of both types shows very small differences. We note again that like laboratory SOA, evaporation of ambient SOA is nearly size-independent and conclude that, even when mixed with sulfate, ambient SOA particles do not behave like liquid droplets.
Fig. 6.

Evaporation kinetics of 100-nm and 151 ambient SOA particles classified into two particle types (43 and 44). The solid lines are biexponential fits to the data and the gray shaded area marks the upper and lower limits of all observed data.
Aerosol loadings for June 28 were a factor of two higher and present a slightly different scenario. Because on this day the SOA particles mass spectra show continuous range of compositions spanning between the two particle types, they were treated as one class. Because the number of 100-nm particles in the evaporation chamber was insufficient to yield usable data, we present for this day data for 151-nm particles only.
In addition to the data points, Fig. 6 displays a gray-shaded region that marks the area between the fastest and slowest evaporation rates observed in this study, and biexponential fits to the data points given in SI Text. Note that there is no reason to expect the evaporation curves on different days to be identical. Variation in temperature, solar flux, cloud cover, wind speed, and wind direction are expected to produce particles with somewhat different compositions and hence evaporation rates. Nevertheless, a comparison of the data from these four days points to a remarkably similar evaporation behavior.
Examination of changes in mass-spectral intensities of ambient SOA particles with evaporation time show that changes are relatively small, and that unlike in the case of laboratory SOA, no increase in the mass-spectral intensity at high m/z is observed. In addition, the intensity of peaks 44 and 73, for both particle types, decrease rapidly with evaporation and then remain constant, which could indicate that some of their intensity belongs to surface coatings (shown in Fig. S3).
Fig. 7 shows a comparison between the results for laboratory-generated SOA shown in Fig. 4, the measured evaporation kinetics of ambient atmospheric SOA particles as presented by the gray-shaded region, and the calculated evaporation kinetics presented in Fig. 3C. Based on Fig. 7 we conclude that the evaporation of atmospheric SOA is very similar to that of laboratory-generated α-pinene SOA coated either with PY, organic mixture, or aged SOA coated with DOP or DOS. This underscores the importance of the laboratory investigations of the coated SOA particles and suggests that the coated SOA is in a sense similar to real-world atmospheric SOA. The differences between the observed evaporation behaviors and that predicted by models are rather extreme. Even when using unreasonable mass accommodation of 0.05, models predict very fast evaporation, whereas the data indicate that evaporation is slow. Assuming liquid-like SOA behavior, models predict that evaporation of small particles is faster than that of larger ones, but the data clearly show that evaporation is nearly size-independent indicating that these particles cannot be liquids.
Fig. 7.

Comparison between the observed evaporation kinetics of ambient organic particles (gray area), the laboratory-generated α-pinene SOA particles with and without various coatings and aging, and evaporation kinetics calculated based on current models.
Conclusions
We summarize our findings as follows: (i) Overall, SOA evaporation is more than ∼100 times slower than expected by current models. It proceeds in two stages: a fast stage that takes ∼100 min, followed by very slow evaporation that lasts more than a day. (ii) SOA evaporation is nearly size-independent and does not follow the evaporative behavior of liquid droplets, indicating that SOA particles are not liquids. This conclusion is consistent with the results of our previous study that showed that particles with two reverse morphologies—DOP-coated SOA particles and SOA-coated DOP particles—remain stable for many hours, which would be incompatible with the rapid diffusion rates expected if both DOP and SOA are liquids (20). (iii) SOA particles formed in the presence of vapors of hydrophobic organic compounds exhibit lower evaporation rates. (iv) Aging decreases evaporation rates in all cases; however, its effect on coated particles is significantly more pronounced. (v) The evaporation of ambient SOA is similar to that of coated and aged laboratory-generated α-pinene SOA—it is slow and size-independent. (vi) Changes in mass-spectral intensity with evaporation suggest that ambient SOA acquires coatings, much like laboratory SOA.
Because in our experiments evaporation is induced by sudden and complete removal of the vapor phase, it is expected to be significantly faster than in the atmosphere, where the vapor phase concentrations decrease in response to dilution and chemical processing, both of which occur on a time scale of a few hours. Moreover, in the atmosphere, where a large number of organic vapors are present, as some coatings evaporate, others are adsorbed to form new coatings.
These observations have important implications to the way we view and model the formation and fate of SOA particles. The data show that the assumptions of liquid-like SOA behavior and instantaneous reversible equilibrium gas–particle partitioning are seriously flawed. They show that adsorption of organic vapors, present at pressures equal to or lower than their vapor pressure, increases organic mass and inhibits evaporation, an effect that is enhanced with particles aging. In contrast with current air-quality models, in which SOA particles rapidly evaporate due to dilution, our findings indicate that evaporation due to transport to cleaner environments is almost negligible. This has the effect of increasing the contribution of SOA to organic aerosol in both urban and remote locations. Moreover, the observed size-independent evaporation behavior implies that, in contrast with models, small (∼100 nm) particles do not evaporate faster and thus retain their potential to serve as cloud condensation nuclei. Because SOA formation is modeled on the assumptions that these particles are liquids and that spectator gases play no role, the findings presented here have implications for this aspect of SOA modeling as well. This work could provide an essential missing link to process representation of organics aerosols in models.
Materials and Methods
Particle Generation.
DOP and DOS particles were generated by atomization of the neat liquids. Homogeneous nucleation of SOA was initiated by injecting 200 ppb of α-pinene, ∼500 ppb of O3, and ∼250 ppm of cyclohexane, used as an OH scavenger, into a clean 100-L Teflon bag filled with zero air. Coated particles were generated by carrying out the SOA generation procedure in a Teflon bag that contained a small amount of bulk coating material. Ambient particles were sampled in situ, dried, and characterized using SPLAT II and a DMA, as described elsewhere (31).
Particle Analysis.
Particles were size-selected with a DMA, passed through two charcoal denuders, and loaded in the stainless steel evaporation chamber, partially filled with activated charcoal.
A single-particle mass spectrometer, SPLAT II, was used to measure individual particle composition, vacuum aerodynamic diameter, dva, density, and shape , as described elsewhere (19, 32). Particle evaporation kinetics was quantified by measuring changes in particle dva, with 0.5% precision. A more detailed description of the experimental setup is provided in SI Text.
Supplementary Material
Acknowledgments.
The authors thank Prof. Paul Ziemann, Prof. Pete McMurry, and Dr. Chen Song for helpful discussions. Special thanks to Mr. Mike Ezell for his help with manufacturing Teflon bags and Dr. Ilona Riipinen and Dr. Richard Easter for initial help with model configuration. This work was supported by the US Department of Energy Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences and Office of Biological and Environmental Research. This research was performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research at Pacific Northwest National Laboratory (PNNL). PNNL is operated by the US Department of Energy by Battelle Memorial Institute under Contract DE-AC0676RL0 1830.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013391108/-/DCSupplemental.
References
- 1.Houghton JT, et al. Contribution of working group I to the Third Assessment Report of the Intergovernmental Panel on Climate Change; Climate Change 2001: The Scientific Basis; Cambridge, UK: Cambridge Univ Press; 2001. [Google Scholar]
- 2.Kanakidou M, et al. Organic aerosol and global climate modelling: A review. Atmos Chem Phys. 2005;5:1053–1123. [Google Scholar]
- 3.Kroll JH, Seinfeld JH. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos Environ. 2008;42:3593–3624. [Google Scholar]
- 4.Zhang Q, et al. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys Res Lett. 2007;34:L13801. [Google Scholar]
- 5.de Gouw J, Jimenez JL. Organic aerosols in the Earth’s atmosphere. Environ Sci Technol. 2009;43:7614–7618. doi: 10.1021/es9006004. [DOI] [PubMed] [Google Scholar]
- 6.Dzepina K, et al. Evaluation of recently-proposed secondary organic aerosol models for a case study in Mexico City. Atmos Chem Phys. 2009;9:5681–5709. [Google Scholar]
- 7.de Gouw JA, et al. Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002. J Geophys Res. 2005;110:D16305. [Google Scholar]
- 8.Odum JR, et al. Gas/particle partitioning and secondary organic aerosol yields. Environ Sci Technol. 1996;30:2580–2585. [Google Scholar]
- 9.Robinson AL, et al. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science. 2007;315:1259–1262. doi: 10.1126/science.1133061. [DOI] [PubMed] [Google Scholar]
- 10.Pye HOT, Seinfeld JH. A global perspective on aerosol from low-volatility organic compounds. Atmos Chem Phys. 2010;10:4377–4401. [Google Scholar]
- 11.Donahue NM, Robinson AL, Stanier CO, Pandis SN. Coupled partitioning, dilution, and chemical aging of semivolatile organics. Environ Sci Technol. 2006;40:2635–2643. doi: 10.1021/es052297c. [DOI] [PubMed] [Google Scholar]
- 12.Ravishankara AR. Heterogeneous and multiphase chemistry in the troposphere. Science. 1997;276:1058–1065. [Google Scholar]
- 13.Virtanen A, et al. An amorphous solid state of biogenic secondary organic aerosol particles. Nature. 2010;467:824–827. doi: 10.1038/nature09455. [DOI] [PubMed] [Google Scholar]
- 14.Huffman JA, et al. Chemically-resolved volatility measurements of organic aerosol from different sources. Environ Sci Technol. 2009;43:5351–5357. doi: 10.1021/es803539d. [DOI] [PubMed] [Google Scholar]
- 15.Cappa CD, Jimenez JL. Quantitative estimates of the volatility of ambient organic aerosol. Atmos Chem Phys. 2010;10:5409–5424. [Google Scholar]
- 16.Riipinen I, Pierce JR, Donahue NM, Pandis SN. Equilibration time scales of organic aerosol inside thermodenuders: Evaporation kinetics versus thermodynamics. Atmos Environ. 2010;44:597–607. [Google Scholar]
- 17.Grieshop AP, Donahue NM, Robinson AL. Is the gas-particle partitioning in alpha-pinene secondary organic aerosol reversible? Geophys Res Lett. 2007;34:L14810. [Google Scholar]
- 18.Matsunaga A, Ziemann PJ. Gas-wall partitioning of organic compounds in a Teflon film chamber and potential effects on reaction product and aerosol yield measurements. Aerosol Sci Tech. 2010;44:881–892. [Google Scholar]
- 19.Zelenyuk A, Yang J, Choi E, Imre D. SPLAT II: An aircraft compatible, ultra-sensitive, high precision instrument for in-situ characterization of the size and composition of fine and ultrafine particles. Aerosol Sci Tech. 2009;43:411–424. [Google Scholar]
- 20.Vaden TD, Song C, Zaveri RA, Imre D, Zelenyuk A. Morphology of mixed primary and secondary organic particles and the adsorption of spectator organic gases during aerosol formation. Proc Natl Acad Sci. 2010;107:6658–6663. doi: 10.1073/pnas.0911206107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis EJ, Ravindran P, Ray AK. A review of theory and experiments on diffusion from submicroscopic particles. Chem Eng Commun. 1980;5:251–268. [Google Scholar]
- 22.Zhang S-H, Seinfeld JH, Flagan RC. Determination of particle vapor pressures using the tandem differential mobility analyzer. Aerosol Sci Tech. 1993;19:3–14. [Google Scholar]
- 23.Tang IN, Munkelwitz HR. Determination of vapor pressure from droplet evaporation kinetics. J Colloid Interf Sci. 1991;141:109–118. [Google Scholar]
- 24.Pathak RK, et al. Ozonolysis of alpha-pinene: Parameterization of secondary organic aerosol mass fraction. Atmos Chem Phys. 2007;7:3811–3821. [Google Scholar]
- 25.Cruz CN, Dassios KG, Pandis SN. The effect of dioctyl phthalate films on the ammonium nitrate aerosol evaporation rate. Atmos Environ. 2000;34:3897–3905. [Google Scholar]
- 26.Zelenyuk A, et al. Characterization of organic coatings on hygroscopic salt particles and their atmospheric impacts. Atmos Environ. 2010;44:1209–1218. [Google Scholar]
- 27.Rudich Y, Donahue NM, Mentel TF. Aging of organic aerosol: Bridging the gap between laboratory and field studies. Annu Rev Phys Chem. 2007;58:321–352. doi: 10.1146/annurev.physchem.58.032806.104432. [DOI] [PubMed] [Google Scholar]
- 28.Miet K, Le Menach K, Flaud PM, Budzinski H, Villenave E. Heterogeneous reactions of ozone with pyrene, 1-hydroxypyrene and 1-nitropyrene adsorbed on particles. Atmos Environ. 2009;43:3699–3707. [Google Scholar]
- 29.Gao S, et al. Low-molecular-weight and oligomeric components in secondary organic aerosol from the ozonolysis of cycloalkenes and alpha-pinene. J Phys Chem A. 2004;108:10147–10164. [Google Scholar]
- 30.Vaden TD, Imre D, Beránek J, Zelenyuk A. Extending the capabilities of single particle mass spectrometry: II. Measurements of aerosol particle density without DMA. Aerosol Sci Tech. 2011;45:125–135. [Google Scholar]
- 31.Zelenyuk A, Imre D, Han JH, Oatis S. Simultaneous measurements of individual ambient particle size, composition, effective density, and hygroscopicity. Anal Chem. 2008;80:1401–1407. doi: 10.1021/ac701723v. [DOI] [PubMed] [Google Scholar]
- 32.Zelenyuk A, Imre D. Beyond single particle mass spectrometry: multidimensional characterisation of individual aerosol particles. Int Rev Phys Chem. 2009;28:309–358. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.