

Abstract
Dasatinib is a potent dual Abl/Src inhibitor approved for treatment of Ph-positive leukemias. At a once-daily dose and a relatively short half-life of 3-5 hours, tyrosine kinase inhibition is not sustained. However, transient inhibition of K562 leukemia cells with a high-dose pulse of dasatinib or long-term treatment with a lower dose was reported to irreversibly induce apoptosis. Here, the effect of dasatinib on treatment of Bcr/Abl-positive acute lymphoblastic leukemia (ALL) cells was evaluated in the presence of stromal support. Dasatinib eradicated Bcr/Abl ALL cells, caused significant apoptosis and eliminated tyrosine phosphorylation on Bcr/Abl, Src, Crkl and Stat-5. However, treatment of mouse ALL cells with lower doses of dasatinib over an extended period of time allowed the emergence of viable drug-resistant cells. Interestingly, dasatinib treatment increased cell surface expression of CXCR4, which is important for survival of B-lineage cells, but this did not promote survival. Combined treatment of cells with dasatinib and a CXCR4 inhibitor resulted in enhanced cell death. These results do not support the concept that long-term treatment with low dose dasatinib monotherapy will be effective in causing irreversible apoptosis in Ph-positive ALL, but suggest that combined treatment with dasatinib and drugs such as AMD3100 may be effective.
Keywords: drug stability and suboptimal dose, SFK, tyrosine kinase inhibition, stromal support, Ph-positive ALL, AMD3100, CXCR4, CCL12
Introduction
Philadelphia chromosome positive (Ph-positive) leukemias include chronic myeloid leukemia (CML) and a subset of acute lymphoblastic leukemia (ALL) -around 20–30% of adults and 5–10% of children with ALL are Ph-positive. These leukemias are initiated by the abnormal fusion of two genes, BCR and ABL, which results in the synthesis of a Bcr/Abl protein with constitutive tyrosine kinase activity. A kinase inhibitor with relative specificity for Abl, imatinib, is widely used to treat patients with Ph-positive leukemias. Although it is very effective in the chronic phase of CML, patients in the accelerated phase and blast crisis of CML respond poorly to it. In addition, Ph-positive ALL treated with the same drug has an unfavorable prognosis, suggesting that factors in addition to Bcr/Abl are involved in its progression. 1,2
A number of studies indicate an important contribution of Src-family kinases (SFKs) to Bcr/Abl-induced lymphoblastic leukemogenesis. 3-5 Bcr/Abl-transduced hematopoietic cells from mouse bone marrow lacking Lyn, Hck and Fgr were able to cause myeloid leukemia but not lymphoblastic leukemia in transplant recipients. In these mice, inhibition of Bcr/Abl and SFKs together showed greater therapeutic potency than inhibition of Bcr/Abl alone. 6 The SFK member Lyn is also implicated in the development of Bcr/Abl-independent imatinib resistance. 7,8
Dasatinib (Sprycel, formerly BMS-354825; Bristol-Myers Squibb, New York, NY) is a dual Abl and Src kinase inhibitor that has been developed for the treatment of Ph-positive leukemias. 9 Dasatinib has a 325-fold greater potency than imatinib in inhibiting the activity of non-mutated Bcr/Abl. Moreover, dasatinib, compared with imatinib, is more active against most imatinib-resistant mutant Bcr/Abl isoforms with the exception of the T315I mutant. 10,11 In the START phase II trials, dasatinib was found to be effective and tolerable to CML or Ph-positive ALL patients who were intolerant or resistant to imatinib, and the optimal dasatinib dose ranges were confirmed in phase III randomized trials. 12 In 2009, dasatinib was FDA-approved in the USA as second-line treatment for chronic phase CML at a dose of 100 mg 1× per day.
Dasatinib is relatively unstable, with a half life of approximately 3-5 hours in the circulation 13 and 24 hours after ingestion of 100 mg of dasatinib, concentrations of 1-10 nM were measured in plasma of five CML patients. 14 Therefore, initially, patients were given dasatinib twice a day, but side effects made this regimen undesirable and a once-daily treatment is now standard. 15 Based on in vitro dasatinib treatment, Shah et al 14 argued that temporary inhibition of Bcr/Abl, consisting of either a pulse of a high drug dose or a prolonged drug exposure, using dasatinib or imatinib, is sufficient to elicit cytotoxicity. These experiments challenged the general assumption, that for molecules such as dasatinib to be effective, they need to continually inhibit the target kinase, and provide a rationale for using lower drug doses in a clinical setting. However, these conclusions were almost exclusively based on experiments performed with K562, a cell line derived from a CML patient who developed an erythroleukemic blast crisis.
There have been relatively few preclinical studies that report the use of dasatinib as monotherapy for the treatment of Bcr/Abl positive ALL and none that investigate possible drug resistance against dasatinib in this type of leukemia. In previous studies performed in our lab, we modeled the development of drug resistance using lymphoblastic leukemia cells from BCR/ABL P190 transgenic mice. Using intermediate doses of drugs, a long-term follow-up, and stromal support, we were able to generate Bcr/Abl lymphoblastic leukemia cells resistant to imatinib, nilotinib, lonafarnib and a CKIIα inhibitor. 16-19
In the current study, we have investigated different aspects of dasatinib therapy in vitro in the presence of stromal support, using both mouse and human Bcr/Abl-positive ALL cells. Our results show, that short-term dasatinib treatment indeed generates a significant amount of apoptosis. However, suboptimal therapy, even given over an extended period of time, readily gives rise to viable drug-resistant cells in in vitro models in which stromal support is provided to the leukemic cells.
Materials and methods
Drugs, reagents and cells
Dasatinib (Toronto Research Chemicals, North York, Ontario, Canada) and AMD3100 octahydrochloridehydrate (1,1′-[1,4-phenylene bis (methylene) bis-1, 4,8,11 Tetra azacyclotetradecane octahydrochloride) (Sigma-Aldrich, St.Louis, USA) were diluted in DMSO.
The murine OP9 stromal cell line (CRL-2749) was purchased from the ATCC (Manassas, VA, USA). The 8093 and Bin2 Bcr/Abl P190-expressing mouse lymphoblastic leukemia cells have been previously described 17, 18 and were grown in the presence of E13.5 irradiated mouse embryonic fibroblasts (MEFs) except where indicated. TXL2 Bcr/Abl P210-expressing acute lymphoblastic leukemia cells from a patient at diagnosis were passaged in (NOD/SCID/IL2)r2γ-/- mice (Jackson Labs, Bar Harbor, ME). The sample lacked mutations in the ATP-binding pocket of ABL. Leukemia cells harvested from the spleen were plated on irradiated OP9 feeder layers. Mouse leukemia cells were grown in McCoy's 5A medium (Invitrogen Corporation, Carlsbad, USA) supplemented with 15% FBS, 110 mg/L sodium pyruvate, 1% L-glutamine, 1% penicillin/streptomycin, 10 ng/ml recombinant IL-3 (Invitrogen Corporation) and 50 μmol/L β-mercaptoethanol. Human TXL2 cells were grown in αMEM including 20% F B S, 1 % L-glutamine and 1% penicillin/streptomycin.
In vitro treatment with dasatinib
For drug treatment, lymphoblastic leukemia cells were cultured at 1-3 ×106 cells/ml in wells of a 24-well plate or a 6-well plate, either in the presence of E13.5 irradiated MEFs or OP9 cells. The Trypan blue exclusion assay was used to determine viability of cells in triplicate wells. Dasatinib was added every second day along with a fresh complete change of medium. Apoptotic cells were measured using the Annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit I (BD Pharmingen, San Diego, USA). For cell cycle analysis, cells were washed and fixed in 70% ethanol for one hour. Fixed cells were stained with PI and subjected to flow cytometry (FACScan, Becton Dickinson, USA). For assays of CXCR4 expression, 8093 cells (5×105 /ml) were seeded in 24-well plates in the presence of irradiated MEFs. After overnight incubation, cells were treated with dasatinib or dasatinib and AMD3100 for 4 hrs, 24 hrs and 48 hrs before being stained with FITC-conjugated anti-CXCR4 monoclonal Ab (2B11, BD-Pharmingen). The mean fluorescence intensity was determined by flow cytometry. Sequencing of the DNA encoding the Abl ATP binding site was as described by Hochhaus et al. 20
Western blotting
8093 cells were treated with various concentrations of dasatinib and lysed in 1× RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS, 5 mM EDTA) containing PMSF, aprotinin, leupeptin, pepstatin A, Na Fluoride and Na Orthovanadate for 30 minutes on ice. Cell extracts were subjected to 7.5-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were reacted with phospho-p44/42 MAP kinase, phospho-src family (Tyr416), Src, phospho-Stat5 (Tyr694) (Cell Signaling Technology, USA), PY-20-Horseradish peroxidase (BD-Transduction Laboratories, San Jose, CA), Bcr (N-20), Crkl (Santa Cruz Biotechnology, Santa Cruz, USA), β-actin (Sigma-Aldrich) or GAPDH (Chemicon International/Millipore, Billercia, MA, USA) antibodies using standard procedures.
Statistical analysis
Statistical analysis was performed with SPSS software (Chicago, IL, USA). Data are presented as mean ± SD. Statistical significance of differences between groups was evaluated using one-way-ANOVA (post hoc Scheffe test). A value of p< 0.05 is considered statistically significant.
Results
Dasatinib has significant cytotoxic activity against Bcr/Abl-positive lymphoblastic leukemia cells
Unlike the K562 cell line used by Shah et al14 the ALL cells used in this study, including mouse 8093 and Bin2, and human TXL2 cells, were only grown in tissue culture with stromal support. Because of the continued presence of stroma, their drug sensitivity may be more representative for the in vivo situation. We therefore tested increasing concentrations of dasatinib in the presence of stromal support. As shown in Figure 1a, dasatinib showed a clear dose-dependent effect on the percentage of viable 8093, Bin2 and TXL2 cells. Interestingly, the mouse leukemia cells were much more sensitive than the human TXL2 cells, based on estimated half maximal effective concentrations of 500 nM for TXL2 (Figure 1a, right panel) versus 0.3 nM and 0.5 nM for 8093 and Bin2, respectively, after 3 days of treatment.
Figure 1.
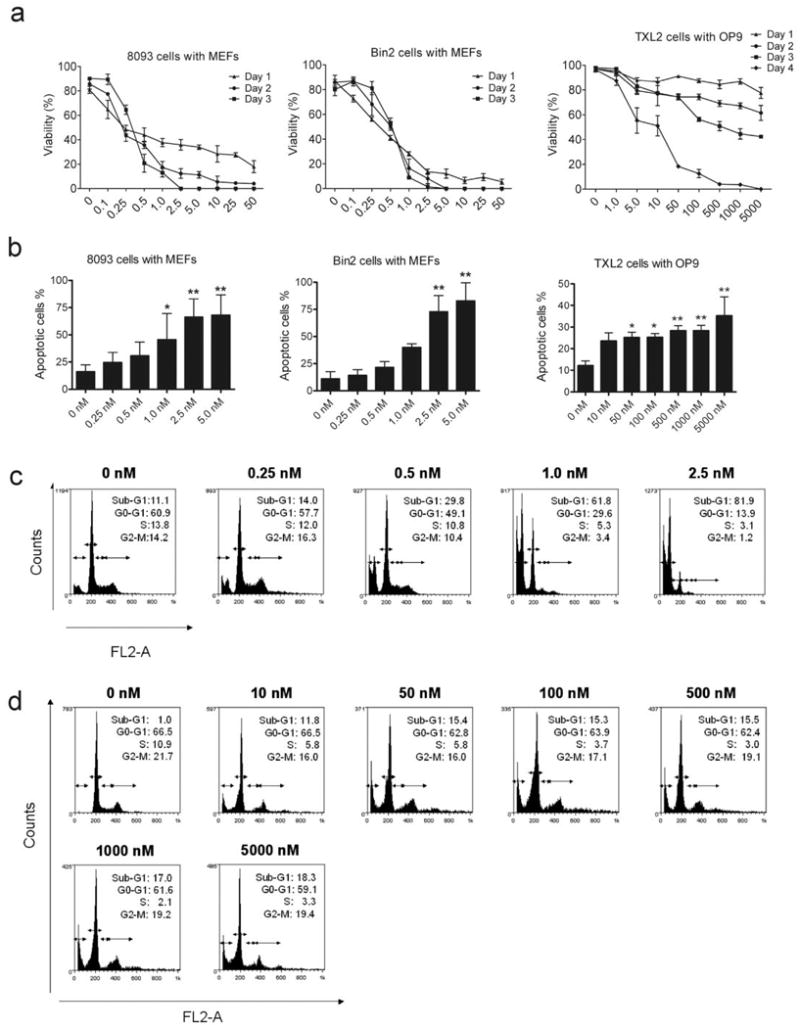
Dasatinib has significant activity against human and mouse Bcr/Abl lymphoblastic leukemia cells grown with stromal support. (a). 8093, Bin2 and TXL2 lymphoblastic leukemia cells (1×106/well) were grown in the presence of irradiated MEFs or OP9 cells. Viable cell counts were performed in triplicate over a 1-4 day period as indicated. Note: scale on X-axis is not linear. (b). 8093, Bin2 and TXL2 cells were treated with different concentrations of dasatinib, as indicated, for 48 hrs. The percentage apoptotic cells was determined by FACS analysis. Error bars indicate SD. Results shown are one of two independently performed experiments with similar results. (*, p < 0.05; **, p < 0.01). (c). 8093 and (d) TXL2 cells were treated with different concentrations of dasatinib for 72 hrs. Cell cycle distribution was determined by flow cytometry. The percentage of cells in the different phases of the cell cycle (sub-G1, G0/G1, S and G2/M) after treatment with dasatinib is indicated.
Next, we investigated whether the inhibitory effect of dasatinib on the proliferation of leukemia cells was associated with induction of apoptosis. As shown in Figure 1b, after 48 hrs of treatment, dasatinib induced apoptosis in 8093, Bin2 and TXL2 cells, as assessed by Annexin V/PI staining. However, perhaps because the TXL2 cells grow more slowly than the mouse cells, the effect of 2 days of drug treatment was much less pronounced. We also examined the effects of 72-hrs dasatinib treatment on cell cycle progression. A significant increase in the numbers of 8093 cells with a DNA content of less than N was seen at concentrations of 0.5 nM and higher, and very few cells treated with 2.5 nM dasatinib had a normal DNA content (Figure 1c). The inhibitory effect of dasatinib on proliferation was associated with a concomitant decrease in the number of cells in S or G2/M phase of cell cycle. TXL2 cells (Figure 1d) were similarly affected, although even at the highest dasatinib concentration, a substantial number of cells with a normal DNA content remained. Overall, these results indicate that dasatinib has growth-inhibitory effects on both human and mouse Bcr/Abl-positive ALL cells and induces apoptosis even when cells are grown in the presence of stromal support.
Dasatinib inhibits Bcr/Abl and Src kinases
Dasatinib targets both Bcr/Abl and SFKs. 10, 21 To compare its effect on these two classes of kinases, we treated 8093 cells with various concentrations of dasatinib for 4 hrs and investigated inhibition of Bcr/Abl, SFKs and downstream targets of these kinases using Western blotting. Figure 2 shows that P190 Bcr/Abl autophosphorylation was eliminated at around 5 nM dasatinib, as judged by the decline in phosphotyrosine. Crkl is a direct Bcr/Abl binding partner and substrate. Whereas a substantial amount of Crkl protein was tyrosine phosphorylated in non-dasatinib treated cells, increasing concentrations of dasatinib progressively shifted Crkl to its non-tyrosine phosphorylated, more rapidly migrating form. Inhibition of phosphorylation of Src Tyr 416 was also greatly reduced. Since Nam et al22 reported that phosphorylation of Stat5 is a readout for dasatinib-induced apoptosis in K562, we also evaluated Stat5 phosphorylation. As shown in Figure 2, Stat5 phosphorylation was reduced to about 50%, at 0.25 nM dasatinib. A dose of 100 nM dasatinib eliminated all visible phosphotyrosine on Bcr/Abl, Src and Crkl.
Figure 2.
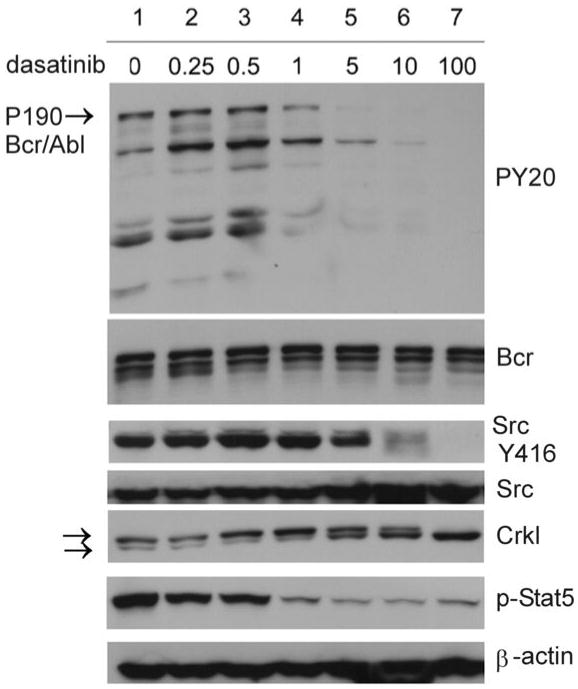
Dasatinib inhibits Bcr/Abl and Src kinase activities. 8093 cells were treated with indicated concentrations of dasatinib for 4 hrs. Western blot analysis was done on total lysates with the antibodies indicated to the right of the panels. Blots were stripped and reprobed with Bcr (N-20), Src and β-actin antibodies to ensure equal loading and transfer of protein. The Crkl panel is from a different set of samples of an experiment with similar outcome. Arrows point to the tyrosine-phosphorylated, more slowly migrating and non-tyrosine phosphorylated, more rapidly migrating Crkl isoforms.
Stroma allows emergence of dasatinib-resistant leukemia cells
The dose-response curve with dasatinib only involved the examination of the 8093 and Bin2 lymphoblastic leukemia cells for a short period. A dose of 0.5 nM eliminated 70% of the 8093 and a dose of 1 nM 80% of the Bin2 cells within 3 days (Figure 1a). We next investigated the longer-term effects of treatment with this dose of dasatinib. Dasatinib was added every second day along with a fresh complete change of medium. As shown in Figure 3a, left panels, consistent with the results in Figure 1, the viability of leukemia cells in the presence of stroma significantly decreased from 85∼95 % to 30∼40 % in the first 3∼4 days of dasatinib treatment.
Figure 3.
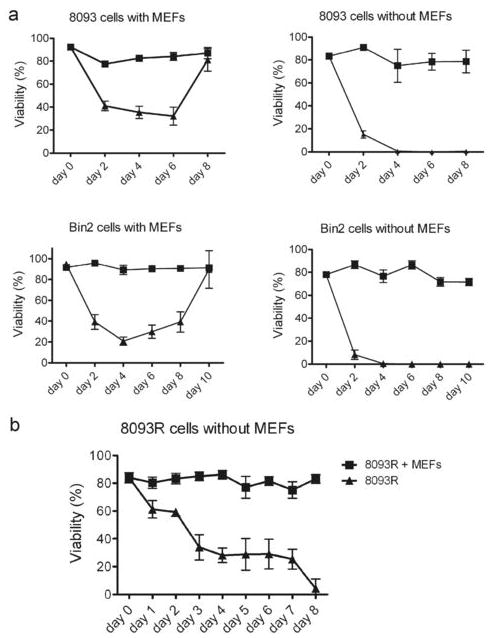
Resistance to dasatinib is facilitated by stroma. (a). Leukemia cells as indicated (3×106/well) were treated with DMSO (■) or with 0.5 nM (8093) or 1.0 nM (Bin2) dasatinib (▲) with or without MEFs as indicated. (b). 8093 cells grown on MEFs that had become resistant to 0.5 nM dasatinib (8093-R) on day 8 were cultured in the presence or absence of MEFs, as indicated, for an additional 8 days. Fresh dasatinib was added every second day with a complete medium change. Viability was assessed by the Trypan Blue exclusion method and is expressed as percentage. Each point represents the average of triplicate values ± SD.
However, viability began to increase in the following days. On the eighth or tenth day, the viability of the cells recovered to a level similar to that of the DMSO treatment group. Thus, the cells were able to proliferate again in the presence of stroma and in concentrations of dasatinib that initially killed most of the cells (Figure 3). To examine the possibility that these dasatinib-resistant cells contained point mutations in the Abl ATP binding domain or activation loop, we isolated RNA at day 8 and performed DNA sequencing of this region. However, since no mutations were detected (not shown) dasatinib-resistance could not be explained by the most commonly detected structural changes in Bcr/Abl.
A very different outcome of dasatinib treatment was obtained in long-term cultures of the leukemia cells without stroma. Similar to the results reported for K562 by Shah et al14, viability of cells without stroma decreased to zero on the fourth day and no drug-resistant cells grew out (Figure 3a right panels).
We then collected the dasatinib-resistant cells generated on stromal co-culture at day 8 (8093-R) and continued treatment with the same dose of dasatinib, but re-plated half of the cells without stromal support. Interestingly, whereas the 8093-R cells in the presence of MEFs and dasatinib retained viability as expected, the viability of drug-treated 8093-R cells without stromal support decreased to 0 over an 8-day period. This supports the concept that the continued presence of microenvironmental support is needed to maintain the drug-resistant phenotype.
To investigate possible changes in signal transduction during the period that 8093 cells developed the resistance to dasatinib, cell lysates were collected at different time points from day 0 to day 8. As shown in Figure 4, in the first six days of dasatinib treatment, when viability was declining, the overall levels of phosphotyrosine (PY20), Src Tyr416, pCrkl and pStat5 also declined. However, by the eighth day, when the percentage of viable cells was around 80%, levels of tyrosine phosphorylation on Bcr/Abl, Src, Crkl and Stat5 were restored to those at day 0.
Figure 4.
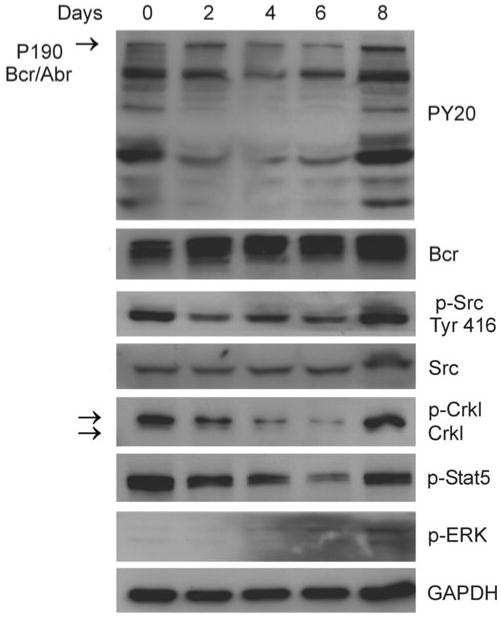
Re-emergence of tyrosine kinase activity after 8093 cells develop resistance to dasatinib. 8093 cells were grown in the presence of MEFs with 0.5 nM dasatinib. Fresh dasatinib was added every second day with the change of medium. 8093 cell lysates were prepared at different time points as indicated. The levels of PY20, p-Src Tyr416, Crkl, p-Stat5 and p-ERK1/2 were determined by Western blot analysis. Blots were stripped and re-probed with Bcr (N-20), Src and GAPDH antibodies to ensure equal loading and transfer of protein.
Dasatinib increases CXCR4 cell-surface expression on leukemia cells
In previous studies, we investigated factors that could contribute to the development of drug resistance in co-culture of mouse Bcr/Abl lymphoblastic leukemia cells with MEFs and reported, that the chemoattractant and hematopoietic survival factor SDF-1α secreted by MEFs contributes to survival of these cells during the emergence of imatinib resistance. 16 Interestingly, Jin et al23 recently reported that imatinib treatment increased expression of CXCR4, the receptor for SDF-1α, in 5 peripheral blood samples of CML patients and in the cell lines K562 and KBM, when the latter were co-cultured with mesenchymal cells.
Therefore, we investigated if dasatinib could affect cell surface expression of CXCR4 on leukemia cells when co-cultured with MEFs. In these experiments, 8093 cells were plated de novo on MEFs (t=0 hours) and the cells were then sampled for up to 48 hrs afterwards. Engagement of the CXCR4 receptor with SDF-1α causes rapid internalization of CXCR4. 24 As expected and as shown in Figure 5a, CXCR4 cell surface expression typically declined in non-drug treated cells after 48 hrs, reflecting binding of SDF-1α produced by the MEFs to CXCR4 on the leukemia cells and subsequent receptor internalization. In concordance with this, levels of cell surface CXCR4 remained much higher in 8093 cells cultured without MEFs for up to 24 hrs. Based on Western blots, there was no evidence that the internalized CXCR4 was catabolized, since total levels of CXCR4 did not dramatically change in the different treatments (not shown). Interestingly, 48 hours after start of treatment with 1.0 or 2.5 nM dasatinib, there was a significantly higher cell surface expression of CXCR4 as compared to non-drug treated cells (Figure 5a).
Figure 5.
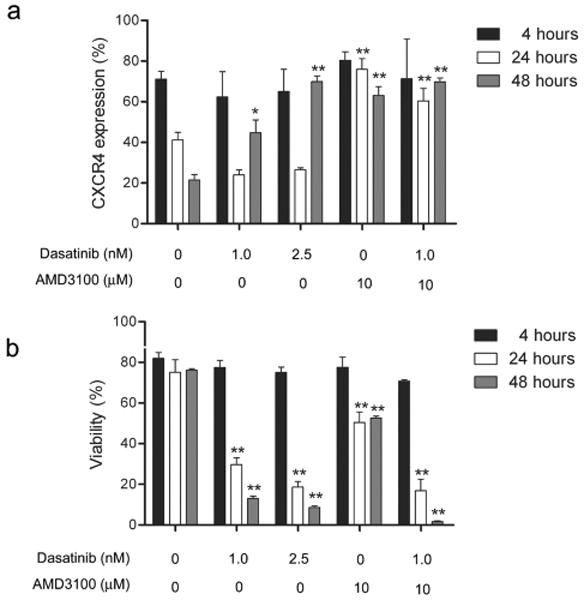
Dasatinib increases cell-surface expression of CXCR4. 8093 cells were treated with dasatinib and AMD3100 at the concentrations indicated below the figures in the presence of irradiated MEFs. (a) Percentage of cells expressing CXCR4 on the surface from 4-48 hrs of the total number of live cells determined by flow cytometry. (b) Viability as assessed by Trypan Blue exclusion. Results shown are from one of three independent experiments with similar results. Error bars, mean values ±SD of triplicate samples. *, p < 0.05; **, p< 0.001 compared to control group at the same time point.
The non-peptide bicyclam AMD3100 blocks the binding of SDF-1α to CXCR4. 25 As shown in Figure 5a, treatment of the ALL cells over a period of 48 hours with 10 μM AMD3100 significantly abrogated the normal time-dependent removal of CXCR4 from the cell surface. A similar high level of cell surface CXCR4 was measured in cells treated with both dasatinib and AMD3100 (Figure 5a).
To investigate how this correlated with survival signals, we also measured the viability of the cells during these treatments (Figure 5b). As expected, control cells that normally internalized CXCR4 maintained normal high viability during the 48 hours. Cells treated with 1.0 or 2.5 nM dasatinib, which had increased cell surface CXCR4 at 48 hours, had significantly decreased viability. Cells that were treated with AMD3100 alone and maintained high CXCR4 cell surface expression were slightly less viable compared to control cells. Interestingly, cells at 48 hours that had been treated with both AMD3100 and dasatinib had very high cell surface expression of CXCR4 (comparable to that of non-drug treated controls), but very low viability. Since the levels of total cellular CXCR4 as assessed by Western blotting of lysates prepared from these cells were not higher than those of control non-treated cells (not shown), we conclude that the dasatinib plus AMD3100-treated cells fail to normally internalize the CXCR4 receptor in the presence of SDF-1α produced by the MEFs.
Discussion
Effective treatment of Ph-positive ALL remains a considerable challenge and dasatinib, because it is a more potent tyrosine kinase inhibitor that targets Bcr/Abl as well as SFK implicated in Ph-positive ALL, is a very promising therapeutic drug. However, most preclinical studies to date focused on the effect of dasatinib on myeloid Ph-positive leukemias. For example, Deguchi et al. 26 compared the growth-inhibitory effects of imatinib, nilotinib and dasatinib against six CML cell lines and reported that dasatinib showed the most potent effects. In addition, it was also effective in imatinib-resistant CML cell lines. 26 Similar results were described in Redaelli et al. who studied the activity of dasatinib, imatinib, and nilotinib against 18 mutated forms of Bcr/Abl expressed in Ba/F3 transfected cells. 27
The first human clinical study with dasatinib 28 reported that nearly all patients with Ph-positive ALL had a relapse within 6 months, although Ottmann et al 29 reported in a phase II study, that dasatinib was safe and effective, and that, with the 8-month follow up, it seemed promising. 29 Using models for Ph-positive leukemias in which mice were transplanted with Bcr/Abl-transduced bone marrow, Hu et al. showed that dasatinib inhibits the proliferation of both myeloid and B-lymphoid cells in vivo. 30 However, even after treatment for 3 months with dasatinib, leukemic cells persisted in both models and gave rise to relapse when treatment was discontinued. This shows that relapse during dasatinib treatment is likely and prompted us to examine early steps in possible emergence of insensitivity to it.
In our studies, we confirm that dasatinib is “effective” against Bcr/Abl positive lymphoblastic leukemia: it clearly inhibits the proliferation of Bcr/Abl P190-expressing mouse ALL cells and, to a lesser extent, that of human Ph-positive ALL cells, causes apoptosis, and knocks down the tyrosine phosphorylation activities of both Bcr/Abl and Src-family kinases. The most critical test of the effectiveness of a drug, however, is its ability to totally eradicate leukemic cells. Using that as criterion, we find resistance against dasatinib can be evoked by providing the leukemia cells with stromal support and using suboptimal doses of dasatinib.
With a half-life of 3-5 hours and a once daily dose regimen, we would expect dasatinib to reach sub-therapeutic levels within 24 hours in patients. Shah et al 14 argue that this should not prevent dasatinib effectiveness, because leukemia cells treated with such tyrosine kinase inhibitors would be put on an irreversible path to apoptosis. We suggest that it may be very difficult to set ALL cells on the pathway to irreversible apoptosis under conditions that resemble those in vivo. Our results show that stroma provides protection against dasatinib and allows viable cells to emerge even during long-term treatment.
The only other study to investigate dasatinib resistance in Ph-positive cells is that of Okabe et al 31, who found decreased expression of Bcr/Abl during emerging drug resistance in K562. In our study, we did not detect changes in levels of Bcr/Abl protein and we therefore sought alternative mechanisms that could provide protection against dasatinib. Stroma is known to secrete factors that can partially protect Bcr/Abl expressing cells from the inhibitory effects of imatinib and nilotinib. 32 Using a stromal MEF co-culture system, we previously showed that SDF-1α is one of the protective factors that provide support to mouse Bcr/Abl cells treated with Imatinib. 16 Indeed, the interaction of the cytokine SDF-1α (CXCL12) with the CXCR4 receptor plays a pivotal role in the earliest stages of normal B-cell development and CXCR4 also helps retain pre-B cells in the bone marrow. 33
Because Jin et al. demonstrated that imatinib increases the expression of CXCR4 on imatinib sensitive KBM-5 and K562 cells 23, we considered the possibility that dasatinib could induce CXCR4 expression and by this mechanism possibly increase the longer-term survival of the ALL cells. Our results show that fully viable ALL cells that are plated de novo on a stromal feeder layer and are stimulated by the stromal-produced SDF-1α remove the cell-surface CXCR4 over a period of 48 hours. We found that Bcr/Abl-ALL cells treated with dasatinib, similar to K562 treated with Imatinib or BV-173 cells treated with Imatinib or nilotinib, significantly increased cell surface expression of CXCR4. 23,34 Since there were no major changes in CXCR4 total cellular protein, our results are consistent with a model in which dasatinib prevents the normal internalization of the SDF-1α-stimulated CXCR4 receptor. Also, treatment with AMD3100 increased CXCR4 cell surface expression on these cells. Interestingly, the increased cell surface expression of CXCR4 did not provide a survival benefit to these cells. In contrast, cells exposed to dasatinib plus AMD3100 were killed effectively while they had amounts of CXCR4 on their surface comparable to that of non-drug treated cells at the 4-hr time point.
In spite of the importance of the CXCR4-CXCL12 axis for many cell types, surprisingly little is known about the trafficking of the CXCR4 receptor and the significance of this for the transduction of survival and other SDF-1α-associated signals. Early studies in the context of HIV research show that, depending on the cell type, there is a varying degree of spontaneous CXCR4 endocytosis. 24, 35 Internalized receptor can be degraded in the lysosome or recycled back to the cell surface. Binding of SDF-1α to CXCR4 causes rapid internalization of CXCR4 24 and activates numerous tyrosine kinases including Lyn, ZAP-70 and Tec in T-cells. 36 Based on our data, it is possible that dasatinib interferes with CXCR4 endocytosis and/or transduction of survival signals downstream of CXCR4 by blocking tyrosine kinase activity needed for this. This raises the interesting possibility that one of the mechanisms by which dasatinib kills ALL cells is by interfering with CXCR4 signal transduction.
Our in vitro results suggest that Ph-positive ALL patients will not be able to achieve a durable response, if dasatinib is used as monotherapy. However, the combination with drugs such as AMD3100 could present an alternative strategy. More than 23 types of cancers other than leukemias express CXCR4 37 and dasatinib is in clinical trials for the treatment of, among others, prostate, pancreatic, non-small lung and metastatic breast cancer (www.clinicaltrials.gov). The combined treatment with dasatinib and a drug that inhibits CXCR4-SDF-1α could therefore be an interesting alternative approach as well for these types of cancers.
Acknowledgments
We would like to acknowledge Reshmi Parameswaran for helpful suggestions and critical reading of the manuscript. This work was supported by PHS grants CA090321 (to NH) and R01CA137060 and RO1CA139032 (MM), by funding from a CHLA RCDA, a Jean Perkins Scholar and a StopCancer award (Y-mK); and by a private donor (Y-mK, MM, NH, JG).
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Faderl S, Garcia-Manero G, Thomas DA, Kantarjian HM. Philadelphia chromosome-positive acute lymphoblastic leukemia- current concepts and future perspectives. Rev Clin Exp Hematol. 2002;6:142–160. doi: 10.1046/j.1468-0734.2002.00066.x. [DOI] [PubMed] [Google Scholar]
- 2.Ren R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. 2005;5:172–183. doi: 10.1038/nrc1567. [DOI] [PubMed] [Google Scholar]
- 3.Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56:3589–3596. [PubMed] [Google Scholar]
- 4.Lionberger JM, Wilson MB, Smithgall TE. Transformation of myeloid leukemia cells to cytokine independence by Bcr-Abl is suppressed by kinase-defective Hck. J Biol Chem. 2000;275:18581–18585. doi: 10.1074/jbc.C000126200. [DOI] [PubMed] [Google Scholar]
- 5.Klejman A, Schreiner SJ, Nieborowska-Skorska M, Slupianek A, Wilson M, Smithgall TE, et al. The Src family kinase Hck couples BCR/ABL to STAT5 activation in myeloid leukemia cells. EMBO J. 2002;21:5766–57674. doi: 10.1093/emboj/cdf562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36:453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- 7.Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–698. doi: 10.1182/blood.V101.2.690. [DOI] [PubMed] [Google Scholar]
- 8.Dai Y, Rahmani M, Corey SJ, Dent P, Grant S. A Bcr/Abl-independent, Lyn-dependent form of imatinib mesylate (STI-571) resistance is associated with altered expression of Bcl-2. J Biol Chem. 2004;27934:227–239. doi: 10.1074/jbc.M402290200. [DOI] [PubMed] [Google Scholar]
- 9.Brave M, Goodman V, Kaminskas E, Farrell A, Timmer W, Pope S, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14:352–359. doi: 10.1158/1078-0432.CCR-07-4175. [DOI] [PubMed] [Google Scholar]
- 10.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 11.O'Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–4505. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 12.Keam SJ. Dasatinib: in chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. BioDrugs. 2008;22:59–69. doi: 10.2165/00063030-200822010-00007. [DOI] [PubMed] [Google Scholar]
- 13.Christopher LJ, Cui D, Wu C, Luo R, Manning JA, Bonacorsi SJ, et al. Metabolism and disposition of dasatinib after oral administration to humans. Drug Metab Dispos. 2008;36:1357–1364. doi: 10.1124/dmd.107.018267. [DOI] [PubMed] [Google Scholar]
- 14.Shah NP, Kasap C, Weier C, Balbas M, Nicoll JM, Bleickardt E, et al. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008;14:485–493. doi: 10.1016/j.ccr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 15.Shah NP, Kantarjian HM, Kim DW, Réa D, Dorlhiac-Llacer PE, Milone JH, et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib-resistant and-intolerant chronic-phase chronic myeloid leukemia. J Clin Oncol. 2008;26:3204–3212. doi: 10.1200/JCO.2007.14.9260. [DOI] [PubMed] [Google Scholar]
- 16.Mishra S, Zhang B, Cunnick JM, Heisterkamp N, Groffen J. Resistance to imatinib of Bcr/abl p190 lymphoblastic leukemia cells. Cancer Res. 2006;66:5387–93. doi: 10.1158/0008-5472.CAN-05-3058. [DOI] [PubMed] [Google Scholar]
- 17.Kaur P, Feldhahn N, Zhang B, Trageser D, Müschen M, Pertz V, et al. Nilotinib treatment in mouse models of P190 Bcr/Abl lymphoblastic leukemia. Mol Cancer. 2007;6:67. doi: 10.1186/1476-4598-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Groffen J, Heisterkamp N. Increased resistance to a farnesyltransferase inhibitor by N-cadherin expression in Bcr/Abl-P190 lymphoblastic leukemia cells. Leukemia. 2007;21:1189–1197. doi: 10.1038/sj.leu.2404667. [DOI] [PubMed] [Google Scholar]
- 19.Mishra S, Pertz V, Zhang B, Kaur P, Shimada H, Groffen J, et al. Treatment of P190 Bcr/Abl lymphoblastic leukemia cells with inhibitors of the serine/threonine kinase CK2. Leukemia. 2007;21:178–180. doi: 10.1038/sj.leu.2404460. [DOI] [PubMed] [Google Scholar]
- 20.Hochhaus A, Kreil S, Corbin AS, La Rosee P, Muller MC, Lahaye T, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia. 2002;11:2190–2196. doi: 10.1038/sj.leu.2402741. [DOI] [PubMed] [Google Scholar]
- 21.Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 22.Nam S, Williams A, Vultur A, List A, Bhalla K, Smith D, et al. Dasatinib (BMS-354825) inhibits Stat5 signaling associated with apoptosis in chronic myelogenous leukemia cells. Mol Cancer Ther. 2007;6:1400–1405. doi: 10.1158/1535-7163.MCT-06-0446. [DOI] [PubMed] [Google Scholar]
- 23.Jin L, Tabe Y, Konoplev S, Xu Y, Leysath CE, Lu H, et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol Cancer Ther. 2008;7:48–58. doi: 10.1158/1535-7163.MCT-07-0042. [DOI] [PubMed] [Google Scholar]
- 24.Signoret N, Oldridge J, Pelchen-Matthews A, Klasse PJ, Tran T, Brass LF, et al. Phorbol esters and SDF-1 induce rapid endocytosis and down modulation of the chemokine receptor CXCR4. J Cell Biol. 1997;139:651–664. doi: 10.1083/jcb.139.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schols D, Struyf S, Van Damme J, Esté JA, Henson G, De Clercq E. Inhibition of T-tropic HIV strains by selective antagonization of the chemokine receptor CXCR4. J Exp Med. 1997;186:1383–1388. doi: 10.1084/jem.186.8.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deguchi Y, Kimura S, Ashihara E, Niwa T, Hodohara K, Fujiyama Y, et al. Comparison of imatinib, dasatinib, nilotinib and INNO-406 in imatinib-resistant cell lines. Leuk Res. 2008;32:980–983. doi: 10.1016/j.leukres.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, et al. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol. 2009;27:469–471. doi: 10.1200/JCO.2008.19.8853. [DOI] [PubMed] [Google Scholar]
- 28.Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 29.Ottmann O, Dombret H, Martinelli G, Simonsson B, Guilhot F, Larson RA, et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: interim results of a phase 2 study. Blood. 2007;110:2309–2315. doi: 10.1182/blood-2007-02-073528. [DOI] [PubMed] [Google Scholar]
- 30.Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic progenitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103:16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okabe S, Tauchi T, Ohyashiki K. Characteristics of dasatinib- and imatinib-resistant chronic myelogenous leukemia cells. Clin Cancer Res. 2008;14:6181–6186. doi: 10.1158/1078-0432.CCR-08-0461. [DOI] [PubMed] [Google Scholar]
- 32.Weisberg E, Wright RD, McMillin DW, Mitsiades C, Ray A, Barrett R, et al. Stromal-mediated protection of tyrosine kinase inhibitor-treated BCR-ABL-expressing leukemia cells. Mol Cancer Ther. 2008;7:1121–1129. doi: 10.1158/1535-7163.MCT-07-2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Glodek AM, Honczarenko M, Le Y, Campbell JJ, Silberstein LE. Sustained activation of cell adhesion is a differentially regulated process in B lymphopoiesis. J Exp Med. 2003;197:461–473. doi: 10.1084/jem.20021477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dillmann F, Veldwijk MR, Laufs S, Sperandio M, Calandra G, Wenz F, et al. Plerixafor inhibits chemotaxis toward SDF-1 and CXCR4-mediated stroma contact in a dose-dependent manner resulting in increased susceptibility of BCR-ABL+ cell to imatinib and nilotinib. Leuk Lymph. 2009 doi: 10.1080/10428190903150847. epub 1-11. [DOI] [PubMed] [Google Scholar]
- 35.Tarasova NI, Stauber RH, Michejda CJ. Spontaneous and ligand-induced trafficking of CXC-chemokine receptor 4. J Biol Chem. 1998;273:15883–15886. doi: 10.1074/jbc.273.26.15883. [DOI] [PubMed] [Google Scholar]
- 36.Patrussi L, Baldari CT. Intracellular mediators of CXCR4-dependent signaling in T cells. Immunol Lett. 2008;115:75–82. doi: 10.1016/j.imlet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 37.Wong D, Kortz W. Translating an antagonist of chemokine receptor CXCR4 from bench to bedside. Clin Can Res. 2008;14:7975–7980. doi: 10.1158/1078-0432.CCR-07-4846. [DOI] [PubMed] [Google Scholar]