


Abstract
The human DNA ligase III gene encodes both nuclear and mitochondrial proteins. Abundant evidence supports the conclusion that the nuclear DNA ligase III protein plays an essential role in both base excision repair and homologous recombination. However, the role of DNA ligase III protein in mitochondrial genome dynamics has been obscure. Human tumor-derived HT1080 cells were transfected with an antisense DNA ligase III expression vector and clones with diminished levels of DNA ligase III activity identified. Mitochondrial protein extracts prepared from these clones had decreased levels of DNA ligase III relative to extracts from cells transfected with a control vector. Analysis of these clones revealed that the DNA ligase III antisense mRNA-expressing cells had reduced mtDNA content compared to control cells. In addition, the residual mtDNA present in these cells had numerous single-strand nicks that were not detected in mtDNA from control cells. Cells expressing antisense ligase III also had diminished capacity to restore their mtDNA to pre-irradiation levels following exposure to γ-irradiation. An antisense-mediated reduction in cellular DNA ligase IV had no effect on the copy number or integrity of mtDNA. This observaion, coupled with other evidence, suggests that DNA ligase IV is not present in the mitochondria and does not play a role in maintaining mtDNA integrity. We conclude that DNA ligase III is essential for the proper maintenance of mtDNA in cultured mammalian somatic cells.
INTRODUCTION
DNA ligases catalyze the formation of phosphodiester bonds and thus play an essential role in both DNA replication and repair. To date, five DNA ligases have been identified in mammalian cells. The DNA ligase I gene encodes a 125 kDa protein involved in the sealing of Okazaki fragments formed during lagging strand synthesis of nuclear genomic DNA (1–3). DNA ligase I protein is also thought to be involved in some DNA repair processes (4,5). The DNA ligase III gene encodes a 100 kDa protein (6) that interacts with XRCC1. It is known to be involved in the repair of certain types of oxidative lesions via base excision repair (7) and may be involved in recombinational DNA repair (8). A testis-specific DNA ligase is encoded by an alternatively spliced DNA ligase III mRNA (6). The 70 kDa DNA ligase II protein (9–11) is now known to be a proteolytic fragment of DNA ligase III (12). The DNA ligase IV gene encodes a 100 kDa protein (13) and is involved in non-homologous DNA end-joining (14,15). Most recently, a 44 kDa enzyme named DNA ligase V has been described; however, the gene encoding this protein has not yet been identified (16).
Analysis of cells with mutant DNA ligase genes has revealed the central role these enzymes play in maintaining the integrity of the nuclear genome. For example, cell line 46BR, which has two missense mutations in different alleles of the DNA ligase I gene was derived from a patient who displayed stunted growth, sun sensitivity and ultimately died of lymphoma (17). Fibroblast cells derived from this patient displayed abnormal lagging strand DNA synthesis, elevated sister chromatid exchange, hypersensitivity to several DNA-damaging agents and delayed re-joining of DNA double-strand breaks (18–22). Chinese hamster ovary-derived EM-9 cells that lack a functional Xrcc1 protein are also deficient in DNA ligase III. EM-9 cells are defective in homologous recombination and hypersensitive to DNA-damaging agents, suggesting that DNA ligase III is involved in recombinational repair of the nuclear genome (23,24). The cell line 180BR was derived from a leukemia patient who over-responded to radiation therapy and died during treatment. These cells display hypersensitivity to ionizing radiation and defective non-homologous DNA end-joining due to a mutation within the DNA ligase IV gene (25). Gene targeting technology was used to inactivate both alleles of the DNA ligase IV gene in human pre-B cells (26). These cells were unable to carry out V(D)J recombination and were hypersensitive to ionizing radiation. This former phenotype was also observed in mouse embryos that lacked a functional DNA ligase IV gene (27). Studies of yeast cells that lack DNL4, the yeast homolog of the DNA ligase IV gene, also provide clear evidence that this gene participates in nuclear DNA double-strand break repair (20,28,29). This conclusion is further supported by the finding that hamster XR-1 cells, which are deficient in DNA ligase IV activity due to mutations in the ligase IV-stabilizing XRCC4 gene, are also deficient in DNA double-strand break repair (30,31).
While considerable information is available on the function of nuclear DNA ligases, far less is known about mtDNA ligase. Over two decades ago DNA ligase activity in the mitochondria was reported (32). Since then, the various forms of DNA ligase in the nucleus have been identified and characterized with no further investigation into the mitochondrial form, until a recent report showed that DNA ligase activity purified from Xenopus laevis mitochondrial extracts was immunologically related to nuclear DNA ligase III (33). Following this report, it was determined that the DNA ligase III gene encodes a mitochondrial form of the protein through translation initiation from an alternative initiation site upstream of that identified previously (34). More recently the yeast DNA ligase I homolog, CDC9, has been shown to encode a mitochondrial form of this enzyme (35).
The presence of DNA ligase III in the mitochondria led us to investigate the role of this enzyme in mtDNA maintenance. In this study we generated stable transfectants that expressed high levels of DNA ligase III antisense mRNA. We show that DNA ligase III antisense mRNA expression leads to reduced DNA ligase III protein levels in the mitochondria. Reduced mtDNA ligase III resulted in a decrease in the cellular mtDNA content. The mtDNA present in these clones contains numerous DNA single-strand breaks, suggesting that these cells are defective in sealing nicks generated during DNA replication and/or repair. In addition, cells expressing DNA ligase III antisense mRNA have a diminished capacity to restore their mtDNA to pre-irradiation levels following exposure to γ-radiation. These results demonstrate that DNA ligase III plays an essential role in mtDNA maintenance.
MATERIALS AND METHODS
Plasmid constructs
The antisense expression plasmid pREP-hLIGIII was constructed by cloning a portion of human DNA ligase III (nt 73–333; 6,12) into the HindIII site of pREP4 (Invitrogen). The antisense expression plasmid pREP-hLIGIV was similarly constructed by cloning a portion of human DNA ligase IV (nt 274–474; 12) into the HindIII site of pREP4.
Cells
The DNA ligase III and DNA ligase IV antisense expression plasmids were separately transfected into human fibrosarcoma-derived HT1080 cells (36) by electroporation (37). Clones of stable transfectants were isolated based on their resistance to hygromycin (Boehringer Mannheim). The full-length DNA ligase III gene, which expresses the mitochondrial form, was cloned in a pEGFP-N1 vector as described earlier (34). AS1 clones were transfected with the above construct, while AS1 cells transfected with an empty vector served as the control. Stable clones were isolated based on their growth in media containing hygromycin and geneticin (200 µg/ml).
Preparation of mitochondrial protein extracts
Mitochondria were prepared from control and DNA ligase III or DNA ligase IV antisense-expressing cells by differential centrifugation as described previously (38). Isolated mitochondria were washed and lysed by hypo-osmotic shock. Proteins were separated from the membrane fraction by centrifugation at 70 000 r.p.m. for 30 min in a Beckman TL 100/3 rotor at 4°C. Protein extracts were dialyzed against 50 mM Tris, pH 7.5, buffer containing 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 10 mM phenylmethylsulfonyl fluoride and 10% glycerol. Protein concentration was measured using the Bradford assay (39).
Preparation of nuclear extracts
Cells were rinsed with phosphate-buffered saline (PBS), scraped and collected into centrifuge tubes. Following two washes of these cells in PBS, they were resuspended in a homogenization buffer containing 10 mM KCl, 10 mM MgCl2, 10 mM Tris, pH 7.4, and 10 mM DTT and homogenized in a dounce homogenizer in the presence of protease inhibitors. Nuclei were sedimented at 1500 g and extracts prepared by hypo-osmotic shock as described earlier (40).
Adenylation reaction
Protein adenylation reactions were carried out as described earlier (41). Mitochondrial protein extract (5 µg) was incubated with 0.5 µCi [α-32P]ATP in the presence of 60 mM Tris, pH 8.0, 10 mM MgCl2, 5 mM DTT and 50 µg/ml BSA at room temperature for 15 min. The reaction was terminated by addition of SDS loading buffer. The adenylated product was resolved on a 10% SDS–polyacrylamide gel. Following electrophoresis, the gel was fixed in 10% acetic acid and exposed to a phosphorimager screen.
Western blot analysis
Aliquots of 5 µg nuclear protein extracts from control and antisense cells were separated by 10% SDS–PAGE and electrophoretically transferred to a nitrocellulose membrane. The membrane was blocked with 5% BSA in Tris-buffered saline for at least 1 h prior to probing with anti-ligase III antibody (Novus Biologicals) or anti-ligase IV antibody (kindly provided by Dr S. Critchlow, UK) for 1 h at room temperature. Following incubation with a 1:5000 dilution of anti-rabbit IgG conjugated to alkaline phosphatase, color was developed using the alkaline phosphatase substrate 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (Sigma).
Northern blot analysis
Cytoplasmic RNA was isolated from mammalian cells as described earlier (42). An aliquot of 10 µg of the RNA from control and antisense-expressing cells was resolved on a denaturing agarose gel containing formaldehyde and transferred to a nylon membrane by capillary action. Membranes were hybridized with a random prime (Gibco BRL) generated [α-32P]dATP-labeled double-stranded DNA probe corresponding to either the DNA ligase III cDNA (nt 73–333) or DNA ligase IV cDNA (nt 274–474). Northern blots of cells transfected with HA-tagged DNA LIG3 were hybridized to an EcoRI–BamHI fragment corresponding to the C-terminal region of DNA ligase III cDNA carrying a hemagglutinin (HA) tag (nt 2435–3423).
Isolation of cellular genomic DNA
Cells were lysed in cell culture plates with 10 mM Tris, 1 mM EDTA, 0.5% SDS. The lysate was incubated with proteinase K for 12 h at 37°C. DNA was extracted with phenol/chloroform, ethanol precipitated and the DNA strand pulled out using a glass pipette. The DNA was washed with 70% ethanol, air dried and dissolved in 10 mM Tris, pH 8.0, 1 mM EDTA (TE buffer). DNA concentration was measured by reaction with diphenylamine (43) with sheared herring sperm DNA as the standard.
Southern blot analysis
DNA was linearized with PvuII and 1 µg linearized DNA resolved on a 0.5% agarose gel. To analyze denatured DNA, the linearized sample was treated with 0.2 N NaOH for 30 min at 37°C, prior to running on a gel as described earlier (44). The samples were transferred to a nylon membrane by capillary action. Blots were hybridized overnight at 50°C in the presence of Church buffer (45).
DNA probe
The entire mouse mitochondrial genome cloned in pSP6 (a gift of Dr S. P. Ledoux, University of South Alabama) was digested with XhoI and BglII. The resulting 1778 bp fragment from the mouse mitochondrial genome was gel purified and labeled with [α-32P]dATP by the random primer method (46). Typically the specific activity of the probes was 5 × 107 c.p.m./µg DNA.
Cell respiration
HT1080 cells were suspended in serum-free Dulbecco’s modified Eagle’s medium (DMEM) at a density of 1 × 106 cells/ml and oxygen consumption measured at 37°C, using a Clark-type oxygen electrode (47).
Generation of ρ0 cells
HT1080 fibroblast cells were grown for 3–4 weeks in medium containing 9% fetal bovine serum and supplemented with 50 ng/ml ethidium bromide, 50 ng/ml uridine and 1 mM pyruvate. Loss of mtDNA was monitored by Southern blot analysis by hybridizing total genomic DNA with a mtDNA probe (42).
Treatment of mtDNA with T4 DNA ligase
Genomic DNA (1 µg) isolated from control and antisense DNA ligase III-expressing cells was treated with 1 µl of T4 DNA ligase (2000 U/µl; New England Biolabs) at 14°C for 3–4 h. Following ligation, the DNA was digested with PvuII for 2 h at 37°C. The samples were denatured with 0.2 N NaOH prior to electrophoresis on a 0.5% agarose gel. DNA was transferred to a nylon membrane and hybridized with a mtDNA probe.
Overexpression of mitochondrial DNA ligase III in AS1 cells
The plasmid pLIG3-FL2, a construct previously shown to generate only the mitochondrial form of DNA ligase III (34), was transfected into AS1 cells. Cells were selected in 200 µg/ml Genetecin (Gibco BRL) in DMEM containing 9% fetal bovine serum and hygromycin. Individual clones were isolated and positive clones identified by northern and western blot analyses.
Irradiation of cells
Human HT1080 cells (1 × 106 cells/ml) were suspended in 3 ml serum-free medium and exposed to 2 Gy (200 rad) γ-radiation in a 137Cs irradiator. The irradiated cells were divided into three equal aliquots. Genomic DNA was isolated from one aliquot immediately. The other two aliquots were plated separately in serum-containing medium and allowed to recover for 24 and 48 h, respectively, after which time genomic DNA was isolated. Equal numbers of untreated cells were used as controls.
RESULTS
Recently our laboratory demonstrated that the mammalian DNA ligase III gene encodes a mitochondrial form of this protein (34). Additional studies suggest that this enzyme may be involved in mtDNA repair processes such as base excision repair and double-strand break repair (33,34,48,49). It appears that DNA ligase III is the sole form of ligase in the mitochondria, thereby playing a central role in mitochondrial DNA dynamics. To study the role of DNA ligase III in mitochondrial DNA maintenance, cells with reduced levels of DNA ligase III activity were created using an antisense strategy. This method was chosen over gene inactivation based on results obtained with Saccharomyces cerevisiae. DNA ligase gene CDC9 has been shown to be essential for mitochondrial function (35). Recently, experiments in our laboratory showed that loss of mitochondrial Cdc9p leads to complete loss of mitochondrial DNA (Donahue et al., submitted for publication). Since DNA ligase III appears to be the sole form of mitochondrial ligase in mammalian cells, we hypothesized that creation of a DNA ligase III null would lead to cells entirely lacking mitochondrial DNA. Therefore, human cells deficient in DNA ligase III were created using an antisense strategy. Human fibrosarcoma-derived HT1080 cells were stably transfected with pREP-hLIG3, a plasmid harboring a portion of the human ligase III gene (nt 73–331) in the antisense orientation. As a control cells were transfected in parallel with an unmodified pREP4 vector. Transfectants were isolated by growth in hygromycin. Clones that expressed DNA ligase III antisense mRNA were identified by northern blot analysis (Fig. 1A) and used for further study.
Figure 1.
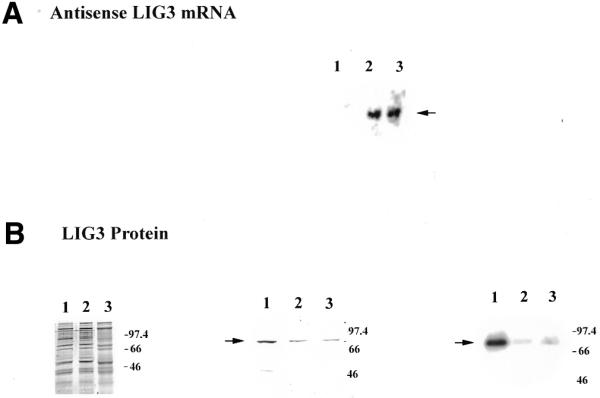
Analysis of DNA ligase III antisense-expressing clones. (A) An aliquot of 10 µg total RNA from control and antisense cells was separated on a denaturing gel and transferred to a nylon membrane. The membranes were hybridized with an [α-32P]dATP-labeled probe corresponding to the DNA ligase III cDNA (nt 73–331). Lane 1, control; lane 2, AS1 cells; lane 3, AS2 cells. (B) (Left) An aliquot of 10 µg mitochondrial extracts from control (lane 1), AS1 (lane 2) and AS2 (lane 3) cell lines. Samples were boiled for 10 min following addition of buffer containing SDS and β-mercaptoethanol and loaded on a 10% SDS–polyacrylamide gel. The gel was stained with Coomassie brilliant blue. The mobility of protein size standards in kilodaltons is indicated. (Middle) Western blot analysis of 10 µg mitochondrial protein extracts from control (lane 1), AS1 (lane 2) and AS2 (lane 3) cell lines, probed with anti-ligase III antibody at a dilution of 1:500. The arrow indicates DNA ligase III protein. (Right) Protein adenylation pattern of 5 µg mitochondrial protein extracts prepared from control cells (lane 1) and from the DNA ligase III antisense-expressing clones AS1 (lane 2) and AS2 (lane 3). The arrow indicates the adenylated mtDNA ligase. The mobility of protein size standards in kilodaltons is indicated.
Analysis of cells expressing DNA ligase III antisense mRNA
To characterize the decrease in mtDNA ligase III as a consequence of antisense expression, mitochondrial protein extracts were prepared from control and DNA ligase III antisense mRNA-expressing clones. Aliquots of 10 µg mitochondrial protein extract from a control cell line transfected with unmodified pREP4 vector (lane 1) and from two DNA ligase III antisense mRNA-expressing clones, named AS1 (lane 2) and AS2 (lane 3), were separated by PAGE and stained with Coomassie brilliant blue (Fig. 1B, left). Western blot analysis of the above mitochondrial extracts with a DNA ligase III-specific antibody (Novus Biologicals) showed reduced amounts of DNA ligase III in lanes 2 and 3, corresponding to AS1 and AS2, compared to the control in lane 1 (Fig. 1B, middle). To further characterize the DNA ligase III antisense mRNA-expressing clones, adenylation experiments were performed. Incubation of DNA ligase with ATP results in the formation of a covalent protein–adenylate complex. The amount of adenylated protein thus provides a direct measure of the amount of DNA ligase protein present. This approach has been used previously to show that XRCC1 mutant hamster cell lines have reduced DNA ligase III levels (50). Mitochondrial protein extracts prepared from cell lines AS1 and AS2 showed a significant decrease in the level of adenylated protein compared to mitochondrial extracts from control cells (Fig. 1B, right). We performed a similar analysis on mitochondrial extracts prepared from an additional control cell line and found that the level of adenylated protein detected in this extract was similar to that seen in the mitochondrial protein extract from the other control cell line. Further, to assess the quality of the mitochondrial extract the mitochondrial marker enzyme cytochrome c oxidase was assayed. The specific activity of cytochrome c oxidase in mitochondrial extracts from control cells was 1.3 ± 0.3 U/min/mg protein while the specific activities in the mitochondrial extracts from AS1 and AS2 cells were 1.4 ± 0.7 and 1.4 ± 0.4 U/min/mg protein, respectively. Since the specific activity of the mitochondrial marker enzyme is equivalent, we conclude that DNA ligase III antisense mRNA expression led to reduced levels of mtDNA ligase III. These results are consistent with our earlier observation that transient expression of DNA ligase III antisense mRNA led to reduced DNA ligase III activity in mitochondrial extracts (34).
Reduced mtDNA content in cells expressing DNA ligase III antisense mRNA
We next examined the effect of antisense DNA ligase III mRNA expression on cellular mtDNA content. Total genomic DNA was isolated from control, AS1 and AS2 cells. The DNA was digested using the restriction enzyme PvuII, which linearizes the human mitochondrial genome (51), then 1 µg total genomic DNA was subjected to agarose gel electrophoresis, transferred to a nylon membrane and hybridized to a radiolabeled mtDNA probe (see Materials and Methods). As a control, following restriction endonuclease treatment and prior to electrophoresis, the DNA was quantified using diphenylamine to ensure that equal amounts of DNA were loaded in each lane. In addition, scanning laser densitometry was performed on the ethidium bromide stained gel following electrophoresis to confirm that equal amounts of DNA had been run in each lane (not shown). Southern blot analysis revealed a hybridizing species of ∼16.5 kb, corresponding to the linearized mtDNA genome. As Figure 2A indicates, the amount of hybridization signal seen in DNA derived from the AS1 (lane 2) and AS2 cells (lane 3) was reduced compared to that seen in control cells (lane 1).
Figure 2.
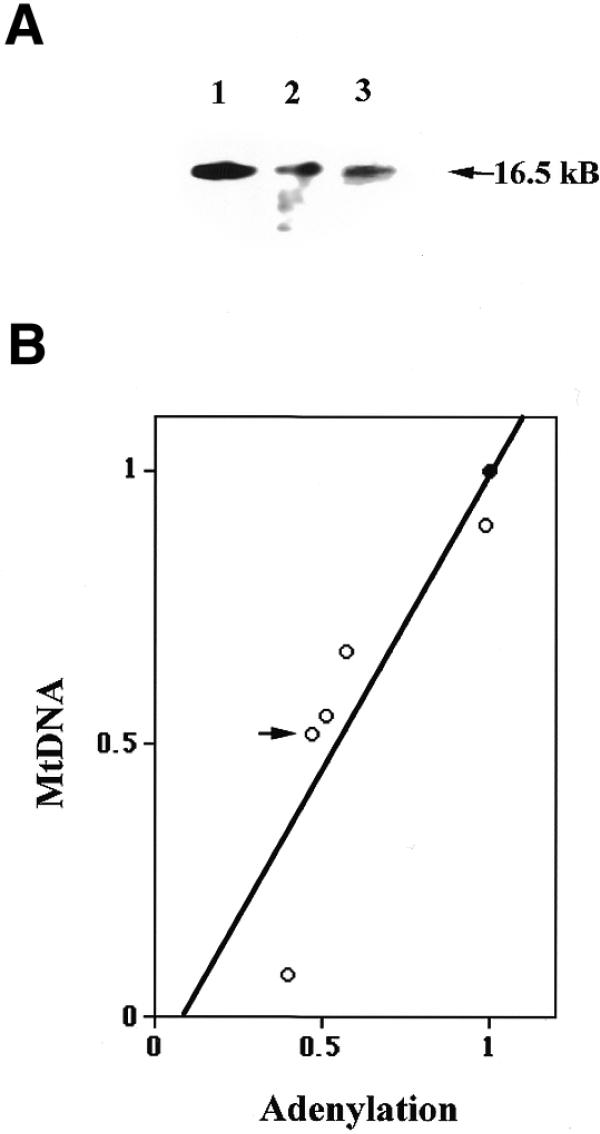
mtDNA contents in DNA ligase III antisense mRNA-expressing cells. (A) Southern blot analysis of 1 µg genomic DNA from control cells (lane 1) and clones AS1 (lane 2) and AS2 (lane 3) digested with PvuII and hybridized with a mtDNA probe. The arrow indicates the position of the 16.5 kb linearized mtDNA. (B) A plot of the adenylation activity of mitochondrial extracts versus the amount of mtDNA in control and DNA ligase III antisense mRNA-expressing cells. Southern blot analysis of 1 µg genomic DNA digested with PvuII and hybridized with a mtDNA probe. The amount of mtDNA and adenylation activity was quantified and the values obtained for AS cells normalized to the value obtained with control cells. The filled circle represents the control cell value and open circles represent the values obtained from DNA ligase III antisense mRNA-expressing cells.
Three additional DNA ligase III antisense mRNA-expressing cell lines were examined to determine more accurately the relationship between mtDNA ligase level and mtDNA content. Southern blot hybridization was used to determine relative mtDNA content, while mtDNA ligase levels were measured using the adenylation assay described above. The mean value obtained from three independent experiments was plotted. Figure 2B represents such a plot of relative mtDNA content versus the relative mtDNA ligase level in the five DNA ligase III antisense mRNA-expressing cell lines (represented as the five open circles), along with the same value obtained from control cells (closed circle). These data reveal that reduced levels of mtDNA ligase III activity correlates well with a corresponding reduction in cellular mtDNA content (R value 0.886, linear regression indicated in Fig. 2B).
DNA ligase III antisense mRNA-expressing cells have reduced respiration rate
In order to study the consequence of altered mtDNA on mitochondrial respiration, oxygen uptakes of control and AS1 cells were measured. Respiration rates were determined in sets of three and mean oxygen uptake is represented as pmol/s/106 cells (52). Whereas control HT1080 cells had a respiration rate of 12.2 pmol/s/106 cells, the respiration rate of AS1 cells was 9.2 pmol/s/106 cells. This seemingly modest reduction in respiration rate is deceptive, however, since similar analysis of HT1080 cells that lack detectable levels of mtDNA, referred to as HT1080 ρ0 cells (53,54), had a respiration rate of 5.7 pmol/s/106 cells. Thus, the respiration rate of AS1 cells is essentially half-way between that seen in the parental HT1080 cells and cells that entirely lack mitochondrial function. This result is not unexpected, given that AS1 cells have approximately half as much mtDNA as control HT1080 cells (Fig. 2B, indicated by an arrow).
Creation and analysis of a cell line expressing DNA ligase IV antisense mRNA
The nucleotide sequence located upstream of the putative initiation codon of the mammalian DNA ligase IV resembles that encoding a mitochondrial targeting sequence. However, we (34) and others (55) have shown that the peptide sequence encoded by this portion of the DNA ligase IV cDNA is not capable of targeting the green fluorescent protein into cultured mammalian somatic cells in vitro. Nevertheless, we wished to further examine whether DNA ligase IV may play an important role in mtDNA maintenance in mammalian cells. A DNA ligase IV antisense mRNA expression vector was constructed and used to transfect HT1080 cells. Transfectants expressing high levels of DNA ligase IV antisense mRNA were identified by northern blot analysis (Fig. 3A). Nuclear extracts were prepared from the control and DNA ligase IV antisense mRNA-expressing cell lines, separated on a SDS–polyacrylamide gel and a western blot analysis performed using a polyclonal antibody specific for DNA ligase IV. Figure 3B depicts the Coomassie blue stained image of the electrophoretically separated proteins (left) and the results of the western blot analysis (right). This result indicates that the nuclear extracts from DNA ligase IV antisense mRNA-expressing cell have reduced levels of DNA ligase IV.
Figure 3.
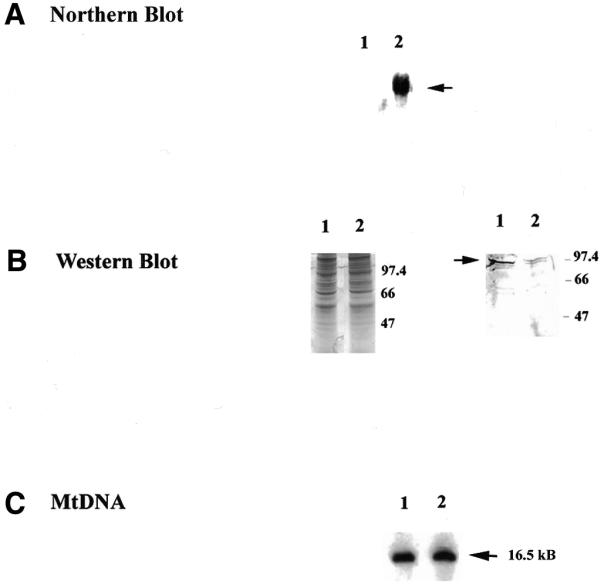
Analysis of DNA ligase IV antisense mRNA-expressing cells. (A) An aliquot of 10 µg total RNA from control and antisense cells was separated on a denaturing gel, transferred to a nylon membrane and probed with an [α-32P]dATP-labeled fragment of DNA ligase IV cDNA (nt 2435–3423). (B) (Left) An aliquot of 5 µg nuclear protein extract made from the control cell line (lane 1) or from an antisense ligase IV-expressing cell line (lane 2) was separated by SDS–PAGE. The gel was stained with Coomassie brilliant blue to ensure equal loading. (Right) Western blot analysis of 5 µg nuclear protein extract from the control cell line (lane 1) or an antisense ligase IV-expressing cell line (lane 2) using polyclonal anti-ligase IV antibody (B) at a dilution of 1:1000. (C) Southern blot analysis using a mtDNA probe of 1 µg genomic DNA isolated from control (lane 1) and antisense ligase IV mRNA-expressing cells (lane 2). The arrow indicates the mobility of intact, linearized mtDNA (16.5 kb).
The mtDNA content of control and DNA ligase IV antisense mRNA-expressing cells was examined. As Figure 3C indicates, DNA ligase IV antisense mRNA expression sufficient to reduce nuclear levels of this protein by 3-fold had no effect on mtDNA content. Taken together the results summarized above suggest that DNA ligase IV is not present in the mitochondria. Consistent with this hypothesis, western blot analysis failed to reveal any evidence for the presence of this protein in mitochondrial extracts (data not shown).
Cells expressing DNA ligase III antisense mRNA have damaged mtDNA
It is known that mtDNA is particularly prone to oxidative damage by oxygen radicals due to its proximity to the inner mitochondrial membrane, which is the center for the electron transport chain (56–59). Given the likely role of DNA ligase III in mitochondrial base excision repair (33), it was hypothesized that antisense-expressing cells would accumulate single-strand nicks in the mtDNA. The effect of antisense DNA ligase III mRNA expression on mtDNA integrity was therefore examined. This analysis was performed on DNA from ligase III antisense mRNA-expressing clone AS1. Genomic DNA from control and AS1 cells was subjected to restriction endonuclease digestion using either PvuII or SacI. To detect single-strand DNA nicks within mtDNA the digested samples were run in parallel under native or denaturing conditions. DNA samples were denatured by treatment with 0.2 N NaOH for a period of 30 min at 37°C prior to electrophoresis. Following electrophoreis and transfer to nylon membranes, duplicate samples were hybridized with radiolabeled probes complementary to nuclear or mtDNA (see Materials and Methods).
Figure 4A–C depicts samples electrophoresed under native conditions. Hybridization to a total genomic DNA probe, presented in Figure 4A, indicates that equivalent amounts of total genomic DNA from each cell line had been loaded. Figure 4B and C confirms that mtDNA content is reduced in AS1 cells, compared to the control. Figure 4D–F presents results obtained when an identical analysis was performed on samples that had been denatured with NaOH prior to electrophoresis. Hybridization of control cell DNA to a mtDNA probe revealed a single strongly hybridizing signal corresponding to intact, single-stranded mtDNA (Fig. 4E and F). In contrast, similar hybridization of both the PvuII- and SacI-digested mtDNA from AS1 cells resulted instead in a lower molecular weight smear. It is noteworthy that this occurred whether the DNA had been digested with PvuII or SacI (Fig. 4E and F). The most reasonable interpretation of these data is that the mtDNA from AS1 cells has accumulated numerous single-strand nicks or gaps that are not present in mtDNA from control cells. The fact that this mtDNA damage was not readily apparent following electrophoresis under native conditions implies that if mtDNA double-strand breaks are present in AS1 cells, they are relatively infrequent.
Figure 4.
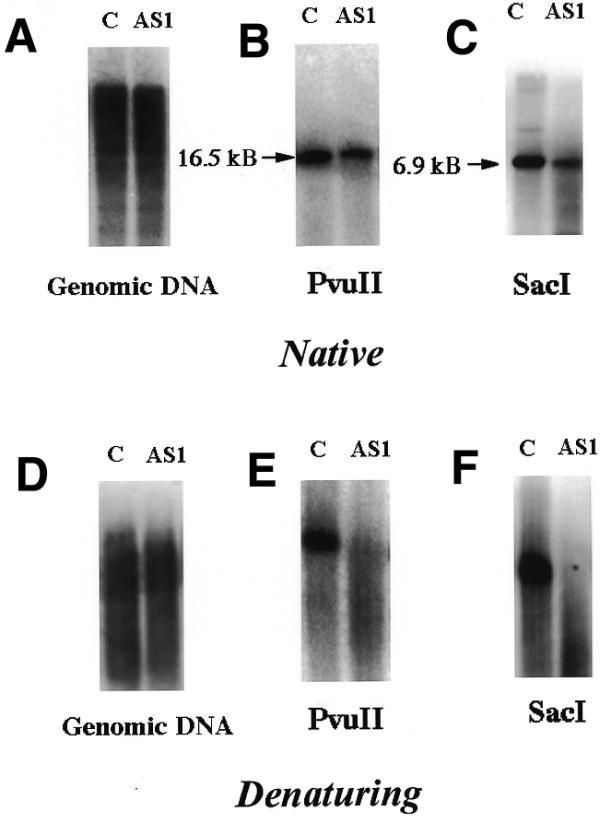
mtDNA from AS1 cells is damaged. Equal amounts of total genomic DNA from control (c) and AS1 (AS1) cells was digested with PvuII and separated on a 0.5% agarose gel under native or denaturing conditions (see Materials and Methods). (A and D) Genomic DNA digested with PvuII hybridized to a total genomic DNA probe. (B and E) Genomic DNA digested with PvuII and hybridized with a mtDNA probe. (C and F) Genomic DNA digested with SacI and hybridized to a mtDNA probe.
If mtDNA from AS1 cells had significant numbers of single-strand DNA nicks, treatment of this DNA with T4 DNA ligase should seal these nicks and retard electrophoretic mobility, causing this DNA to co-migrate with wild-type mtDNA under denaturing conditions. In contrast, if the increased gel electrophoresis migration is due to the presence of single-strand gaps, this treatment should have no effect. To distinguish between these two possibilities, genomic DNA from control and AS1 cells was treated with T4 DNA ligase. The DNA samples were then digested with PvuII, denatured with NaOH, resolved in a 0.5% agarose gel and Southern blot hybridization performed using a mtDNA probe. As Figure 5A indicates, single-stranded mtDNA isolated from control cells appeared as a band both with and without treatment with T4 DNA ligase (control – and +). However, mtDNA from AS1 cells migrated as a broad smear with a greater mobility (AS1 –). The mobility of mtDNA was retarded following treatment with T4 DNA ligase (AS1 +). Scanning densitometric analysis was performed on the Southern blot hybridization as depicted in Figure 5A and the mtDNA band intensity plotted versus the size of the DNA fragment (calculated based on its mobility relative to a marker). The untreated AS1 mtDNA migrated as a broad smear with an average fragment length of 3.5 kb. This represents an average of 4.7 nicks per mitochondrial genome. However, following treatment with T4 DNA ligase the electrophoretic mobility of this mtDNA was reduced to an average fragment length of 7.5 kb (Fig. 5B, right), equivalent to 2.2 nicks per mitochondrial genome. In contrast, the electrophoretic mobility of mtDNA from control cells was unaffected by treatment with T4 DNA ligase. The mtDNA from control cells migrated with an average fragment length of 15 kb, irrespective of whether it had been treated with T4 DNA ligase or not (Fig. 5B, left). This analysis strongly supports the conclusion that mtDNA from AS1 cells has large numbers of single-strand DNA nicks that are not present in mtDNA from control cells.
Figure 5.
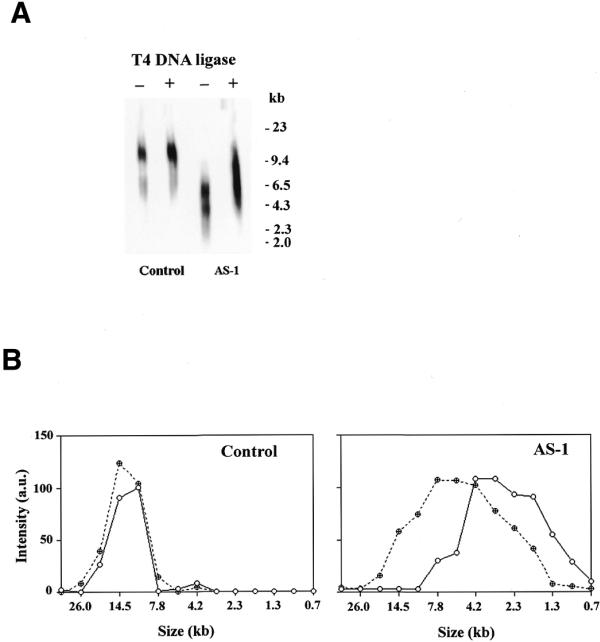
T4 DNA ligase treatment of mtDNA from control and AS1 cells. (A) Genomic DNA control and AS1 cells electrophoresed under denaturing conditions and hybridized with a mtDNA probe. –, no pretreatment; +, purified DNA treated with T4 DNA ligase prior to denaturation and electrophoresis. (B) Scanning densitometry was performed on the data presented above. (Left) DNA from control cell line; (right) DNA from AS1 cell line. Filled circles, pre-treatment with T4 DNA ligase; open circles, no pre-treatment with T4 DNA ligase. The sizes of the fragments were calculated based on the mobility of the band compared to a molecular weight standard.
The mitochondrial DNA is under constant attack by reactive oxygen species (ROS) that leak from the electron transport chain. The DNA repair enzymes in the mitochondria efficiently challenge such oxidative damage to the mitochondrial genome. Deficiency of DNA ligase III, a key component in this process, leads to accumulation of DNA repair intermediates such as nicks in AS1 cells.
A number of experiments were performed in which AS1 mtDNA was treated with increasing concentrations of T4 DNA ligase. In no case were we able to cause AS1 mtDNA to co-migrate with mtDNA from control cells during denaturing gel electrophoresis (not shown). Thus, it appears that lesions other than simple single-strand DNA nicks are also present in the mtDNA from these cells, such as, for example, single-strand gaps. It seems likely that these gaps form as a consequence of nucleolytic processing of nicks. However, it is unclear whether this process occurs within intact cells or instead occurs during purification of genomic DNA from lysed cells.
In order to further establish that the nicks formed in AS1 cells were a consequence of reduced ligase activity, AS1 cells were transfected with mtLIG3-pEGFPmut. This plasmid construct specifically encodes the mitochondrial form of DNA ligase III and its localization in the mitochondria has been confirmed by immunocytochemistry (34). Clones that overexpressed the mitochondrial DNA ligase were identified and rescue of mitochondrial DNA ligase level was measured by western blot analysis (Fig. 6A). Mitochondrial DNA from such cells was analyzed further. Under native conditions AS1 cells have only 50% mtDNA relative to control cells, while AS1 cells transfected with full-length human ligase III showed a partial recovery in the amount of mtDNA to 75% relative to control (Fig. 6B). Under denaturating conditions control AS1 cells failed to show any intact mtDNA band while AS1 cells that overexpress mitochondrial DNA ligase III showed a hybridization signal at 16.5 kb, corresponding to full-length mtDNA (Fig. 6C). The above results allow us to conclude that loss of integrity of mitochondrial DNA is a direct consequence of reduced levels of mitochondrial DNA ligase III.
Figure 6.

Partial rescue of mtDNA amount and integrity of AS1 cells by overexpression of mitochondrial DNA ligase III. (A) Western blot analysis of mitochondrial extracts prepared from control cells, AS1 cells and AS1 cells overexpressing mitochondrial DNA ligase III. Aliquots of 10 mg mitochondrial extracts were separated by 10% SDS–PAGE and transferred to a nylon membrane. Blots were probed with anti-ligase III antibody at a 1:500 dilution. (B) The amount of mtDNA in control, AS1 cells and AS1 cells overexpressing mitochondrial DNA ligase III was measured. Genomic DNA digested with PvuII was separated on a 0.5% agarose gel under native conditions, transferred to a nylon membrane and probed with a portion of mtDNA. The mtDNA was quantified using IP Labgel. Three independent experiments were carried out and the average value obtained plotted as a graph. Standard errors of the mean are represented as error bars. (C) PvuII-digested genomic DNA from control cells, AS1 cells and AS1 cells overexpressing mitochondrial DNA ligase III was denatured in the presence of 0.2 N NaOH prior to electrophoresis on a 0.5% agarose gel. The blot was treated similarly to as described above.
Effect of ionizing radiation on cells expressing DNA ligase III antisense mRNA
The results described above suggest that AS1 cells have a diminished ability to repair spontaneous mtDNA damage. It was therefore of interest to examine the mtDNA of these cells following treatment with a DNA-damaging agent. Control and AS1 cells were therefore subjected to 2 Gy γ-radiation from a 137Cs source. Total genomic DNA was then harvested from the cells immediately after damage or following a recovery period at 37°C of 24 or 48 h. The DNA harvested from the samples was quantitated and an equivalent amount of DNA from each sample was separated on a native 0.5% agarose gel and hybridized with a mtDNA probe. A series of independent irradiation/recovery experiments were performed and the pooled data from three such experiments are presented in Figure 6. In both control and AS1 cells there is an immediate reduction in mtDNA content upon irradiation. In control cells mtDNA content was immediately reduced by ∼80% of intact mtDNA content (Fig. 7A). A similar treatment of AS1 cells reduced mtDNA levels to below the limit of detection (Fig. 7B). Following a 24 h recovery period the mtDNA content in control cells had recovered to ∼75% that of pre-treatment levels. In marked contrast, AS1 cellular mtDNA content remained nearly undetectable for over 24 h. Allowing a longer period of recovery (48 h) led to an ∼40% restoration of mtDNA content in the antisense-expressing cells. By this time control cells had restored their mtDNA to pre-irradiation levels.
Figure 7.

Analysis of mtDNA following exposure of control and AS1 cells to γ-radiation. mtDNA content of control and AS1 cells at various times before and after exposure to 2 Gy radiation. Genomic DNA was isolated and Southern hybridization performed using a mtDNA specific probe as described (see Materials and Methods). Scanning densitometry was performed on data from three independent experiments. The mtDNA content present in each cell line at each time point was normalized to that present in untreated cells. (A) Control cells; (B) AS1 cells. Error bars indicate the standard error of the mean, n = 3.
We have determined that DNA ligase III antisense mRNA expression reduces homologous recombination activity and renders these cells slightly more sensitive to the cytotoxic effects of ionizing radiation, presumably due to reduced levels of nuclear DNA ligase III (U.Lakshmipathy and C.Campbell, manuscript in preparation). We therefore wanted to be certain that the reduced capacity of AS1 cells to restore mtDNA content to pre-irradiation levels was not due to their increased sensitivity to the cytotoxic effects of this agent. Using cell counting and trypan blue exclusion, the cell number and cell viability of both control and AS1 cells was measured before, immediately after and 24 h after exposure to 2 Gy radiation. We saw that there was a slightly greater decrease in the number of AS1 cells (59%) present 24 h following irradiation compared to control cells (40%). However, there was no difference between the two cell populations in the percentage of non-viable cells present at any of the time points tested, suggesting that cells killed by irradiation had come off the dish and were lost. Thus the reduced capacity of AS1 cells to restore their mtDNA content to pre-irradiation levels is not due to increased cell death, but is instead a consequence of reduced levels of mtDNA ligase III.
DISCUSSION
Using antisense expression technology a series of cell lines that contained reduced levels of mtDNA ligase III was generated. Detailed inspection of a number of these clones revealed a direct relationship between reduced levels of mtDNA ligase III and reduced cellular mtDNA content. The residual mtDNA present in antisense-expressing cells harbored significant numbers of single-strand DNA breaks. In addition, cells with reduced levels of mtDNA ligase III were deficient in restoring mtDNA to pre-irradiation levels following exposure to ionizing radiation.
These findings support the conclusion that mtDNA ligase III plays an essential role in the maintenance of mtDNA in mammalian somatic cells in culture. Our results suggest that DNA ligase III is probably the only mtDNA ligase in mammalian cells. While it had been proposed (33) that the DNA ligase IV gene could encode a mitochondrial form of DNA ligase, our results indicate that this is unlikely. First, we found that expression of antisense DNA ligase IV mRNA had no effect on cellular mtDNA content. Second, western blot analysis of mitochondrial extracts failed to detect the presence of DNA ligase IV protein. Third, we and others (34,55) have shown that a fusion protein of DNA ligase IV and green flourescent protein fails to localize to the mitochondria. These data suggest that if DNA ligase IV is present in mammalian mitochondria, it is present only at very low levels. Likewise, if other, as yet unidentified, DNA ligase proteins are present in mammalian mitochondria they must be present at very low levels, since adenylation experiments performed on mitochondrial extracts detected only a single DNA ligase protein, which corresponded to DNA ligase III. Taken together, these results are most consistent with the hypothesis that DNA ligase III is the sole form of DNA ligase present in mammalian mitochondria.
It appears overwhelmingly likely that DNA ligase III participates in mitochondrial base excision repair. This conclusion is supported by the finding that a DNA ligase III-like molecule participates in cell-free base excision repair catalyzed by mitochondrial extracts prepared from Xenopus oocytes (33). The presence of elevated numbers of mtDNA single-strand breaks in DNA ligase III-deficient cells is consistent with a role of this enzyme in base excision repair of spontaneous DNA damage. The reduced rate of recovery of mtDNA in DNA ligase III-deficient cells following irradiation suggests that this protein could also participate in base excision repair of induced mtDNA damage. Based on the apparent absence of additional forms of mtDNA ligase, it seems likely that mtDNA ligase III also seals nicks formed during replication of the mitochondrial genome (60). It is known that mammalian nuclear DNA ligases function by interacting with other DNA repair proteins (12). We are thus interested in identifying mitochondrial proteins that interact with DNA ligase III, in order to gain more insight into the molecular mechanisms of mtDNA repair.
Acknowledgments
ACKNOWLEDGEMENTS
We gratefully acknowledge Dr Nick Broustovetski (Department of Neuroscience, University of Minnesota) for his help in cellular respiration studies. Polyclonal ligase IV antibody was a kind gift from Dr Susan Critchlow (Wellcome/CRC Institute, UK). We thank Dr Timothy F. Walseth, Dr Richard Lundberg and Greg Coffey for editorial help. Grants from the American Heart Association (9601390), the American Cancer Society (DHP 171) and the National Institutes of Health (CA61906 and AG11678) supported this work. U.L. was supported by a Postdoctoral Fellowship Award from the American Heart Association Minnesota Affiliate.
References
- 1.Applegren N., Hickey,R.J., Kleinschmidt,A.M., Zhou,Q.Q., Cell,J., Wills,P., Swaby,R., Wei,Y.T., Quan,J.Y., Lee,M.Y.W.T. and Malkas,L.H. (1995) Further characterization of the human cell multiprotein DNA replication complex. J. Cell. Biochem., 59, 91–107. [DOI] [PubMed] [Google Scholar]
- 2.Turchi J.J., Huang,L., Murante,R.S., Kim,Y. and Bambara,R.A. (1994) Enzymatic completion of mammalian lagging-strand DNA replication. Proc. Natl Acad. Sci. USA, 91, 9803–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waga S., Bauer,G. and Stillman,B. (1994) Reconstitution of complete SV40 replication with purified replication factors. J. Biol. Chem., 69, 10923–10934. [PubMed] [Google Scholar]
- 4.Montecucco A., Savini,E., Biamonti,G., Stefanini,M., Focher,F. and Ciarrocchi,G. (1995) Late induction of human DNA ligase I after UV-C irradiation. Nucleic Acids Res., 23, 962–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aboussekhra A., Biggerstaff,M., Shivji,M.K.K., Vilpo,J.A., Moncollin,V., Podust,V.N., Protic,M., Hubscher,U., Egly,J.M. and Wood,R.D. (1995) Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell, 80, 859–868. [DOI] [PubMed] [Google Scholar]
- 6.Chen J.W., Tomkinson,A.E., Ramos,W., Mackey,Z.B., Danehower,S., Walter,C.A., Schultz,R.A., Besterman,J.M. and Husain,I. (1995) Mammalian DNA ligase III: molecular cloning, chromosomal localization and expression in spermatocytes undergoing meiotic recombination. Mol. Cell. Biol., 15, 5412–5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubota Y., Nash,R.A., Klugland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 8.Caldecott K.W., Mckeown,C.K., Tucker,J.D., Ljungquist,S. and Thompson,L. (1994) An interaction between the mammalian repair protein Xrcc1 and DNA ligase III. Mol. Cell. Biol., 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y.C., Burkhart,W.A., Mackey,Z.B., Moyer,M.B., Ramos,W., Husain,I., Chen,J.W., Besterman,J.M. and Tomkinson,A.E. (1994) Mammalian DNA ligase II is highly homologous with vaccinia DNA ligase: identification of the DNA ligase II active site for enzyme adenylate formation. J. Biol. Chem., 269, 31923–31928. [PubMed] [Google Scholar]
- 10.Roberts E., Nash,R.A., Robins,P. and Lindahl,T. (1994) Different active sites of mammalian DNA ligases I and II. J. Biol. Chem., 269, 3789–3792. [PubMed] [Google Scholar]
- 11.Husain I., Tomkinson,A.E., Burkhart,W.A., Moyer,M.B., Ramos,W., Mackey,Z.B., Besterman,J.M. and Chen,J.W. (1995) Purification and characterization of DNA ligase III from bovine testis. J. Biol. Chem., 270, 9683–9690. [DOI] [PubMed] [Google Scholar]
- 12.Tomkinson A.E. and Levin,D.S. (1997) Mammalian DNA ligases. Bioessays, 19, 893–901. [DOI] [PubMed] [Google Scholar]
- 13.Wei Y.F., Robins,P., Carter,K., Caldecott,K., Pappin,D.J.C., Yu,G.L., Wang,R.P., Shell,B.K., Nash,R.A., Schar,P., Barnes,D.E., Haseltine,W.A. and Lindahl,T. (1995) Molecular cloning and expression of human cDNAs encoding a novel DNA ligase IV and DNA ligase III, an enzyme active in DNA repair and genetic recombination. Mol. Cell. Biol., 15, 3206–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chu G. (1997) Double strand break repair. J. Biol. Chem., 272, 24097–24100. [DOI] [PubMed] [Google Scholar]
- 15.Labhart P. (1999) Nonhomologous DNA end-joining in cell-free systems. Eur. J. Biochem., 265, 847–861. [DOI] [PubMed] [Google Scholar]
- 16.Johnson A.P. and Fairman,M.P. (1997) The identification and purification of a novel mammalian ligase. Mutat. Res., 383, 205–212. [DOI] [PubMed] [Google Scholar]
- 17.Webster A.D.B., Barnes,D.E., Arlett,C.F., Lehmann,A.R. and Lindahl,T. (1992) Growth retardation and immunodeficiency in a patient with mutations in the ligase I gene. Lancet, 339, 1508–1509. [DOI] [PubMed] [Google Scholar]
- 18.Lonn U., Lonn,S., Nylen,U. and Winblad,G. (1989) Altered formation of DNA replication intermediates in human 46 BR fibroblast cells hypersensitive to 3-aminobenzamide. Carcinogenesis, 10, 981–985. [DOI] [PubMed] [Google Scholar]
- 19.Prigent C., Satoh,M.S., Daly,G., Barnes,D.E.and Lindahl,T. (1994) Abberant DNA repair and DNA replication due to an inherited defect in human DNA ligase I. Mol. Cell. Biol., 14, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teo S.H. and Jackson,S.P. (1997) DNA ligase IV: involvement in DNA double strand break repair. EMBO J., 16, 4788–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson L.M., Arlett,C.F., Harcourt,S.A., Lehmann,A.R. and Broughton,B.C. (1985) Cells from an immunodeficient patient (46BR) with a defect in DNA ligation are hypomutable but sensitive to the induction of sister chromatid exchange. Proc. Natl Acad. Sci. USA, 82, 2044–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lehmann A.R., Willis,A.E., Broughton,B.C., James,M.R., Steingrimsdottir,H., Harcourt,S.A., Arlett,C.R. and Lindhal,T. (1988) Relation between the human fibroblast strain 46BR and cell lines representative of Bloom’s syndrome. Cancer Res., 48, 6343–6347. [PubMed] [Google Scholar]
- 23.Hoy C.A., Fuscoe,J.C. and Thompson,L.H. (1987) Recombination and ligation of transfected DNA in CHO mutant EM9, which has high levels of sister chromatid exchange. Mol. Cell. Biol., 7, 2007–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thacker J. (1989) The use of integrating DNA vectors to analyze the molecular defects in ionizing radiation-sensitive mutants of mammalian cells including ataxia telangiectasia. Mutat. Res., 220, 187–204. [DOI] [PubMed] [Google Scholar]
- 25.Riballo E., Critchlow,S.E., Teo,S.H., Doherty,A.J., Priestley,A., Broughton,B., Kysela,B., Beamish,H., Plowman,N., Arlett,C.F., Lehmann,A.R., Jackson,S.P. and Jeggo,P.A. (1999) Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol., 9, 699–702. [DOI] [PubMed] [Google Scholar]
- 26.Grawunder U., Zimmer,D., Fugmann,S., Schwarz,K. and Lieber,M.R. (1998) DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell, 2, 477–484. [DOI] [PubMed] [Google Scholar]
- 27.Frank K.M., Sekiguchi,J.M., Seidl,K.J., Swat,W., Rathbun,G.A., Cheng,H.L., Davidson,L., Kangaloo,L. and Alt,F.W. (1998) Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature, 396, 173–177. [DOI] [PubMed] [Google Scholar]
- 28.Schar P., Herrmann,G., Daly,G. and Lindahl,T. (1997) A newly identified DNA ligase of Saccharomyces cerevisiae involved in RAD52-independent repair of DNA double-strand breaks. Genes Dev., 11, 1912–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson T.E., Grawunder,U. and Lieber,M.R. (1997) Yeast DNA ligase IV mediates non-homologous DNA end-joining. Nature, 388, 495–498. [DOI] [PubMed] [Google Scholar]
- 30.Bryans M., Valenzano,M.C. and Stamato,T.D. (1991) Absence of DNA ligase IV protein in XR-1 cells: evidence for stabilization by Xrcc4. Mutat. Res., 433, 53–58. [DOI] [PubMed] [Google Scholar]
- 31.Grawunder U., Zimmer,D., Kulesza,P. and Lieber,M.R. (1998) Requirement for an interaction of XRCC4 with DNA ligase IV for wild type V(D)J recombination and DNA double strand break repair in vivo. J. Biol. Chem., 273, 24708–24714. [DOI] [PubMed] [Google Scholar]
- 32.Levin C.J. and Zimmerman,S.B. (1976) A DNA ligase from mitochondria of rat liver. Biochem. Biophys. Res. Commun., 69, 5614–5620. [DOI] [PubMed] [Google Scholar]
- 33.Pinz K.G. and Bogenhagen,D.F. (1998) Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol. Cell. Biol., 18, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lakshmipathy U. and Campbell,C. (1999) The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol., 19, 3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Willer M., Rainey,M., Pullen,T. and Stirling,C.J. (1999) The yeast cdc9 gene encodes both nuclear and a mitochondrial form of DNA ligase I. Curr. Biol., 9, 1085–1094. [DOI] [PubMed] [Google Scholar]
- 36.Rasheed S., Nelson-Rees,W.A., Toth,E.M., Arnstein,P. and Garnder,M.B. (1974) Characterization of a newly derived human sarcoma cell line (HT-1080). Cancer, 33, 1027–1033. [DOI] [PubMed] [Google Scholar]
- 37.Keown W.A., Campbell,C.R. and Kucherlapati,R.S. (1990) Methods for introducing DNA into mammalian cells. Methods Enzymol., 185, 527–357. [DOI] [PubMed] [Google Scholar]
- 38.Darley-Usmar V.M., Rickwood,D. and Wilson,M.T. (1987) Mitochondria: A Practical Approach. IRL Press, New York, NY.
- 39.Bradford M.M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of proteins utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 40.Jessberger R. and Berg,P. (1991) Repair of deletions and double-strand gaps by homologous recombination in a mammalian in vitro system. Mol. Cell. Biol., 11, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomkinson A.E., Totty,N.F., Ginsburg,M. and Lindahl,T. (1991) Location of the active site for the enzyme-adenylate formation in DNA ligases. Proc. Natl Acad. Sci. USA, 88, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 43.Abraham G.N., Scaletta,C. and Vaughan,J.H. (1972) Modified diphenylamine reaction for increased sensitivity. Anal. Biochem., 49, 547–549. [DOI] [PubMed] [Google Scholar]
- 44.LeDoux S.P., Wilson,G.L., Beecham,E.J., Stevsner,T., Wassermann,K. and Bohr,V.A. (1992) Repair of mitochondrial DNA after various types of DNA damage in chinese hamster ovary cells. Carcinogenesis, 13, 1967–1973. [DOI] [PubMed] [Google Scholar]
- 45.Church G.M. and Gilbert,W. (1984) Genomic sequencing. Proc. Natl Acad. Sci. USA, 81, 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feinberg A.P. and Vogelstein,B. (1983) A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem., 132, 6–13. [DOI] [PubMed] [Google Scholar]
- 47.Broustovetski N. and Dubinsky,J.M. (2000) Dual responses of CNS mitochondria to elevated calcium. J. Neurosci., 20, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lakshmipathy U. and Campbell,C. (1999) Double strand break rejoining by mammalian mitochondrial extracts. Nucleic Acids Res., 27, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Coffey G., Lakshmipathy,U. and Campbell,C. (1999) Mammalian mitochondrial extracts possess DNA end-binding activity. Nucleic Acids Res., 27, 3348–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Caldecott K.W., McKeown,C.K., Tucker,J.D., Ljungquist,S. and Thompson,L. (1994) An interaction between the mammalian repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol., 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson S., Bankier,A.T., Barrell,B.G., De Bruijn,M.H.L., Coulson,A.R., Drouin,J., Eperon,I.C., Nierlich,D.P., Roe,B.A., Sanger,F., Schreier,P.H., Smith,A.J.H., Staden,R. and Young,I.G. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- 52.Wiseman A. and Attardi,G. (1978) Reversible tenfold reduction in mitochondrial DNA content of human cells treated with ethidium bromide. Mol. Gen. Genet., 167, 51–63. [DOI] [PubMed] [Google Scholar]
- 53.King M.P. and Attardi,G. (1988) Injection of mitochondria into human cells leads to rapid replacement of the endogenous mitochondrial DNA. Cell, 52, 811–819. [DOI] [PubMed] [Google Scholar]
- 54.King M.P. and Attardi,G. (1989) Respiratory enzymes and mitochondrial morphology of HeLa and L cells treated with chloramphenicol and ethidium bromide. Science, 246, 500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bogenhagen D.F. (1999) Repair of mtDNA in vertebrates. Am. J. Hum. Genet., 64, 1276–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritcher C., Park,J.-W. and Ames,B.N. (1988) Normal oxidative damage to mitochondria and nuclear DNA is extensive. Proc. Natl Acad. Sci. USA, 85, 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shigenaga M.K., Hagen,T.M. and Ames,B.N. (1994) Oxidative damage and mitochondrial decay in aging. Proc. Natl Acad. Sci. USA, 91, 10771–10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yakes F.M. and Van Houten,B. (1997) Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl Acad. Sci. USA, 94, 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skowronek P., Haferkamp,O.and Rodel,G. (1992) A fluorescence microscopic and flow-ctyometric study of HeLa cells with an experimentally induced respiratory deficiency. Biochem. Biophys. Res. Commun., 187, 991–998. [DOI] [PubMed] [Google Scholar]
- 60.Holt I.J., Lorimer,H.E. and Jackobs,H.T. (2000) Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell, 100, 515–524. [DOI] [PubMed] [Google Scholar]