


Abstract
Toward the goal of identifying complete sets of transcription factor (TF)-binding sites in the genomes of several gamma proteobacteria, and hence describing their transcription regulatory networks, we present a phylogenetic footprinting method for identifying these sites. Probable transcription regulatory sites upstream of Escherichia coli genes were identified by cross-species comparison using an extended Gibbs sampling algorithm. Close examination of a study set of 184 genes with documented transcription regulatory sites revealed that when orthologous data were available from at least two other gamma proteobacterial species, 81% of our predictions corresponded with the documented sites, and 67% corresponded when data from only one other species were available. That the remaining predictions included bona fide TF-binding sites was proven by affinity purification of a putative transcription factor (YijC) bound to such a site upstream of the fabA gene. Predicted regulatory sites for 2097 E.coli genes are available at http://www.wadsworth.org/resnres/bioinfo/.
INTRODUCTION
Understanding the regulation of gene expression, and transcription regulation in particular, is one of the grand challenges of molecular biology. While transcription is regulated by several mechanisms, the binding of transcription factors (TFs) to their cognate sites is the dominant mechanism and the identification of these sites is indispensable to a comprehensive understanding of gene expression. The experimental methods for TF-binding site identification that have been developed include electrophoretic mobility shift and nuclease protection assays. Despite the fact that gene regulation has been intensely studied in the gamma proteobacterium Escherichia coli, experimental methods have identified TF-binding sites for only a fraction of the estimated 300–350 TFs (1) in the promoters of only a few hundred E.coli genes (2,3).
The three main computational methods that have been developed to identify and characterize TF-binding sites in promoters are: a consensus building greedy algorithm (4), an expectation maximization algorithm (5–8) and a Bayesian Gibbs sampling algorithm (9,10). These methods all identify a collection of aligned sites from multiple sequences and a corresponding site model called a motif. Until recently these methods required the identification of a set of genes for which there is experimental evidence of co-regulation (4,11–13). These computational methods have also been useful for predicting additional binding sites for known, characterized TFs in recently sequenced genomes (3,11,12). With the advent of whole genome sequencing, computational phylogenetic footprinting methods, involving cross-species comparison of DNA sequences, have emerged (12–14). This method allows for the identification of a TF-binding site(s) upstream of a single gene given the promoter sequence of that gene from a number of species, thus eliminating the need for the identification of a set of co-regulated genes. The recent availability of genomic sequence data for several gamma proteobacteria encouraged us to examine the utility of genomic scale phylogenetic footprinting.
MATERIALS AND METHODS
Identification of data sets
We applied TBLASTN (15) with stringent criteria to identify probable orthologous genes in nine gamma proteobacterial species (listed below) for which at least partial genomic sequence data were available. Orthologous gene sets were identified using the E.coli ORF translations from GenBank (U00096) as the queries against a database consisting of the available genome sequence data for all nine species. Selection of the orthologous sequence in each species from a collection of significant TBLASTN hits (which may contain strong paralogs) employed a number of heuristics. The most significant TBLASTN hit from each species was considered the true ortholog if it satisfied the following constraints: (i) the expectation value was <10–20; (ii) the expectation value was less than, and the raw BLAST score more than, the second best hit in E.coli (i.e. true orthologs should have a score more significant than any paralogs present in E.coli); (iii) the TBLASTN hit must start within the first 20 amino acids of the E.coli query sequence. The promoter data sets consisted of the regions upstream of the identified orthologs. For E.coli these data were limited to the intergenic region, with a minimum of 50 bp and up to a maximum of 500 bp. We also used TBLASTN to determine if gene order was conserved in the other species and, if so, limited the upstream data for those species to intergenic regions. If, however, TBLASTN did not reveal a similar gene order in other species, 500 bp upstream of the orthologous gene were used in the data.
The availability of genomic sequence data for several related species was important for several reasons. (i) The genomic sequence data were incomplete for several of the species, so even for a gene with an ortholog in every species it was possible that sequence data for that gene may have only been available for a few species. (ii) Ortholog identification is difficult, and by including data for several species we allowed some uncertainty that true orthologs had been identified in every species for every gene (i.e., even if some orthologs were identified incorrectly, we had enough data from species with correctly identified orthologs for a reliable prediction of the TF-binding site). (iii) Orthologous genes may be regulated differently in some species. By using data from many species we increased the likelihood of having data from enough species with similar gene regulation that TF-binding sites could be identified.
Bayesian Gibbs sampling
An advanced Gibbs motif sampler (9,10) with the following important extensions was utilized. (i) A motif model that accounts for palindromic patterns in TF-binding sites was employed (5). (ii) Because DNA sequences tend to have varying composition (e.g. regions that are G-C rich or A-T rich), a position-specific background model, estimated with a Bayesian segmentation algorithm (16), was used to decide whether a given segment should be judged as being a binding site or as belonging to the background. (iii) The empirical distribution of spacing between TF-binding sites and the translation start site, observed from the E.coli genome sequence, was incorporated, to improve the algorithm’s focus on more probable locations of binding sites (W.Thompson, unpublished results). (iv) The algorithm was configured to detect 0, 1 or 2 sites (repeats) in each upstream region in a data set (W.Thompson, unpublished results).
Iterations of the Gibbs sampler were performed under four conditions: with even (16 bases) or odd (17 bases) palindromic models and with or without the distribution of spacing model. The models were allowed to fragment up to a total width of 24 bases (17). Additionally, after each iteration of the sampler under a given condition the identified site(s) was replaced with Ns and the data set re-analyzed for additional sites. The predicted motifs for each data set were then ordered according to the maximum a posteriori probability (MAP) value to determine the most probable motif. The MAP value is measured relative to an empty or ‘null’ alignment. Therefore, a MAP value >0 indicates that the alignment is more likely than the unaligned random background. A more detailed description of the MAP value is available at http://bayesweb.wadsworth.org/gibbs/gibbs.html.
Affinity chromatography and mass spectrometry
For each site complementary oligonucleotides were synthesized with a duplex region of 16–18 bp carrying the predicted binding site and 5–7 bp of flanking sequence. In addition, each oligonucleotide had a 5′-GAAC single-stranded extension to facilitate coupling to Sepharose beads via the amino groups of those bases. The top strand for each duplex is shown with the predicted binding site underlined and the single-stranded extension in bold. Predicted sites: fabA, 5′-GAACTTGTTCAGCGTACACGTGTTAGCTATCCTG-3′; fabB, 5′-GAACTTGTTCGGCGTACAAGTGTACGCTATTGTG-3′; yqfA, 5′-GAACTATTTTAGCTAACAGGTGTTCACTGGAACT-3′. Control sites: FadR site upstream of fadB, 5′-GAACGACTCATCTGGTACGACCAGATCACCTAA-3′; PurR site upstream of purH, 5′-GAACGCATTGTAACGAAAACGTTTGCGCAACG-3′.
The oligonucleotides were annealed and coupled to CNBr-activated Sepharose beads (Amersham Pharmacia Biotech, Piscataway, NJ), essentially as described by DiRusso et al. (18) except that no aminoethyl group was added to the oligonucleotides. For each column 54 nmol DNA duplex was coupled to ∼2.5 g (wet) Sepharose beads to generate a bed volume of ∼3 ml. Crude extracts were prepared from soluble cell lysates of E.coli MG1655 grown to mid-log phase in LB medium. Cell pellets were resuspended in 20 mM Tris–HCl, pH 7.5, 10 mM NaCl, 1 mM EDTA, 1 mM DTT, sonicated and clarified by centrifugation. Proteins were precipitated with 60% saturated ammonium sulfate and the precipitate was dissolved and dialyzed against column buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.1 mM DTT, 10 mM NaN3). Extracts from up to 6 l of cultured cells were passed through a 20 ml pre-column containing an unrelated control sequence (purH) to reduce the presence of non-specific DNA-binding proteins and thereby increase the yield of specific TFs. Extracts were then passed over 3 ml experimental columns and DNA-binding proteins were eluted sequentially with TE buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA) containing 0.2 or 0.8 M NaCl.
Column fractions were subjected to SDS–PAGE, from which protein bands were subjected to in-gel tryptic digestion and MALDI-TOF mass spectrometry analysis (19,20). Comparison of tryptic peptide masses to predicted peptide masses of all of the E.coli proteins in the SWISS-PROT database was done with the ProteinProspector MS-Fit software at the University of California at San Francisco Mass Spectrometry Facility (http://prospector.ucsf.edu/).
Genome sequence data
Escherichia coli genome sequence data (U00096) were obtained from GenBank (http://www.ncbi.nlm.nih.gov/Genbank/index.html). Complete genome sequence data for Haemophilus influenzae and preliminary sequence data for Shewanella putrefaciens, Thiobacillus ferrooxidans and Vibrio cholerae were obtained from The Institute for Genomic Research (http://www.tigr.org/). Salmonella typhi (ftp://ftp.sanger.ac.uk/pub/pathogens/st/) and Yersinia pestis (ftp://ftp.sanger.ac.uk/pub/pathogens/yp/) preliminary genome sequence data were produced and obtained from the respective Sequencing Groups at the Sanger Centre (http://www.sanger.ac.uk/Projects/). Actinobacillus actinomycetemcomitans preliminary genome sequence data were obtained from the Actinobacillus Genome Sequencing Project at the University of Oklahoma (http://www.genome.ou.edu/act.html). Pseudomonas aeruginosa preliminary genome sequence data were obtained from the Pseudomonas Genome Project (http://www.pseudomonas.com).
Availability
A web server for the Gibbs motif sampler with the extensions described, a list of the genes used in our study set with a reference for the known TF-binding sites and our results for all data sets are available at our web site (http://www.wadsworth.org/resnres/bioinfo/).
RESULTS AND DISCUSSION
Analysis of the study set
Using data available from DPInteract (11; http://arep.med.harvard.edu/dpinteract/), RegulonDB (21; http://www.cifn.unam.mx/Computational_Biology/regulondb/) and the literature, we identified a study set of 190 genes in the E.coli genome for which TF-binding sites have been identified by nuclease protection or mobility shift experiments. Sequences upstream of the orthologous genes were extracted into 190 data sets for analysis (see Materials and Methods). For six of the 190 E.coli genes no orthologs were detected. For these we could not perform cross-species comparisons, leaving 184 data sets in our study set. The upstream regions for these 184 E.coli genes contained documented binding sites for 53 different TFs (Table 1).
Table 1. Known E.coli TF-binding sites represented in the study set and predictions.
| Transcription factor |
No. of genes in the study set
with known sitesa |
Total no. of known sites in study
setb |
No. of sites detected in study
set predictionsc |
No. of additional probable sites in predictionsd |
| Ada | 3 | 3 | 3 | 3 |
| AraC | 4 | 6 | 3 | 3 |
| ArcA | 9 | 13 | 5 | 7e |
| ArgR | 7 | 15 | 13 | 7 |
| CarP | 1 | 2 | 0 | – |
| CpxR | 5 | 6 | 3 | 4 |
| Crp | 42 | 63 | 27 | 22e |
| CspA | 2 | 3 | 1 | 0 |
| CynR | 1 | 2 | 1 | 1 |
| CysB | 2 | 3 | 2 | 0 |
| CytR | 8 | 8 | 3 | 2 |
| DeoR | 1 | 1 | 0 | – |
| DnaA | 3 | 5 | 0 | – |
| FadR | 5 | 7 | 3 | 5 |
| FhlA | 3 | 3 | 2 | 0 |
| Fis | 9 | 25 | 5 | 1 |
| FlhCD | 3 | 3 | 0 | – |
| Fnr | 9 | 14 | 6 | 13 |
| FruR | 10 | 11 | 5 | 7 |
| Fur | 6 | 9 | 8 | 18 |
| GalR | 4 | 5 | 4 | 1 |
| GcvA | 2 | 4 | 3 | 0 |
| GlpR | 3 | 11 | 7 | 6e |
| IciA (ArgP) | 1 | 2 | 1 | 1 |
| IclR | 1 | 1 | 1 | 1 |
| Ihf | 14 | 22 | 5 | 1 |
| IlvY | 1 | 2 | 2 | 3 |
| LacI | 1 | 2 | 0 | – |
| LexA | 15 | 18 | 15 | 6 |
| Lrp | 6 | 29 | 5 | 2 |
| MalT | 3 | 10 | 1 | 0 |
| MarR | 1 | 2 | 2 | 5 |
| MelR | 1 | 5 | 2 | 2 |
| MetJ | 5 | 15 | 15 | 9 |
| MetR | 5 | 8 | 1 | 1 |
| Mlc (DgsA) | 4 | 5 | 5 | 3 |
| ModE | 3 | 3 | 2 | 4 |
| NagC | 3 | 6 | 5 | 5e |
| NarL | 8 | 11 | 2 | 5 |
| NarP | 4 | 4 | 0 | – |
| NtrC | 3 | 5 | 4 | 5 |
| OmpR | 2 | 6 | 2 | 5 |
| OxyR | 3 | 3 | 3 | 4 |
| PdhR | 1 | 2 | 1 | 6 |
| PhoB | 5 | 12 | 8 | 3 |
| PurR | 16 | 16 | 9 | 4 |
| RhaR | 1 | 1 | 1 | 2 |
| RhaS | 2 | 2 | 0 | – |
| SoxR | 1 | 1 | 1 | 1 |
| SoxS | 4 | 7 | 1 | 3 |
| TorR | 1 | 4 | 3 | 5 |
| TrpR | 3 | 3 | 3 | 1 |
| TyrR | 8 | 16 | 8 | 3 |
| Stem–loopsf | 4 | 7 | 5 | – |
aThe number of genes in our study set with documented binding sites for each TF. Of the 184 genes in the study set 118 had only one type of TF-binding site and the remaining 66 genes had multiple types of TF-binding sites in the data analyzed (i.e. the upstream intergenic regions).
bThe total number of documented binding sites for each TF in our data for the study set.
cThe total number of predicted sites that corresponded to documented sites in the study set. The most probable motif predictions for 146 genes in the study set corresponded to documented sites. Because many of these genes had multiple documented TF-binding sites and up to two E.coli sites could be identified in each motif prediction, these results included a total of 187 predicted sites that corresponded to documented sites. Additionally, it is known that in some instances different TF-binding sites overlap and their cognate TFs compete for binding, therefore, some sites identified in our predictions overlapped two or more different TF binding sites. Overlap to all documented TF-binding sites was counted.
dThe number of probable sites detected by scan (10) for these known TFs from among the 2627 E.coli sites that did not correspond to documented TF-binding sites (the most probable motif predictions for the 2097 genes included a total of 2814 E.coli sites: 2627 undocumented and 187 documented). An additional 187 were identified by scan as probable sites for these 46 TFs.
eThe most probable motif predictions for two genes (gcd and nanA) were detected by the Crp and GlpR models and the site for one gene (yjiT) was detected by both the ArcA and NagC models. Because different TF-binding sites can overlap the same sequence, particularly global TFs like Crp and ArcA, this was not unexpected and these sites were counted in both categories.
fFour genes in our study set (ilvB, ilvG, trpE and ompA) have upstream sequences that form stem–loop structures in the mRNA involved in attenuation or mRNA stability. These genes were in the study set because they also have known TF-binding sites in their upstream intergenic regions.
A subset consisting of 24 data sets was used as the training set to tune the parameters of a Gibbs sampling strategy (9,10,17) to identify TF-binding sites in these data. We performed several iterations of the Gibbs sampler in order to identify the most probable motif for each data set, i.e. the motif with the highest MAP value (see Materials and Methods). Using these parameters 20 of the 24 most probable motif predictions (83%) corresponded to previously documented TF-binding sites. Although it is not uncommon for a gene to be regulated by more than one TF, and different TF-binding sites were identified during multiple iterations of the Gibbs sampler, we restricted the analysis described below to the most probable motif for each data set.
For the full study set (184 data sets), 146 of the most probable motif predictions corresponded with documented transcription regulatory sites (Table 1). A single ortholog was identified for 18 of the 184 genes, which allowed only limited cross-species comparison. For these data the predictions were less reliable, as evidenced by lower MAP values and a lower correspondence with previously documented TF-binding sites (12 of 18, or 67%). However, when at least two orthologous genes were identified (166 data sets) 81% of the most probable motif predictions corresponded with previously documented transcription regulatory sites: 131 corresponded to TF-binding sites and an additional three corresponded to known stem–loop structures involved in attenuation or RNA stability. The remaining 32 data sets contained several predictions with large MAP values, suggesting the presence of undocumented regulatory sites in these data. The documented TF-binding sites for these data were frequently detected as the second or third most probable motif.
Because the majority of known prokaryotic TFs bind as homodimers and recognize palindromic sites, the Gibbs sampling parameters used to generate the results described above specified palindromic models. Interestingly, Gibbs sampling analysis of the same study set data without palindromic models also performed well, detecting documented sites in 138 of the 184 data sets. While our results with these data clearly benefited from using palindromic models, the fact that a significant number of sites were detected without them indicates the power of this type of cross-species comparison and suggests that this approach is applicable to eukaryotic data.
Identification of YijC-binding sites
Among the 32 undocumented sites identified in the study set were several strongly predicted sites, including one upstream of the fabA gene. A scan (10) of the E.coli genome with the motif model revealed two additional occurrences of this site in intergenic regions: one upstream of fabB and one upstream of yqfA. To identify and characterize the transcription factor(s) that binds to these predicted sites we used DNA sequence-specific affinity chromatography of crude E.coli extracts from exponentially growing cells (see Materials and Methods). A protein bound specifically and with varying affinity to all three of the sites, fabA, fabB and yqfA (Fig. 1, lanes 1–3), that did not bind to affinity columns containing binding sites for FadR, a transcriptional regulator of fatty acid metabolism genes (22,23), or PurR, which negatively regulates genes involved in purine nucleotide biosynthesis (24; Fig. 1, lane 4, and data not shown). The protein bound to each of the predicted sites (fabA, fabB and yqfA) was identified by mass spectrometry analysis as YijC, an uncharacterized member of the TetR family of transcription factors (25).
Figure 1.
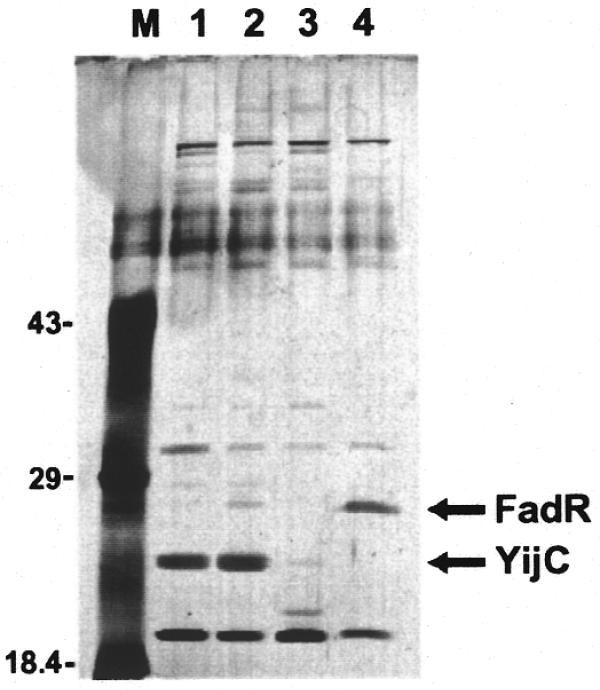
SDS–PAGE gel showing affinity purification of YijC. Escherichia coli MG1655 extracts, passed over a purH pre-column, were fractionated on DNA affinity columns carrying sequences predicted to be TF-binding sites upstream of the fabA, fabB or yqfA genes or a control column carrying a known FadR site upstream of fadB (see Materials and Methods). A silver stained SDS–PAGE gel of representative fractions eluted from the columns with 0.8 M NaCl is shown. M, molecular weight markers; lane 1, fabA column; lane 2, fabB column; lane 3, yqfA column; lane 4, fadB column. Mass spectrometry analysis identified the 26 kDa protein bound specifically to the fabA, fabB and yqfA columns as YijC and the protein bound to the fadB column as FadR.
FadR is a known repressor of fatty acid degradation (fad) genes and an activator of fatty acid biosynthesis (fab) genes (23,26). Indeed, expression from the fabA promoter is known to be activated 20-fold upon binding FadR (23). However, FadR regulation does not completely explain the decrease in transcription of fabA upon entry of cells into stationary phase or that fadR mutant strains contain only one third less unsaturated fatty acids than the wild-type (23). DiRusso and Nystrom (26) have proposed that complex regulatory activities responding to growth rate, growth phase and stringent response must exist to coordinate fatty acid biosynthesis with phospholipid synthesis and turnover. In addition, control of the relative levels of fabA and fabB may be necessary to establish correct saturated to unsaturated fatty acid ratios (26). The YijC-binding sites we have identified upstream of fabA and fabB are positioned between the –10 and –35 regions of these promoters, suggesting that YijC represses expression of these genes. The role of YijC as a repressor is supported by the position of its helix–turn–helix motif at the N-terminus, the position typical for repressors (1). Based on these data we propose renaming this repressor FabR (fatty acid biosynthesis regulator).
Genomic scale phylogenetic footprinting
We proceeded to apply our phylogenetic footprinting procedure genome wide. We identified 2113 E.coli open reading frames (ORFs) that were suitable for phylogenetic footprinting of their upstream regions (Table 2); this group includes the study set described above. Table 2 indicates the number of orthologs detected by our criteria in each of the nine species, as well as how frequently data from each species contributed to the most probable motif predictions. Sites identified by our Gibbs sampling strategy for 2097 orthologous sets are reported on our web site; for the remaining 16 data sets no site was predicted in E.coli (Table 2). Figure 2 illustrates the information available for each gene at our web site. For every motif prediction two sequence logos are given, one representing the motif model and one representing the sites that were predicted. Specifying palindromic models during Gibbs sampling effectively doubled the amount of data by including in the model the reverse complements of the sites, leading to correspondingly tighter confidence intervals. The palindromic models were perfectly symmetrical, as illustrated in Figure 2B. The sites detected by the palindromic models were not necessarily symmetrical, however (Fig. 2C). A sequence alignment of the sites in each motif prediction is also given, with an indication of which positions (*) contributed to the model (Fig. 2D).
Table 2. Number of orthologs and representation in the most probable motif predictions for each species.
| Species |
No. of orthologs detecteda |
No. of times represented in the most probable
motif predictionsb |
| Enterobacteriaceae | ||
| Escherichia coli | 2113 | 2097 |
| Salmonella typhi | 1962 | 1835 |
| Yersinia pestis | 1442 | 1041 |
| Pasteurellaceae | ||
| Haemophilus influenzae | 677 | 472 |
| Actinobacillus actinomycetemcomitans | 585 | 376 |
| Vibrionaceae | ||
| Vibrio cholerae | 984 | 614 |
| Alteromonadaceae | ||
| Shewanella putrefaciens | 869 | 534 |
| Pseudomonas group | ||
| Pseudomonas aeruginosa | 963 | 363 |
| Unclassified | ||
| Thiobacillus ferrooxidans | 460 | 169 |
aOf the 4289 predicted ORFs in the E.coli genome (27) we eliminated from our analysis 1719 ORFs that are encoded by IS elements or transposons or have upstream intergenic regions of <50 bp. For an additional 457 ORFs no orthologs were detected using our TBLASTN criteria, therefore leaving 2113 ORFs for analysis by cross-species comparison. The number listed for each of the other species is the number of orthologs detected in that species for those 2113 E.coli ORFs.
bThe number of times that data from each species contributed to the most probable motif prediction for each gene. For 16 of the 2113 data sets analyzed no site was predicted in E.coli. The identified sites in all species for the remaining 2097 data sets can be viewed at http://www.wadsworth.org/resnres/bioinfo/.
Figure 2.
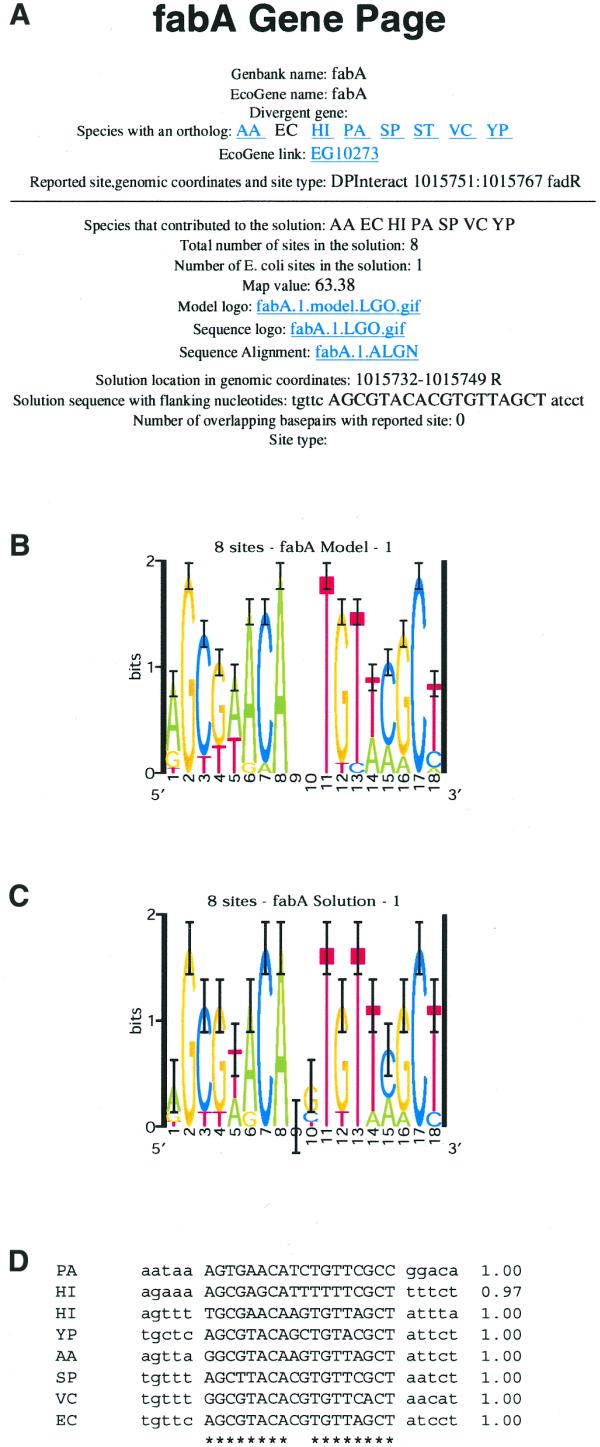
Snapshot from the fabA Gene Page on our web site illustrating the data available. (A) At the top of each Gene Page are given the gene name as it appears in both the E.coli genome GenBank entry (27) and in EcoGene (28; http://bmb.med.miami.edu/EcoGene/EcoWeb/index.html), as is the name of the divergently transcribed gene when one exists. The species in which orthologs were detected for the gene are indicated (EC, E.coli; ST, Salmonella typhi; YP, Yersinia pestis; HI, Haemophilus influenzae; AA, Actinobacillus actinomycetemcomitans; VC, Vibrio cholerae; SP, Shewanella putrefaciens; PA, Pseudomonas aeruginosa; TF, Thiobacillus ferrooxidans). For those genes with a documented regulatory site(s), the reference(s), the genomic coordinates of the site(s) and the site type(s) are given. Information from up to three predictions (ordered by MAP value) are then described. For each prediction the species in which a site was predicted are indicated, as are the total number of sites and the number of sites in the E.coli data, followed by the MAP value of the motif. Links are provided to the motif model (B), represented as a sequence logo (29), and to two representations of the sites that were identified: a sequence logo (C) and a sequence alignment with site probabilities (D). The E.coli genomic coordinates of the site (an R indicates that the solution sequence given is the reverse complement of that in the GenBank entry), as well as the site sequence plus 5 flanking bp, are given. When a predicted site overlaps a previously documented site the site type (TF name or stem–loop) is indicated. If a predicted site overlaps an E.coli intergenic repeat (28), that is also reported. While analysis of the study set for correlation to documented TF-binding sites was confined to the most probable motif predictions, up to three predictions (ordered by MAP value) are described on our web site for each gene, since many genes are regulated by more than one transcription factor. The most probable motif (the YijC-binding site) detected in the fabA data is shown.
Figure 3 compares the distribution of MAP values for the most probable motifs predicted in our study set, which was a subset of the full set, to those of the full set; the distributions include only those data for which a site was predicted in E.coli (183 of the 184 in the study set and 2097 of the 2113 in the full set). The mode of the MAP values for the full set of 2097 predicted sites was shifted somewhat to the left (lower MAPs) relative to the mode for the 183 sites (Fig. 3A). This shift was primarily the result of the presence of proportionally more genes with low numbers of orthologs in the full set compared to the study set (Fig. 3B and C). Two factors contributed to this effect. (i) For a significant number of genes (472 of the 2113; Fig. 3C) only one ortholog was detected, frequently from Salmonella typhi, a close relative of E.coli. This was in part due to our use of several partial genome sequences. We also expected a significant number of genes to be unique to E.coli or to have diverged sufficiently such that no ortholog was detectable using our criteria. Accordingly, more reliable predictions could be made for the 1641 genes with data for E.coli and at least two orthologs than for those 472 genes (for which, as expected, the predicted sites had lower than average MAP values). (ii) The study set of 184 genes with known TF-binding sites reflected a bias in the literature toward genes that are more likely to be present in many species, i.e. those involved in carbon and nitrogen metabolism, amino acid biosynthesis, nucleotide biosynthesis, etc. Historically, genes involved in these common metabolic pathways have been the subject of intense research.
Figure 3.

Distributions of the MAP values for the most probable motifs from the study set (183 data sets) and the full set (2097 data sets). (A) The distribution of MAP values for the full set compared to the study set, illustrating the shift to the left (toward lower MAPs) for the full set (see text) and indicating the relative number in the study set of genes that have experimentally identified sites compared to the full set. (B) The distribution of MAP values for the study set broken down according to the number of orthologs detected for each gene. (C) The distribution of MAP values for the full set broken down according to the number of orthologs detected for each gene. Comparison of (B) and (C) again illustrates the shift toward lower MAP values for the full set compared to the study set, as well as the observation that when data were available from only two species the predictions typically had lower MAP values.
Additionally, the mode of the MAP values was likely influenced somewhat by the inclusion of data sets from within operons. Genes with upstream intergenic regions of <50 bp were excluded from our analysis (see Materials and Methods and Table 2) in order to limit the likelihood of including many genes that are coded within operons and therefore less likely to have a promoter or TF-binding sites immediately upstream. In a subset of genes for which the operon structure is known, our selection criteria excluded 63% of the intra-operon genes: of 75 operons encoding 267 total genes, upstream sequence data from all 75 first genes and 71 intra-operon genes were included in our analysis. For this subset the mode of the MAP values was shifted toward lower MAPS for the predictions made within operon regions as compared to the predictions made upstream of operons (10.5 versus 18.9).
To determine how many of the E.coli sites predicted by this genome-wide analysis were likely additional sites for known TFs we constructed transcription factor motif models for each of 46 known TFs (Table 1) to scan the E.coli sites identified in the predictions. Specifically, the predicted sites from the study set that corresponded to known TF-binding sites were grouped (using data from all the species) and common models for each TF made using the Gibbs site sampler (9). These models were then used to scan (10) a data set consisting of the unknown E.coli sites present in the most probable motif predictions from the genome-wide analysis. Using a stringent expectation value cut-off of 0.5 an additional 187 sites were identified as probable sites for these 46 known TFs (Table 1). Under these stringent conditions the remaining E.coli sites (and the motif models) are expected to represent binding sites for the >250 uncharacterized transcription factors predicted in E.coli.
Conclusions
Some caveats are appropriate to these findings. Analysis of the results from our study set revealed that regulatory stem–loops are also conserved across species. While stem–loops are a source of transcription regulation and therefore of considerable interest, they are also a source of false positive results when searching for TF-binding sites. Because sequences within the coding region of orthologous genes are highly conserved, phylogenetic footprinting was restricted to intergenic regions; therefore, TF-binding sites that occur within ORFs were not detected. It should also be noted that intergenic regions between divergently transcribed genes in E.coli were analyzed with respect to both genes because gene order is frequently not conserved across species. If, however, gene order for a given pair of divergent genes was conserved in the other species, the most probable motif predictions for both data sets often identified the same site (of 432 total divergent gene pairs, 267 identified the same site in E.coli), despite the distribution of spacing models focusing at opposite ends of these intergenic regions. This does not necessarily imply that the predicted site is a regulatory site that affects the expression of both genes. Finally, because the available experimental data are biased toward common metabolic pathways, results for the study set may not be representative of all E.coli genes, even after adjustment for the number of orthologs. The TF-binding site data we collected from DPInteract, RegulonDB and the literature are also incomplete, resulting in a bias toward underestimation of the reliability of predictions from our study set. Indeed, our identification of the YijC-binding site upstream of the fabA gene proves that previously undetected TF-binding sites are present even in well-studied promoters.
Previous efforts to identify TF-binding sites in the complete genome of E.coli required that information be provided as to known or likely sets of co-regulated genes. Most efforts have focused on identifying additional binding sites for known TFs (3,11,12). This approach requires that a set of binding sites for a TF have been experimentally identified; these sites are then aligned and weight matrices constructed to search the genome or upstream regions for matches. Additional binding sites for ∼50 characterized E.coli TFs have been predicted in this manner, albeit with typically high false positive rates. The approach of McGuire et al. (13) used cross-species data, but also required the prediction of regulons to provide sets of co-regulated genes. Most of the highly significant motifs predicted in E.coli in this manner were identified as sites for known TFs and lower scoring motifs were prone to high false positive rates. Strategies to reduce the number of false positives have varied from combining string matching with the weight matrix search (3) to filtering the results by position in the coding or non-coding regions (3,11) and base composition (11,13). Our method eliminates the need to identify known or likely sets of co-regulated genes, requiring only genome sequence data for a set of related species, in this case the gamma proteobacteria. In addition, the results presented here benefited from directly incorporating into the Gibbs sampling algorithm the distribution of spacing model to focus on the most probable locations for TF-binding sites and the position-specific background composition to account for heterogeneous base composition.
Identification of motif models and the sites upstream of individual genes is the first step toward understanding the transcription regulatory network of E.coli. Clustering these models to identify sets of co-regulated genes (regulons) is the next critical step. Motif models identified from orthologous data sets are typically more specific (i.e. have more highly conserved positions) than motif models from data sets of co-regulated genes. Therefore, clustering of these models is not straightforward and we are currently developing a Bayesian clustering algorithm to address these issues.
Cross-species comparison involves analyzing sets of intergenic sequences which are expected to have similar regulation without having to assay for gene expression. By using this type of data-driven approach that does not rely on prior knowledge of co-regulation we have shown that such comparisons, applied to a set of nine genomic sequences from gamma proteobacteria, yielded footprint sites for thousands of genes with significant accuracy. These results will also aid the prediction of gene function for the >1600 uncharacterized ORFs in the E.coli genome (27); based on the results presented here, we predict a function for yqfA related to fatty acid metabolism that requires its co-regulation with fabA and fabB. Furthermore, as illustrated by the identification of YijC using DNA sequences derived from these predictions, this approach promises to open a new avenue for the identification of not only TF-binding sites but also their cognate TFs. Finally, the large number of predicted sites with significant MAP scores (Fig. 3A) suggests that perhaps the core transcription regulatory network of E.coli is now within reach.
Acknowledgments
ACKNOWLEDGEMENTS
We thank The Institute for Genomic Research, the Sanger Centre, the University of Oklahoma and the Pseudomonas Genome Project for making partial genome sequence data available and the Computational Molecular Biology and Statistics, Biological Mass Spectrometry and Molecular Genetics Core Facilities at the Wadsworth Center for their assistance. We are grateful to Concetta DiRusso and Howard Zalkin for providing bacterial strains, Ivan Auger for helpful suggestions throughout this project, Bill Albano for expert technical assistance and Linda Mayerhofer for assistance with the literature. This work was supported by NIH grants RO1HG01257 and R21RR14036 to C.E.L.
References
- 1.Perez-Rueda E. and Collado-Vides,J. (2000) The repertoire of DNA-binding transcriptional regulators in Escherichia coli K-12. Nucleic Acids Res., 28, 1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gralla J.D. and Collado-Vides,J. (1996) Organization and function of transcription regulatory elements. In Neidhardt,F.C. (ed.), Escherichia coli and Salmonella: Cellular and Molecular Biology. ASM Press, Washington, DC, pp. 1232–1245.
- 3.Thieffry D., Salgado,H., Huerta,A.M. and Collado-Vides,J. (1998) Prediction of transcriptional regulatory sites in the complete genome sequence of Escherichia coli K-12. Bioinformatics, 14, 391–400. [DOI] [PubMed] [Google Scholar]
- 4.Stormo G.D. and Hartzell,G.W. (1989) Identifying protein-binding sites from unaligned DNA fragments. Proc. Natl Acad. Sci. USA, 86, 1183–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawrence C.E. and Reilly,A.A. (1990) An expectation maximization (EM) algorithm for the identification and characterization of common sites in unaligned biopolymer sequences. Proteins, 7, 41–51. [DOI] [PubMed] [Google Scholar]
- 6.Cardon L.R. and Stormo,G.D. (1992) Expectation maximization algorithm for identifying protein-binding sites with variable lengths from unaligned DNA fragments. J. Mol. Biol., 223, 159–170. [DOI] [PubMed] [Google Scholar]
- 7.Bailey T.L. and Elkan,C. (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. ISMB, 2, 28–36. [PubMed] [Google Scholar]
- 8.Lawrence C. and Reilly,A. (1996) Likelihood inference for permuted data with application to gene regulation. J. Am. Stat. Assoc., 91, 76–85. [Google Scholar]
- 9.Lawrence C.E., Altschul,S.F., Boguski,M.S., Liu,J.S., Neuwald,A.F. and Wootton,J.C. (1993) Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science, 262, 208–214. [DOI] [PubMed] [Google Scholar]
- 10.Neuwald A.F., Liu,J.S. and Lawrence,C.E. (1995) Gibbs motif sampling: detection of bacterial outer membrane protein repeats. Protein Sci., 4, 1618–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robison K., McGuire,A.M. and Church,G.M. (1998) A comprehensive library of DNA-binding site matrices for 55 proteins applied to the complete Escherichia coli K-12 genome. J. Mol. Biol., 284, 241–254. [DOI] [PubMed] [Google Scholar]
- 12.Mironov A.A., Koonin,E.V., Roytberg,M.A. and Gelfand,M.S. (1999) Computer analysis of transcription regulatory patterns in completely sequenced bacterial genomes. Nucleic Acids Res., 27, 2981–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGuire A.M., Hughes,J.D. and Church,G.M. (2000) Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res., 10, 744–757. [DOI] [PubMed] [Google Scholar]
- 14.Gelfand M.S., Koonin,E.V. and Mironov,A.A. (2000) Prediction of transcription regulatory sites in Archaea by a comparative genomic approach. Nucleic Acids Res., 28, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu J.S. and Lawrence,C.E. (1999) Bayesian inference on biopolymer models. Bioinformatics, 15, 38–52. [DOI] [PubMed] [Google Scholar]
- 17.Liu J.S., Neuwald,A.F. and Lawrence,C.E. (1995) Bayesian models for multiple local sequence alignment and Gibbs sampling strategies. J. Am. Stat. Assoc., 90, 1156–1170. [Google Scholar]
- 18.DiRusso C., Rogers,R.P. and Jarrett,H.W. (1994) Novel DNA-Sepharose purification of the FadR transcription factor. J. Chromatogr., 677A, 45–52. [DOI] [PubMed] [Google Scholar]
- 19.Stone K.L. and Williams,K.R. (1996) Enzymatic digestion of proteins in solution and in SDS polyacrylamide gels. In Walker,J.M. (ed.), The Protein Protocols Handbook. Humana Press, Totowa, NJ, pp. 415–425.
- 20.Williams K.R., Samandar,S.M., Stone,K.L., Saylor,M. and Rush,J. (1996) Matrix assisted-laser desorption ionization mass spectrometry as a complement to internal protein sequencing. In Walker,J.M. (ed.), The Protein Protocols Handbook. Humana Press, Totowa, NJ, pp. 541–555.
- 21.Salgado H., Santos-Zavaleta,A., Gama-Castro,S., Millan-Zarate,D., Blattner,F.R. and Collado-Vides,J. (2000) RegulonDB (version 3.0): transcriptional regulation and operon organization in Escherichia coli K-12. Nucleic Acids Res., 28, 65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiRusso C.C., Heimert,T.L. and Metzger,A.K. (1992) Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli. J. Biol. Chem., 267, 8685–8691. [PubMed] [Google Scholar]
- 23.Cronan J.E.,Jr and Subrahmanyam,S. (1998) FadR, transcriptional co-ordination of metabolic expediency. Mol. Microbiol., 29, 937–943. [DOI] [PubMed] [Google Scholar]
- 24.Zalkin H. and Dixon,J.E. (1992) De novo purine nucleotide biosynthesis. Prog. Nucleic Acid Res. Mol. Biol., 42, 259–287. [DOI] [PubMed] [Google Scholar]
- 25.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Howe,K.L. and Sonnhammer,E.L. (2000) The Pfam protein families database. Nucleic Acids Res., 28, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiRusso C.C. and Nystrom,T. (1998) The fats of Escherichia coli during infancy and old age: regulation by global regulators, alarmones and lipid intermediates. Mol. Microbiol., 27, 1–8. [DOI] [PubMed] [Google Scholar]
- 27.Blattner F.R., Plunkett,G., Bloch,C.A., Perna,N.T., Burland,V., Riley,M., Collado-Vides,J., Glasner,J.D., Rode,C.K., Mayhew,G.F., Gregor,J., Davis,N.W., Kirkpatrick,H.A., Goeden,M.A., Rose,D.J., Mau,B. and Shao,Y. (1997) The complete genome sequence of Escherichia coli K-12. Science, 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- 28.Rudd K.E. (2000) EcoGene: a genome sequence database for Escherichia coli K-12. Nucleic Acids Res., 28, 60–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider T.D. and Stephens,R.M. (1990) Sequence logos: a new way to display consensus sequences. Nucleic Acids Res., 18, 6097–6100. [DOI] [PMC free article] [PubMed] [Google Scholar]