

Summary
Understanding the diversification of dendritic cell (DC) lineages is one of the last frontiers in mapping the developmental hierarchy of the hematopoietic system. DCs are a vital link between the innate and adaptive immune responses, thus elucidating their developmental pathways is crucial for insight into the generation of natural immunity and for learning how to regulate DCs in clinical settings. DCs arise from hematopoietic stem cells through specialized progenitor subsets under the direction of FMS-like tyrosine kinase 3 ligand (Flt3L) and Flt3L receptor (Flt3) signaling. Recent studies have revealed important contributions from granulocyte-macrophage colony-stimulating factor (GM-CSF) and type I interferons (IFNs) in vivo. Furthermore, DC development is guided by lineage-restricted transcription factors such as IRF8, E2-2, and Batf3. A critical question centers on how cytokines and lineage-restricted transcription factors operate molecularly to direct DC diversification. Here we review recent findings that provide new insight into the DC developmental process.
Keywords: dendritic cells, cytokines, transcription factors, hematopoiesis, lineage commitment/specification
Introduction
Pathogen recognition, activation of immediate and long-term immunity and preserving tolerance to self-antigens are central to maintaining health. These key functions of the immune system are performed by dendritic cells (DCs), named for their unique morphology, which is characterized by dendrite-like extensions that mediate cell:cell contacts to regulate lymphocytes via antigen-presentation (1). In the nearly 40 years since the discovery of DCs as the major antigen-presenting cells (APCs) of the immune system by Steinman, Cohn, and colleagues (2–5), many DC subsets have been identified, and various regulatory events controlling their production and function have been elucidated. Gaps remain, however, in our understanding of the molecular mechanisms that dictate DC subset specification and development. Here we discuss recent advances in this area, focusing on the interplay between cytokines and transcription factors that control DC lineage development.
DC subsets
DCs are defined by cell surface marker protein expression, anatomical location, and functional responses. In mouse, three major types of DCs exist in steady state conditions, the plasmacytoid DCs (pDCs), resident lymphoid organ DCs (resident DCs), and peripheral tissue migratory DCs (migratory DCs) (reviewed in 1, 6–8) (Fig. 1). As their name implies, resident DCs exist in lymphoid tissue, while migratory DCs are present in nonlymphoid tissues and transit to lymphoid organs upon activation. Infection or inflammation boosts DC numbers, although the ‘inflammatory DCs’ produced in these conditions do not fully resemble DCs found in steady state and utilize a distinct developmental pathway, and thus can be considered a separate class (9, 10). Resident DCs, also termed ‘conventional DCs’, are defined in spleen by the surface phenotypes CD11c+CD11b − major histocompatibility complex class II (MHC II)+CD205+CD4−CD8α+ (resident spleen CD8α+ DCs) and CD11c+CD11b+MHC II+33D1+CD4+CD8α − (resident spleen CD4+CD8α − DCs) (6, 11). A minor population of CD11c+CD11b+CD4−CD8α−DCs (resident spleen CD11c+CD11b+ DCs) is also observed. Resident spleen CD8α+ DCs are found in the T-cell zones and exhibit higher Toll-like receptor 3 (TLR3) expression, interleukin-12 (IL-12) production, MHC class I presentation, and cross-presentation activities versus other splenic DC subsets, while resident spleen CD4+CD8α − DCs are localized to marginal zones and exhibit stronger MHC class II presentation activity compared to other DC types (11–16, reviewed in 6, 17). Subsets with similar marker characteristics and functional traits are found in lymph nodes (LNs), e.g. CD11chiMHC II+CD205+CD8α+ (resident LN CD8α+ DCs) and CD11chiMHC II+33D1+CD8α − (resident LN CD8α − DCs) (18, 19). LNs also contain migratory DC populations that share certain markers with resident LN DCs (e.g. low CD8α, CD205) (18) yet originate from the peripheral tissues, and thus the total LN DC repertoire is more extensive than that of spleen.
Fig. 1.
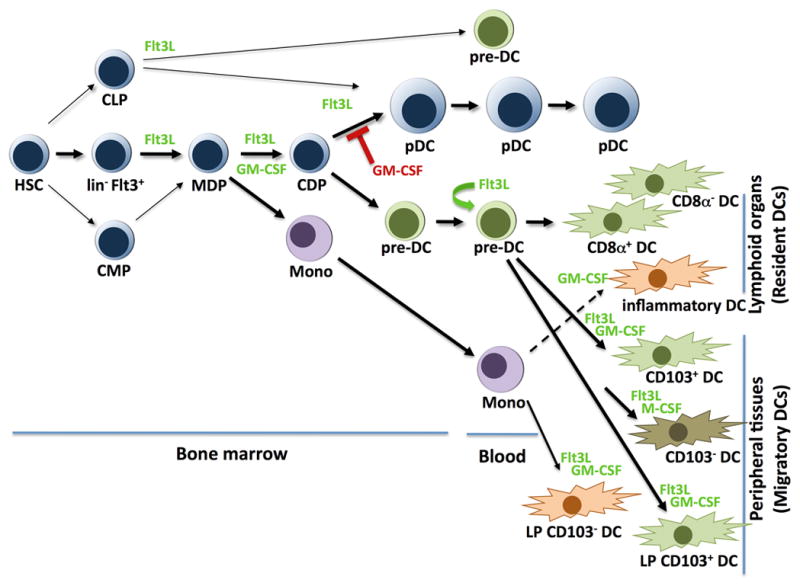
Roles for cytokines in pDC and DC development.
Migratory DCs, also referred to as ‘tissue DCs’, are positioned in several organs including skin, lung, and intestine. Skin contains epidermal Langerhans cells (LCs) and two dermal DC populations, CD103+langerin+CD11blo (dermal CD103+ DCs) and CD11c+CD11b+CD103 − subsets (dermal CD103 − DCs) (20–23, reviewed in 24, 25) (Fig. 1). Dermal CD103+ and CD103 − DCs express distinct transcription factor profiles and utilize different developmental pathways, with dermal CD103+ DCs being closely related to resident lymphoid organ CD8α+ DCs as well as CD103+ DCs in other tissues including the lamina propria (LP) CD103+CD11b − DCs (26). By contrast, dermal CD103−DCs appear to be a heterogeneous population (26, 27). In LP, CD103 − DCs were shown to descend from monocytes (28) (Fig. 1). Regardless of their developmental origin, migratory DCs share the properties of antigen uptake in peripheral tissues and migration via afferent vessels to the lymph nodes. Resident DCs, migratory DCs, and inflammatory DCs are inherently efficient antigen-presenting cells (APCs) capable of eliciting lymphocyte responses, including activation of CD4+ T helper 1 (Th1) and Th2 cells (reviewed in 29–31). Lymphocyte responses are dependent upon the intrinsic antigen presentation machinery within DCs, which mediates antigen capture and regulated secretion of antigen-containing MHC I and II molecules to the DC cell surface (32), as well as production of an array of cytokines from activated DCs (e.g. IL-12, IFNγ) (29–31). Excellent reviews on DC subset localization, phenotype, and function have been published recently (10, 24, 33). From this point forward, we use the term ‘DC’ to refer to the collective assortment of resident and migratory DC subsets, while excluding reference to the pDC lineage, which is classified separately. Further, in some cases we delineate specific DC subsets by category (e.g. resident or migratory) and/or anatomical location where clear indications exist in the literature. As DC subsets are still being resolved and most studies have not been able to comprehensively investigate all DC populations, the information provided here is a snapshot of an emerging area of intense investigation.
Plasmacytoid DCs
By contrast with DCs, pDCs are the major type I interferon (IFN)-producing cells in the body and are considered to be a primary defense against viral pathogens (34–36) (reviewed in 8, 37, 38). Recently, pDCs have been implicated in wound healing (39), indicating a significant role in tissue repair processes. pDCs are activated by triggering of endosomal TLRs, e.g. TLR7 and TLR9, which recognize nucleic acids derived from infectious virus (34–36) or, in tissue injury and autoimmune conditions, host cellular debris (40, 41). pDCs mature to an APC-like state following TLR triggering. The maturation process is characterized by downregulation of IFN secretion and induction of MHC II, costimulatory molecules, and cytokine production (42). In mouse, pDCs are defined by their surface phenotype of CD11c+CD11b−B220+PDCA1+SiglecH+, their plasma cell-like morphology, and their ability to rapidly (e.g. within 4 h) secrete abundant amounts of type I IFNs upon TLR triggering (35, 43). pDCs reside in bone marrow, blood, thymus, and T-cell rich areas of lymphoid organs in steady state conditions and can be localized to skin and other tissues in inflammation or autoimmunity (36, 44, 45).
pDCs and DCs originate from hematopoietic stem cells (HSCs) (46) (Fig. 1), yet the factors that regulate lineage specification, commitment, and development are only now being defined. A major distinction occurs between migratory subsets that self-renew in situ in steady state conditions (e.g. epidermal DCs) (47), compared to pDCs and DC subsets that are continuously regenerated from bone marrow progenitors. A second bifurcation exists between DC subsets derived from monocytes (e.g. inflammatory DCs, LP CD103 − DCs) and DCs that originate from specialized pDC/DC progenitor populations (28, 48–51) (Fig. 1). These specialized pDC/DC progenitors arise within the lin−Flt3+ progenitor population in the bone marrow (52), and proceed sequentially through stages termed the macrophage-DC progenitor (MDP), i.e. lin−c-kithiCD115+CX3CR1+Flt3+ cells, and the common DC progenitor (CDP), i.e. lin−c-kitintCD115+Flt3+ cells (48, 50, 51, 53). In this review, we focus on the developmental pathways of pDCs and DC populations that are replenished from dedicated pDC/DC bone marrow progenitors in steady state, while referring to recent reviews on locally renewing migratory DC subsets and monocyte-derived DC populations (25, 54).
Cytokine control of pDC and DC generation
GM-CSF
DCs were first grown in vitro in blood or bone marrow cultures supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) (55, 56); results suggested this important myeloid cytokine might regulate DC generation in vivo. Surprisingly however, genetic deletion of GM-CSF or the GM-CSF receptor β chain (GM-CSFRβ) results in only a modest decrease in resident DCs in steady state conditions (57, 58). Since GM-CSFRβ is required for IL-3 and IL-5 signaling, these data also indicate that development of resident DCs is largely independent of IL-3 and IL-5 function in vivo. Migratory DCs in the skin are also reduced in GM-CSF-null mice but not completely ablated (57, 59), indicating a partial requirement for GM-CSF. By contrast, overexpression of GM-CSF in transgenic animals or mice receiving daily injections of a modified form of recombinant GM-CSF designed to increase serum half-life showed a significant expansion in DC amounts in spleen and thymus (58, 60, 61), with the expanded DC populations most likely representing inflammatory DCs. It is now evident that GM-CSF drives inflammatory DC development from monocytes (49) (Fig. 1), and this monocyte-DC pathway can be observed in vivo in GM-CSF-dependent inflammatory conditions or thioglycollate-induced peritonitis (62, 63) (Fig. 1). GM-CSF- driven inflammatory DCs resemble a tumor necrosis factor-α (TNFα)- and inducible nitric oxide synthase (iNOS)-producing DC subset (Tip DCs) that appears during L. monocytogenes infection (64); however, neither inflammatory DCs nor Tip DCs appear to correspond to DC subsets found in steady state (62). As GM-CSF amounts are increased naturally during infection or inflammation (65), the production of inflammatory DCs from monocytes and potentially other precursor subsets may serve as an important means to increase APC function during conditions of physiologic demand. For purposes of developmental and functional DC studies, however, it is important to keep in mind that DCs derived ex vivo in GM-CSF cultures most likely represent DCs formed during inflammatory but not homeostatic conditions.
Flt3 ligand
FMS-like tyrosine kinase 3 ligand (Flt3L) and its receptor Flt3, a member of the tyrosine-kinase receptor family, comprise the major extracellular signaling pathway regulating steady state pDC and DC generation from bone marrow progenitors in vivo (57, 66) (Fig. 1). The unique role of Flt3L versus GM-CSF in the differentiation of both pDCs and DCs from hematopoietic progenitor cells was first recognized in ex vivo human bone marrow cultures (67) and was later confirmed in mouse bone marrow cultures (68). The importance of Flt3L in vivo was established by injection of the recombinant cytokine into mice, which stimulates a massive expansion of pDCs and resident DCs in spleen (60, 61, 69, 70). Conditional expression of Flt3L in transgenic mice expressing a tetracycline-inducible Flt3l cDNA expands DC amounts in blood, bone marrow, lymphoid organs (e.g. spleen, thymus, lymph node) as well as other tissues including liver and intestine (71), confirming that Flt3L provides an important stimulus for resident DC production in vivo and indicating its importance in migratory DC generation. Correspondingly, Flt3l−/− or Flt3−/− mice show reduced amounts of resident DCs and dermal DCs under homeostatic conditions (57, 66, 72). Subsequent studies have shown Flt3L treatment induces MDP numbers approximately 10-fold in bone marrow (72), indicating cytokine-sensitivity at an early progenitor stage. Moreover, in steady state, Flt3L and Flt3 control the abundance of the MDP in bone marrow (57, 72), although MDP amounts are not completely ablated in the absence of Flt3L-Flt3 signaling (30–40% reduction), indicating redundant pathways support MDP maintenance. In addition, homeostatic regulation of DC precursors in the spleen requires Flt3 (72, 73). Collectively, these results indicate that supraphysiological amounts of Flt3L expand early pDC/DC progenitors and promote pDC and DC production from the bone marrow compartment, while in steady state conditions Flt3 signals maintain bone marrow MDPs and resident DC numbers (Fig. 1). Studies with mice deficient in both GM-CSF (CSF2) and Flt3L (i.e. Csf2−/− Flt3l−/− mice) show that GM-CSF is also involved in regulating CDPs, resident DCs, and dermal DCs (57); however, as MDPs, pDCs, and DCs are still detected in Csf2−−/−Flt3l−/− mice, these data also suggest the existence of additional factors that control pDC/DC progenitor maintenance as well as pDC and DC generation. One such factor appears to be M-CSF, which is able to stimulate pDC and DC development from bone marrow progenitors in vitro in the absence of Flt3L signaling. In vivo, M-CSF treatment induces pDC and resident spleen DC generation in mice lacking Flt3L (74); however, the development of resident lymphoid organ DCs in homeostatic conditions does not appear to require this cytokine. M-CSF is also important for certain migratory DC subsets, including CD103 − DCs in gut and peripheral tissues, and Langerhans cells in skin (26, 28, 75) (Fig. 1). Whether M-CSF serves a redundant function in the generation of resident DCs in the absence of both GM-CSF and Flt3L signaling requires examination of DC amounts in mice that lack all three growth factors.
The availability of Flt3L in recombinant form has enabled ex vivo expansion of pDCs and DCs from bone marrow or spleen in tissue culture (68, 76–78), facilitating developmental and functional studies. Importantly, DCs that develop in Flt3L cultures (Flt3L-generated DCs) appear to correspond to resident DC subsets that are present in spleen under homeostatic conditions (76), in contrast to the inflammatory DCs formed in GM-CSF cultures. While in vitro Flt3L-generated DCs lack the full repertoire of resident spleen DC marker proteins (e.g. deficient in CD8α), expression and functional assays solidified the relationship between steady state spleen DC subsets and DC populations generated in Flt3L cultures in vitro by demonstrating close similarity in transcription factor and cell surface marker profiles, cytokine secretion patterns, and efficient MHC class I-mediated antigen presentation by classical and cross-presentation routes (76). Flt3L cultures have also led to the identification of spleen DC precursors (62), which are capable of generating resident spleen CD8α+ DCs and CD4+CD8α − DCs. Moreover, pDCs produced in bone marrow cultures with Flt3L show a potent type I IFN response upon TLR triggering by CpGA (TLR9 triggering) or viral infection, indicating functional equivalence to pDCs produced in vivo under homeostatic conditions (68).
GM-CSF has the interesting property of suppressing Flt3L-dependent pDC generation in bone marrow cultures (68). Initially, it was not clear if GM-CSF induced pDC apoptosis or blocked pDC maturation. We found that the inhibitory effect of GM-CSF is exerted at an early stage in the pDC developmental pathway. Using purified bone marrow progenitors, we showed that GM-CSF dominantly inhibits Flt3L-driven pDC development from the lin−Flt3+ bone marrow compartment in vitro (79) (Fig. 1). Furthermore, pDC numbers are decreased by ~75% in bone marrow and ~40% in spleen in response to upregulated GM-CSF expression mediated by hydrodynamic gene transfer (HGT) (Li H, Gelbard, A, Martinez, GJ, Esashi, E, Zhang, H, Ngyuen-Jackson, H., Liu, YJ, Overwijk, WW and Watowich, SS, unpublished results). Since steady state pDC numbers are unaffected in Csf2−/− mice relative to wildtype animals (57), these studies are consistent with a role for GM-CSF in demand but not homeostatic conditions. The resulting effect of GM-CSF induction is a dramatic skewing of the pDC to DC/inflammatory DC ratio, i.e. ~1:5 vs. 1:27 pDC:DC/inflammatory DC in homeostatic conditions versus GM-CSF HGT (Li et al., unpublished results). While this reduction in pDCs upon GM-CSF treatment might be expected to increase susceptibility to viral infection, patients receiving GM-CSF therapy do not appear to suffer more frequent or potent viral infections (80). We speculate that the response to GM-CSF may be in place to dampen innate production of IFN-α via pDCs, as type I IFNs can strikingly reduce DC numbers in certain settings (81) (Fig. 2).
Fig. 2.
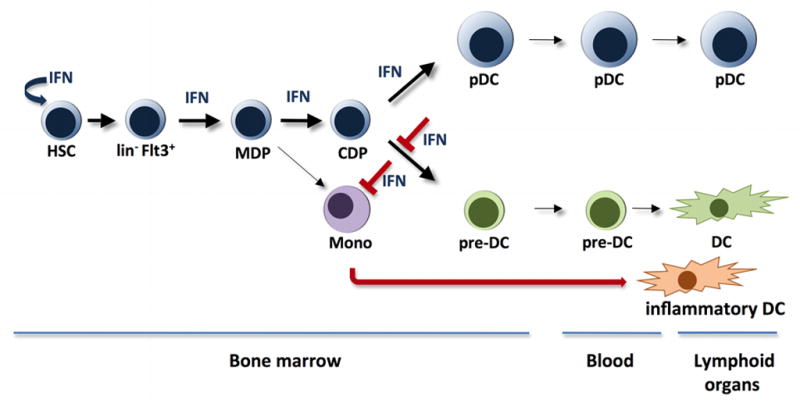
Influence of type I IFNs on pDC and DC generation.
Type I IFNs
Measles virus (MV) or lymphocytic choriomeningitis viral infection was shown to suppress DC development via type I IFN signaling (i.e. IFNAR-dependent), indicating a novel immune-evading mechanism utilized by these pathogens (81). Both GM-CSF-driven DC development in vitro and Flt3L-driven generation of splenic CD8α+ DCs in vivo was blocked by viral infection, suggesting that formation of inflammatory DCs as well as resident spleen CD8α+ DCs is sensitive to type I IFN signaling (81). This idea was confirmed by data showing a decrease in total splenic CD11c+ cells following IFN-β treatment in vivo (81). By contrast, neonatal mice infected with an attenuated strain of vaccinia showed induction of serum Flt3L, a corresponding rise in resident spleen DCs and pDCs, and enhanced defense against heterologous viral infection; these responses were dependent upon an intact IFN system in vivo (82). Furthermore, in studies to determine if the suppressive action of GM-CSF on pDC development could be negated, we identified IFN-α and IFN-β as acting to promote the generation of cells that show many phenotypic features of pDCs except the ability to produce type I IFN upon TLR9 triggering (Li et al., unpublished results). IFN-α HGT stimulates the accrual of cells resembling pDCs in vivo, while simultaneously reducing DC numbers in spleen and bone marrow (Li et al., unpublished results) (Fig. 2). Thus, type I IFNs appear to have distinct effects on pDC and DC development, namely induction of pDC numbers and general suppression of DCs. However, the negative effects of type I IFNs on DCs does not extend to all conditions (82), suggesting the function of type I IFNs in vivo may rely in part upon the specific cytokine profile in the infected animal and/or the age or developmental stage of the host. As different pathogens are likely to induce distinct cytokine profiles within specific microenvironments, we suggest that unique cytokine milieus that arise during infection may tailor local and/or systemic pDC vs. DC ratios to manage pathogen-specific host immune responses. However, this topic is relatively understudied and additional work is necessary to clarify the relevance of local and systemic cytokine production during infection and its effect upon pDC and DC generation as well as pDC/DC-regulated immune responses. Studies that utilize important human pathogens, along with natural dosages and routes of delivery, will be particularly valuable in elucidating why viral infections and type I IFNs regulate DC and pDC abundance in vivo.
To understand the functional consequence of IFN-α on pDCs, we analyzed cells generated in response to IFN-α in vitro or in vivo; these studies showed that IFN-α-elicited pDCs express the full repertoire of pDC cell surface marker proteins (i.e. CD11c+CD11b−B220+PDCA1+Siglec H+) and exhibit the unique pDC ultrastructural morphology, characterized by a well-developed endoplasmic reticulum (35, Li et al., unpublished results), and therefore closely resemble traditional Flt3L-generated pDCs. However, the IFN-α-elicited pDC population lacks TLR-triggered type I IFN production, and thus the cells do not directly correspond to pDCs. We refer to this population as IFN-pDCs to distinguish them from classical pDCs and to reflect the fact that they have been generated or ‘conditioned’ under the guidance of IFN-α. At this point, it is unclear whether IFN-pDCs are a discrete subset that is closely related to pDCs or if they represent a distinct developmental stage of the pDC lineage (e.g. functionally arrested or further differentiated). Importantly, cells with the characteristics of IFN-pDCs (e.g. pDC marker profile, deficiency in type I IFN production) were upregulated during vesicular stomatitis virus (VSV) infection, which is accompanied by high levels of endogenous IFN-α production. By contrast, VSV infection suppressed bone marrow and spleen DC numbers (Li et al., unpublished results). These data suggest that VSV-induced IFN-α may stimulate a feedback response to the bone marrow to boost production of IFN-pDCs and simultaneously inhibit DC generation and/or maintenance. As IFN-pDCs are efficient APCs (Li et al., unpublished results), their induction may compensate for the loss of DCs during VSV infection. Interestingly, the response to VSV resembles the increase in pDCs observed during infection with attenuated vaccinia virus (82); however, VSV and vaccinia have distinct effects on DC generation (suppressive vs. stimulatory, respectively). These results further support the idea that cytokine profiles and/or developmental stage of the host is likely to factor significantly in the response of pDCs and DCs during infection. IFN-α is traditionally considered to be an inflammatory, anti-viral, and anti-proliferative factor, due to its well-documented effects including induction of host genes that interfere with viral replication and cellular proliferation (83), thus induction of IFN-pDCs by this cytokine was somewhat unexpected. Recently, however, IFN-α was shown to stimulate HSC proliferation through direct and indirect mechanisms, indicating it elicits intracellular and extracellular signals to drive dormant HSCs into cell cycle (84). This pathway may contribute to the generation of IFN-pDCs in vivo.
Origins of pDCs and DCs
The importance of Flt3L-Flt3 signals in pDC and DC production led to the identification of a lin−Flt3+ bone marrow progenitor population, which is capable of generating pDCs and resident DCs following adoptive transfer into lethally irradiated hosts (52). These studies provided initial evidence for a specialized bone marrow progenitor of pDC and DCs. Subsequent refinement of progenitor populations identified MDPs, which give rise to monocyte-macrophage-lineage cells, DCs and pDCs (48, 53), and the more restricted and developmentally successive CDP subset (48, 50, 51), which generates pDCs and DCs but not monocyte/macrophage lineage cells (Fig. 1). Progenitors with restricted developmental potential for lymphoid or myeloid lineages [the common lymphoid progenitor (CLP) or common myeloid progenitor (CMP)] can also generate pDCs and resident DCs in vivo, although DC generation is restricted to the Flt3+ subset of the CLP and CMP populations (52, 85). Fully developed pDCs are found within the bone marrow; however, a relative paucity of DCs is observed, supporting the notion that DC differentiation involves the release of a DC-restricted progenitor from the marrow. This concept was verified by the identification of DC precursors in spleen (62) as well as studies using mice with experimentally manipulated shared circulatory systems (parabiotic mice), which showed that resident DCs are continuously regenerated by a short-lived circulating DC progenitor, the pre-DC (72, 86). Pre-DCs are localized in bone marrow, blood, and lymphoid organs (48) and have recently been shown to generate dermal CD103+ DCs in addition to resident DCs (26). While a restricted pDC progenitor has yet to be defined, the existence of such would be consistent with successive lineage restriction events that govern hematopoiesis. Thus, commitment to the pDC and DC lineages occurs within the bone marrow compartment, involving specification events and, potentially, differential retention (pDC) or migration (pre-DC) signals that remain poorly understood.
Molecular cues regulating DC and pDC lineage formation
Lineage-regulatory transcription factors
Cell fate decisions in the hematopoietic system involve the actions of a small number of regulatory transcription factors, which establish specification in pluripotent progenitors and induce commitment and differentiation to the blood/immune cell lineages (87–91). Hematopoietic cell specification and lineage commitment is accomplished in part by the induction and maintenance of transcriptional autoregulatory loops that sustain the expression of a particular factor (89, 91). This permits transcriptional programs that depend upon the autoregulated factor to be enacted to drive commitment and differentiation. Antagonistic interactions can occur among regulatory transcription factors; these often function to reject alternative cell fates (89, 91). Moreover, precise control of transcription factor expression or the activity level of a given factor is a crucial part of hematopoietic lineage decisions, due to autoregulatory or opposing actions, or because regulators for a particular cell type operate in conjunction with one another to affect lineage-specific gene expression.
Insight into the mechanisms controlling pDC and DC lineage specification has benefitted from our understanding of these general concepts governing hematopoiesis. For example, transcription factor expression patterns in pDCs or DCs have accurately predicted roles in their developmental decisions (92–95). Overall, however, our knowledge of pDC/DC transcriptional control mechanisms is less refined than those guiding development of T and B lymphocytes or the major myeloid cell types such as monocytes, granulocytes, erythrocytes, and megakaryocytes. One reason for this difference is the relatively recent identification of DCs and pDCs, compared to other blood cell lineages, coupled with the fact that many DC subsets are still being characterized at phenotypic and functional levels, and developmental relationships between DCs subsets remain poorly understood. Moreover, the diversity of DCs, the similarity between DCs and monocytes, and monocyte-DC conversion complicated analyses. The advent of molecular profiling, in vivo reporters and advanced anatomical studies has and will continue to distinguish DC subsets, enable their molecular characterization and ultimately provide a more complete understanding of the transcriptional networks regulating their development.
At this juncture, it appears that factors controlling pDC and DC production in vivo fall into roughly two classes: (i) those that function broadly across several DC lineages and pDCs as well as myeloid or lymphoid cell types (e.g. PU.1, Gfi1, Ikaros) (96–102) and thus are likely to regulate multipotent and/or pDC/DC progenitor (e.g. MDP or CDP) maintenance and/or differentiation potential, versus (ii) transcription factors that have specific roles in one or more (but not all) of the DC lineages and/or in pDCs. Factors in the latter category include the basic helix-loop-helix (bHLH) transcription factor (or E protein) E2-2/Tcf4, which is exclusively required for pDC development but not other blood lineages (95), and the AP-1-like protein Batf3, which controls resident CD8α+ DCs and dermal CD103+ DCs but does not regulate other DC subsets (27, 94). Here we focus on transcriptional control of the pDC and resident CD8α+ DC subsets, as these lineages have specific (e.g. E2-2 and Batf3) as well as overlapping [e.g. interferon response factor (IRF8), inhibitor of differentiation/DNA binding protein 2 (Id2)] transcription factor requirements and may therefore provide a paradigm for understanding mechanisms of pDC/DC subset specification and commitment.
E2-2
E2-2/Tcf4 expression is highly restricted within the hematopoietic system to pDCs, and analysis of E2-2-deficient mice confirmed an essential role for E2-2 in pDC generation in vivo (95) (Fig. 3). Since Tcf4−/− animals die at birth, Tcf4−/− fetal liver progenitors were used in bone marrow transplantation experiments to examine E2-2 function. These studies showed that Tcf4−/− progenitors fail to reconstitute pDCs in bone marrow or spleen; however, other blood lineages were formed in normal abundance (95). Animals with conditional Tcf4 deletion have a specific loss of pDCs but not resident DCs or other immune subsets (e.g. B cells, T cells, macrophages) (95). Moreover, E2-2 deficiency results in the accumulation of a bone marrow population that expresses some but not all pDC marker proteins (i.e. CD11c+Ly-6C+PDCA1 − cells), suggesting accrual of a progenitor subset that fails to undergo terminal differentiation. The CD11c+Ly- 6C+PDCA1 − progenitor population is observed in bone marrow from wildtype animals, although at reduced amounts relative to Tcf4−/− chimeras (95), implying E2-2 deficiency prevents pDC maturation from a lineage-committed progenitor. These results are interesting, as they point to a possible pre-pDC population (CD11c+Ly-6C+PDCA1− cells) and, furthermore, as they suggest E2-2 may not be necessary for specification of the pDC lineage from multipotent progenitors. If E2-2 were dispensable for pDC specification, this would indicate that other factors dictate pDC cell fate in multipotent progenitors such as MDPs or CDPs. Further analysis of the role of E2-2 in hematopoietic progenitor subsets, as well as studies that assess developmental options of the putative pre-pDC subset (CD11c+Ly-6C+PDCA1 − cells), is required to understand whether E2-2 is involved in pDC specification. Regardless, E2-2 has an essential and non-redundant role in guiding terminal pDC differentiation and in establishing pDC functional responsiveness. Precise E2-2 expression levels are crucial, as indicated by Tcf4 haploinsufficiency for pDC production and IFN secretion (95). Humans with Pitt-Hopkins syndrome, who contain only one wildtype Tcf4 allele, also show Tcf4 haploinsufficiency, as pDCs isolated from these individuals are unable to produce type I IFN in response to nucleic acid or viral challenge. In mice and presumably humans, E2-2 directly regulates genes that are necessary for pDC function, including those encoding IRF7, TLR7, and TLR9. Interestingly, E2-2 also appears to control other pDC-related transcription factors such as SpiB and IRF8 (95). Thus, E2-2 is an essential component of the pDC transcriptional network and may be involved at multiple stages of pDC lineage development, including initiation/reinforcement of commitment, by regulating pDC-related transcription factors, and terminal differentiation, by controlling genes that are required for pDC functional responses.
Fig. 3.
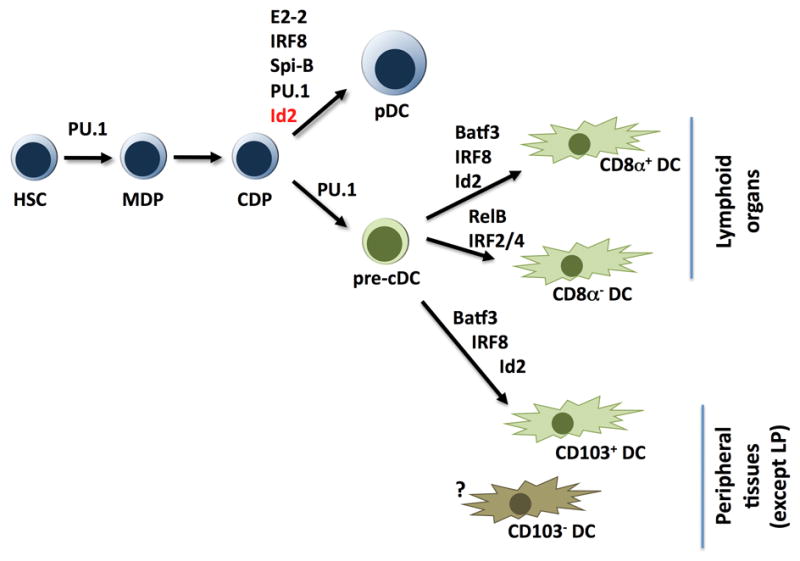
Lineage-restricted transcription factors and their role in pDC and DC development.
Id2
The Id proteins Id2 and Id3 are established antagonists of bHLH E proteins such as E2-2 (103). Id proteins contain an HLH domain that binds the bHLH domain of E proteins, forming non-functional dimers that are unable to bind DNA. Id2 is an interaction partner for E2-2 (104, 105). Accordingly, Id2 deletion augments pDC amounts approximately 1.5-fold in vivo and enhances pDC IFN production 2–4 times, relative to IFN secretion from wildtype pDCs (93) (Fig. 3). Thus, Id2 is a natural antagonist for the pDC lineage and appears to function in direct opposition to E2-2, i.e. Id2 inhibits pDC generation and IFN secretion while E2-2 orchestrates pDC development and IFN production.
The effects of Id2 on pDC production suggest Id2 has a role in suppressing E2-2 activity in pDC/DC progenitors, hence rejecting and/or limiting commitment to the pDC lineage. In agreement with this notion, Id2 is necessary for formation of resident CD8α+ DCs in spleen and dermal CD103+ DCs in vivo (Fig. 3). Thus, E2-2 antagonism via Id2 might be required for specification and/or commitment to these resident and migratory DC subsets. Total splenic CD11c+ numbers are equivalent between wildtype and Id2−/− mice, however, and overrepresentation of splenic CD4+CD8α − DCs appears to compensate for loss of splenic CD8α+ DCs in Id2−/− animals (93). These results suggest that Id2 may also regulate lineage commitment decisions in spleen DC precursors, between CD8α+ DC and CD4+CD8α − DC fates, in addition to decisions in a shared pDC/DC progenitor (e.g. CDPs). Alternatively, insufficient generation of resident spleen CD8α+ DCs in Id2−/− animals may allow expanded colonization by splenic CD4+CD8α−DCs and/or pDCs in niches that are normally occupied by CD8α+ DCs, via in situ proliferation of splenic DC precursors, increased influx/retention of bone marrow precursors and/or enhanced repopulation by differentiated pDCs. Regardless of which mechanism controls splenic DC subset proportions, the regulatory pathways that affect Id2 expression in pDC/DC multipotent progenitors would be expected to have key roles in modulating their transcriptional and developmental programs. Signals inducing Id2 may enable/favor resident spleen CD8α+ DC and dermal CD103+ DC generation and consequently limit pDC output from common pDC/DC progenitors (e.g. MDPs, CDPs), while upregulation of Id2 expression in spleen precursors may induce resident spleen CD8α+ DC development at the expense of splenic CD4+CD8α − DCs. These developmental outcomes could be elicited by direct restraint of transcriptional programs that support pDC commitment (e.g. via inhibition of E2-2 function) and/or resident spleen CD4+CD8α − DC generation (via an unknown factor), thus allowing activation of gene expression patterns that reinforce commitment to the resident spleen CD8α+ DCs and dermal CD103+ DC subsets. Elucidating the signals that regulate Id2 expression is of great interest; in addition, the mechanisms by which Id2 controls pDC/DC lineage decisions are important and will be clarified by determining a precise roadmap of DC precursor-product relationships, and a deeper understanding Id2 function in pDC/DC progenitor developmental potential.
IRF8
Unlike Id2, IRF8 is required for pDC, resident spleen CD8α+ DC, and migratory CD103+ DC development (Fig. 3), as these subsets are depleted in Irf8−/− mice (26, 106–108). Myeloid differentiation is also dependent upon IRF8, and Irf8−/− animals develop a myeloproliferative/pre-leukemic phenotype characterized by excessive granulocyte production and failure to generate adequate monocyte numbers (109). A spontaneous point mutation (R294C) of IRF8 in BHX2 mice also causes myeloproliferative disease and, interestingly, affects resident spleen CD8α+ DC development and IL-12 production without impairing pDC generation (110, 111). IRF8 residue 294 is located within the IRF association domain, which mediates protein:protein interactions, and the R294C mutation abolishes IRF8-partner-dependent DNA binding in vitro and in vivo. For example, the interaction between IRF8 and PU.1 is lost with the R294C mutation, resulting in a failure to recruit IRF8 to the endogenous target gene cystatin C in vivo (111). Since pDC development remains intact in BHX2 animals, these results imply that IRF8 utilizes distinct partner proteins in pDCs versus resident spleen CD8α+ DCs (111). The identity of IRF8 partner proteins is of great interest for further understanding the molecular regulation of the pDC and resident spleen CD8α+ DC developmental programs.
The loss of monocytes at the expense of granulocytes and additional hematopoietic alterations including excess erythropoiesis in Irf8−/− mice was originally interpreted as a role for IRF8 in a multipotent progenitor subset (109). The recent identification of a progenitor subset with monocyte, DC, and pDC developmental potential (MDPs) (48, 53) suggests that IRF8 may be crucial for MDP maintenance and/or lineage specification from the MDP. Importantly, the abundance of CLPs and CMPs was normal in Irf8−/− animals (92), while MDP or CDP amounts have not been determined on the IRF8-deficient background to our knowledge. Studies investigating progenitor amounts in Irf8−/− mice as well as the developmental potential of Irf8−/−progenitor subsets are necessary for a complete picture of IRF8 function during pDC/DC lineage commitment.
In addition to regulating pDC and DC subset generation in vivo, IRF8 also controls genes that have important roles in pDC and resident spleen CD8α+ DC functional responses, including TLR9 expression and type I IFN production in pDCs, and IL-12 production from CD8α+ DCs (112–115). These data indicate IRF8 transcriptional activity is necessary in terminally differentiated pDCs and resident CD8α+ DCs. Since IRF8 and E2-2 both participate in pDC differentiation, it is possible that these factors work cooperatively to regulate pDC-specific genes. In resident spleen CD8α+ DCs, IRF8 appears to collaborate with PU.1 (111), and this may extend to dermal CD103+ DCs, given their functional and developmental similarities with resident CD8α+ DCs. IRF2 has also been reported as an IRF8 binding partner (111); however, IRF8 and IRF2 control different resident spleen DC subsets, i.e., IRF2 is required for spleen CD4+CD8α− DCs, while IRF8 regulates generation of CD8α+ DCs (92, 106, 116), indicating that IRF2:IRF8 binding complexes, if observed in vivo, are not likely to have essential function in the formation of resident spleen DC subsets.
Spi-B
The Ets family member Spi-B has an important role in pDC development from human pDC/DC progenitors (117) (Fig. 3). Spi-B has been reported to bind together with IRF8 on composite oligonucleotides containing ISRE and Ets/IRF elements using in vitro systems (111), although it is not clear if this interaction is found at endogenous pDC gene promoters in vivo. Spib−/− mice show normal T and B cell amounts but impaired B-cell function and defective T-cell-dependent humoral immune responses (118), while to our knowledge pDC and DC populations have not yet been examined in Spib−/− mice. Spib−/− lymphocytes have cell autonomous defects; however, the impaired lymphocyte responses in Spib−/− mice in vivo may also imply an underlying pDC and/or DC deficiency, in accord with the role for Spi-B in human pDCs (117). This possibility can be addressed directly by characterization of DC subset representation and function in Spib−/− mice. The established role for Spi-B in human pDCs is significant, since our understanding of human DC generation is much less sophisticated relative to murine counterparts. Importantly, human pDCs and mouse pDCs share a requirement for E2-2 (95) and potentially Spi-B, suggesting close similarity between the human and murine pDC developmental pathways.
Batf3
The basic leucine zipper transcription factor Batf3 was the first factor identified with an exclusive role in a resident spleen DC subset. Batf3 has been reported to heterodimerize with c-Jun and function as a transcriptional repressor for IL-2 production (119), although its molecular target(s) in DCs remains unknown. Batf3 is necessary for development of resident lymphoid organ CD8α+ DCs and dermal CD103+ DCs but not pDCs or resident lymphoid organ CD8α − DC subsets (27, 94) (Fig. 3). Importantly, the selective effect of Batf3 deletion allowed for functional analysis of lymphoid tissue CD8α+ DCs. Splenic DCs isolated from Batf3−/− mice were unable to mediate antigen cross-presentation, demonstrating conclusively that resident CD8α+ DCs perform MHC class I presentation of exogenous antigen (94). Moreover, Batf3−/− mice lack virus- specific CD8+ T-cell responses and show defective rejection of immunogenic tumors, suggesting antigen cross-presentation by resident CD8α+ DCs elicits CD8+ cytotoxic T- cell responses to virus and tumors (94). However, the generation of migratory CD103+ DCs that populate several tissues including lung, intestine, and dermis is also dependent upon Batf3 (27). These cells have established roles in the cross-presentation of exogenous or cell-associated antigens and thus are also likely to mediate anti-viral and anti-tumor cytotoxic T-cell responses in vivo. Furthermore, the shared requirement for Batf3 between migratory CD103+ DCs and resident lymphoid organ CD8α+ DCs in vivo suggested these populations have a common developmental origin. This concept was recently bolstered by the observation that migratory CD103+ DCs are generated from circulating DC precursors that give rise to resident lymphoid organ CD8α+ DCs, as well as the fact that both DC populations require IRF8 and Id2 for efficient development in vivo (26, 27) (Fig. 3). Overall, studies of DC transcriptional regulators have provided important insights that have enabled developmental relationships between resident and migratory DC subsets to be further elucidated, although in general knowledge in this area is limited. Further analysis of factors participating in the DC and pDC transcriptional networks should contribute toward a better understanding of pDC/DC developmental origins and lineage associations.
STAT function in DC and pDC developmental pathways
For pDCs and DCs as well as other hematopoietic lineages, a key question centers on the mechanisms that control the expression and/or activity of lineage-specific transcriptional regulators. Our attention has focused on cytokine signaling pathways, with the question of how they might influence transcription factor expression during pDC and DC development. Cytokines elicit multiple intracellular signaling cascades, and signal transducers and activators of transcriptions (STATs) are considered to be principal mediators of cytokine signal transduction. STATs are triggered by tyrosine phosphorylation, which is activated by cytokine receptor-associated Jak kinases following cytokine engagement. Tyrosine phosphorylation causes STATs to dissociate from the receptor, dimerize, and accumulate in the nucleus, where they regulate cytokine-responsive gene expression (120–122). The STAT family comprises 7 members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Numerous cytokines activate STAT1, STAT3, and STAT5, while STAT2, STAT4, and STAT6 are relatively specific for type I IFNs, IL-12, and IL-4, respectively (120, 121). The paradigm for STAT activation is now known to be overly simplified; STATs undergo additional post-translational modifications that control their function (e.g. acetylation, serine phosphorylation), STATs can be stimulated by non-receptor kinases (e.g. Src), and STATs can operate as transcriptional regulators in the absence of tyrosine phosphorylation (120, 123). Moreover, Jak2 kinase directly affects nuclear transcriptional competency by phosphorylating histone H3, which leads to dissociation of the chromatin-condenser HP1 (124). These exciting results suggest the possibility of Jaks directly regulating nuclear gene expression in pDCs and/or DCs and add a layer of complexity to cytokine-mediated transcriptional control. In the context of immune cell development, STATs have well-established roles in initiating and sustaining CD4+ T-cell lineage commitment and differentiation, B-cell generation, erythropoiesis, and granulopoiesis (125–129). Whether STATs participate in most hematopoietic lineage commitment events is less clear; however, work from our laboratories as well as others has elucidated STAT function in pDC and DC lineage differentiation (79, 130).
STAT3
STAT3 can be activated by Flt3L signaling and therefore has been considered a likely candidate to mediate pDC and DC development (130). Since complete deletion causes embryonic lethality (131), conditional ablation of Stat3 in the hematopoietic system has been used to investigate STAT3 function in pDCs and DCs. The first report with hematopoietic STAT3-deficient mice [Tg(Tek-cre)12Flv, Stat3f/f] showed a severe reduction in resident spleen CD11c+ DCs in steady state as well as impaired responses in lipopolysaccharide (LPS)-mediated recruitment of tissue (migratory) DCs to spleen (130). In addition, STAT3 is necessary for efficient generation of pDCs (defined by CD11c+CD11b − surface expression) from Flt3L bone marrow cultures in vitro (130). In response to Flt3L treatment in vivo, STAT3-deficient mice fail to upregulate DC precursor and lymphoid organ DC numbers; however, CLPs and CMPs accumulate 2-3-fold relative to wildtype animals (130). Taken together, these data indicate that STAT3 controls both homeostatic amounts and Flt3L-driven production of resident DCs from pDC/DC progenitors; Flt3L responses in the progenitor compartment may be mediated by a STAT3-dependent transition from multipotent cells to DC-committed precursors. STAT3 also appears to control the amount of migratory DCs and/or their response to inflammatory signals (130).
We used Tg(Tek-cre)12Flv, Stat3f/Δ mice to explore the cellular and molecular mechanisms by which STAT3 controls pDC and DC generation. We found that STAT3 was required for Flt3L-dependent proliferation of lin−Flt3+ bone marrow progenitors as well as accumulation of pDCs and CD11c+CD11b+ DCs in ex vivo Flt3L cultures (79). Recently, we determined that pDC numbers are reduced in bone marrow and spleen of hematopoietic STAT3-deficient mice compared to wildtype control animals (Li et al., unpublished results), therefore indicating that STAT3 has an obligatory role in the pDC developmental pathway in homeostatic conditions in vivo and in response to Flt3L signaling in vitro. Surprisingly, however, the low numbers of pDCs that develop from STAT3-deficient bone marrow progenitors respond to CpG-A stimulation by producing IFN-α at similar amounts compared to wildtype pDCs (79). Therefore, the data indicate that STAT3 controls Flt3L-responsive pDC/DC progenitor proliferation and resulting expansion of downstream lineages, e.g. pDCs, DCs, while it is dispensable for terminal pDC differentiation. The function of STAT3 in mediating Flt3L-responsive progenitor proliferation in vitro parallels the induction of MDPs by Flt3L treatment in vivo (72), underscoring the importance of the Flt3L-Flt3-STAT3 axis in driving expansion of the pDC/DC progenitor compartment. The role of STAT3 in MDP and CDP maintenance under steady state conditions is less clear, however, as homeostatic progenitor amounts have not been reported in hematopoietic STAT3-deficient mice. This issue is important to examine, as redundant factors could serve to maintain pDC/DC progenitor amounts in steady state conditions; identification of such factors would be of great interest in learning how to manipulate pDCs and DCs in vitro and in vivo.
In our analyses of STAT3-deficient mice, we did not find a role for STAT3 in regulating CD11c+CD11b+ DC numbers in bone marrow or spleen under homeostatic conditions (Li et al., unpublished results). These results differ from the previous analysis of DC amounts in STAT3-deficient mice (130), although both studies used the Tg(Tek-cre)12Flv transgenic mice to direct STAT3 deletion from floxed STAT3 alleles. The observed differences suggest additional factors such as strain backgrounds or environmental conditions may affect DC maintenance in the absence of STAT3 signaling. Moreover, since Flt3 is required for the local proliferation of DC precursors in spleen (72), our data suggest Flt3 may utilize signaling cascades beyond STAT3 to control steady state DC amounts in lymphoid organs. STAT3 is not necessary for GM-CSF-driven production of inflammatory DCs in vitro or GM-CSF-mediated suppression of Flt3L-dependent pDC development (79), indicating alternative pathways exist for DC and pDC regulation. However, a detailed analysis of resident spleen DC subsets as well as migratory DC amounts in STAT3-deficient animals must be undertaken to fully elucidate the action of this important transcriptional regulator in vivo and assess potential contributions of alternative signaling cascades in DC maintenance.
The phenotype of hematopoietic STAT3-deficient mice must be interpreted with caution and, if possible, validated by specific ablation of Stat3 in pDC/DC progenitors. We find that conditional Stat3 deletion is not 100% effective using the Tg(Tek-cre)12Flv transgene (Tie2cre), and mice with the Tg(Tek-cre)12Flv, Stat3f/Δ genotype can show significant amounts of STAT3 protein in bone marrow cell lysates (Panopoulos, AD, and Watowich, SS, unpublished data). This has prompted us to analyze protein expression in all animals with the Tg(Tek-cre)12Flv, Stat3f/Δ genotype and utilize only those that exhibit efficient ablation of STAT3 protein for our experiments (127). In addition, STAT3-deficient mice have numerous immune impairments and develop severe inflammatory disease with age (132), which may have indirect effects on pDC and/or DC homeostasis.
While STAT3 is typically considered to be a growth-promoting signal transducer and is, in fact, oncogenic if constitutively activated (133), STAT3 overexpression in Flt3 bone marrow progenitors instructed differentiation into pDCs and DCs in vivo (134). These observations suggest that STAT3 drives the expression of genes that induce pDC/DC lineage commitment. Similarly, we found that Flt3L induced the expression of key pDC transcriptional regulators (PU.1, IRF7, IRF8, SpiB) in lin−Flt3+ progenitors (79); however, it remains unclear whether Flt3L utilizes STAT3 in activating and/or sustaining the transcriptional network required for pDC development in vivo. Moreover, PU.1 overexpression in Flt3 bone marrow progenitors directed pDC and DC commitment, and upregulation of PU.1 as well as pDC and DC target genes was found in STAT3-overexpressing bone marrow progenitors (134). These data collectively suggest Sfpi1 (PU.1) may be a target gene of the Flt3L-Flt3-STAT3 signaling cascade. Studies from our laboratory (Yoon, D and Watowich, SS, unpublished results) as well as Dr. Pamela Hankey’s group (135) implicated STAT3 in direct regulation of PU.1 in myeloid progenitors in response to G-CSF signaling or transformation with the erythroleukemia-inducing spleen focus-forming virus. STAT3 was found to interact with an upstream region of the Sfpi1 (PU.1) locus, which is located ~10kb in the 5′ direction relative to the transcriptional start site, via chromatin immunoprecipitation assays (135, Yoon, D and Watowich, SS, unpublished results). These results collectively suggest Sfpi1 as a target gene for STAT3 in pDC/DC progenitors, a notion that can be examined directly by comparing PU.1 regulation in progenitors from STAT3-deficient and wildtype animals. Additional target genes with involvement in pDC/DC lineage commitment may also be regulated by STAT3. This question requires further investigation to determine the mechanism(s) of STAT3 action and could be addressed by genome-wide STAT3 ChIP-sequencing experiments and analysis of gene expression patterns in STAT-sufficient vs. STAT3-deficient MDPs or CDPs.
STAT5
The term STAT5 actually refers to two proteins, STAT5A and STAT5B, which are encoded by adjacent genes found in the vicinity of the Stat3 gene (136). The first mouse model in which both genes were targeted for ablation was later found to be a STAT5 hypomorph (Stat5abΔN/ΔN), as an N-terminally truncated STAT5 protein with partial activity was detected in certain tissues including lymphoid cells (137). The Stat5abΔN/ΔN animals provide a valuable model for understanding the function of the STAT5 N-domain, e.g. revealing important role in leukemia progression (137); however, the mice cannot be considered a full deletion or null strain. Fortunately for our understanding of pDC/DC development, studies did not begin in earnest with this hypomorphic strain as DC subsets were still being resolved when the mice were generated. Therefore timing was appropriate when the complete Stat5a−/−Stat5b−/− (Stat5−/−) mice became available through Dr. Lothar Hennighausen’s laboratory (138).
Using Stat5−/− mice, we asked whether STAT5 was required for GM-CSF-mediated suppression of pDC development, GM-CSF-responsive production of inflammatory DCs in vitro, and DC subset generation in vivo. DC and pDC generation in vivo was determined following adoptive transfer of Stat5−/− or Stat5+/+ fetal liver progenitors into lethally irradiated mice, as complete Stat5 deletion causes neonatal lethality (138). These studies revealed that STAT5 is not essential for repopulation of spleen CD11c+CD11b+ DCs (79), consistent with the fact that GM-CSF is largely dispensable for DC maturation in vivo (58). However, CD11c+CD11b+ DC numbers were reduced in spleen, and total spleen cell counts were dramatically suppressed, in accord with the role for STAT5 in lymphoid development (139). Thus, complete reconstitution of CD11c+CD11b+ DC numbers in spleen requires STAT5, although whether DC repopulation is directly mediated by STAT5 signaling in DC precursors or depends on the lymphoid architecture of the organ remains to be determined. Analysis of resident spleen DC subsets revealed that Stat5−/− chimeras have a higher proportion of CD8α+ DCs and a reduced frequency of CD4+CD8α − DCs, suggesting STAT5 signaling regulates balanced production of these splenic DC subsets in vivo (79). When provided as the sole exogenous growth factor, GM-CSF is completely dependent on STAT5 to direct inflammatory DC production from lin−Flt3+ progenitors (79). Further studies are necessary to evaluate the role of STAT5 in GM-CSF-mediated inflammatory DC and migratory DC subset generation in vivo, and to resolve whether STAT5- activating cytokines are involved in regulating resident spleen CD8α+ and CD4+CD8α− DCs.
We showed that GM-CSF and STAT5 override Flt3L signals in early stages of the pDC developmental pathway (i.e. in lin−Flt3+ progenitors) to block pDC formation (79). GM-CSF was unable to induce the conversion or maturation of terminally differentiated pDCs to DCs, confirming it operates upon a progenitor subset with pDC potential. We speculate that GM-CSF and STAT5 may act on the CDP, which expresses the GM-CSF receptor and would be found within the lin−Flt3+ bone marrow fraction (51). In agreement, pDC repopulation following bone marrow ablation was negatively regulated by STAT5 (79). Hence, the pDC/DC progenitor compartment may respond to STAT5-activating cytokines to control balanced production of the pDC and DC lineages in vivo. The identity of such STAT5-activating cytokines and their effect on pDC/DC subset generation and the outcome of active immune responses require further investigation.
The ability of STAT5 to inhibit pDC generation from bone marrow progenitors suggested to us that STAT5 might affect pDC/DC cell fate decisions. This idea is reminiscent of the lymphoid to myeloid lineage conversion function for GM-CSF in CLPs and early T-cell progenitors (140, 141) as well as IL-2- and STAT5-mediated inhibition of Th17 development (142), which depends upon STAT3 and STAT3-activating cytokines (143, 144). Furthermore, GM-CSF was recently shown to direct lineage commitment in GMPs (145). While the latter studies did not examine STAT5 function, they add to the growing body of evidence demonstrating that GM-CSF signals have instructive activity. Within the pDC/DC progenitor subset (lin−Fl3+ progenitors), we found that GM-CSF and STAT5 signaling had a broad effect on Flt3L-responsive pDC gene expression, e.g. GM-CSF-STAT5 signals suppress Flt3, Tlr9, Irf7, Irf8, and Spib and induce Irf4 expression (79). To understand STAT5 suppressive mechanisms, we inspected the promoter regions of genes encoding the lineage-specific transcription factors IRF4, IRF8 and SpiB. Consensus STAT sites were found in the proximal promoter regions of Irf8 and Irf4; between these two genes, we identified Irf8 as a direct target gene of GM-CSF-activated STAT5 (79). Since IRF8 is crucial for pDC maturation (106, 107), inhibition of its expression by GM-CSF-dependent STAT5 signaling most likely enforces the blockade in pDC production. In addition, suppressed IRF8 expression may impact the expression of other regulatory transcription factors (e.g. SpiB, IRF7) (Fig. 4) and receptors (e.g. Flt3, TLR9) that are necessary for pDC development and function. It is not yet known if GM-CSF-dependent STAT5-mediated suppression of IRF8 operates in other hematopoietic lineages to regulate their development and/or function, although our preliminary studies with the D2SC/1 (DC) and J774 (macrophage) cell lines suggested this pathway may be restricted to pDC/DC progenitors (Esashi et al., unpublished results). Interestingly, we found that STAT5 also negatively regulates the repopulation of splenic CD8α+ DCs (79), which rely on IRF8, suggesting STAT5-mediated suppression of IRF8 influences resident DC subset generation in vivo. Resident DC and migratory DC subsets must be more thoroughly characterized in Stat5−/− chimeras to evaluate this possibility. In addition, the molecular pathways by which STAT5 controls pDC and DC production should be determined by analysis of STAT5 target genes in MDPs and CDPs.
Fig. 4.
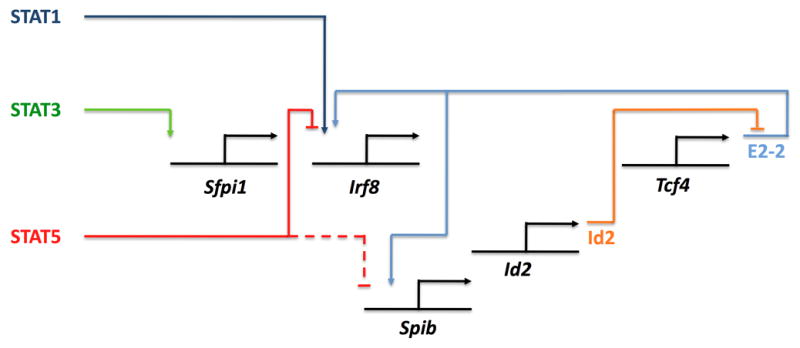
Pathways interconnecting STATs and pDC/DC lineage regulators.
STAT1
We recently found that IFN-α stimulated the generation of a pDC-related subset (IFN-pDCs) with morphologic and phenotypic similarity to classical Flt3L-generated pDCs in all aspects except TLR-triggered type I IFN production (Li et al., unpublished results). STAT1 is a principal type I IFN signal transducer; therefore, we hypothesized it would be involved in mediating IFN-pDC production. This idea was supported by a recent study showing that IFN-α stimulates growth of HSCs and this occurs through a STAT1- dependent pathway (84). While IFN-α activates STAT3 and STAT1 in lin−Flt3+ progenitors, development of IFN-pDCs was equivalent in STAT3-deficient and wildtype bone marrow progenitor cultures (Li et al., unpublished results), indicating that STAT3 is dispensable for IFN-pDC generation. By contrast, STAT1 was critical for IFN-pDC production in vitro and in vivo. Moreover, IFN-responsive STAT1 upregulates Irf8 transcription in the DC cell line D2SC1, implicating STAT1-mediated induction of IRF8 as a mechanism contributing to production of IFN-pDCs (Li et al., unpublished results). IFN-α also induced the number of lin−Flt3+ bone marrow progenitors, suggesting a role in pDC/DC progenitor proliferation (Li et al., unpublished results). These results, taken together with studies in HSCs (84), suggest that IFN-α/STAT1 signaling may direct the expansion of the HSC population as well as enhanced commitment to the pDC/DC progenitor subset, resulting in increases in pDC/DC progenitors and IFN-pDC numbers. In addition, the data showing that IFN-α and STAT1 have hematopoietic activities (84) indicates that the use of poly (I:C) to induce conditional gene deletion with the IFN-responsive Mx-cre transgenic mouse may not be ideal for studying blood cell development.
Type I IFN-mediated suppression of DCs appears to involve a distinct pathway to control IFN-pDC generation. Specifically, inhibition of DCs by IFN signaling was shown to require STAT2 but not STAT1 (81). As STAT2 has been generally considered to operate in conjunction with STAT1 and IRF9 in type I IFN signaling (122), these data provide an important clue suggesting the potential for genes controlled by STAT2 alone. Moreover, the results indicate that separate transcriptional responses regulate IFN-dependent production of IFN-pDCs versus IFN-mediated suppression of DCs in vivo; however, the identity of these regulatory cascades requires continued investigation.
Conclusion and future perspectives
The available data suggest a network of cytokine-responsive STAT factors intersecting with pDC and DC lineage regulators to affect developmental decisions. Fig. 4 presents the beginning of such a model for pDCs and IFN-pDCs, along the lines of models for lymphoid cell specification (146), in which STATs positively or negatively influence pDC/DC transcriptional regulators. In turn, these transcription factors may induce autoregulatory loops that are yet to be characterized, cross-regulate one another to instruct or sustain developmental options, and/or initiate antagonistic interactions that reject alternative cell fates (e.g. Id2-E2-2). Compared with pDCs, our understanding of resident and migratory DC lineage specification and diversification is at a more primitive stage, although we expect similar mechanisms will be utilized as those described for pDCs and other hematopoietic lineages. Future studies should be aimed at connecting extrinsic signals with cell autonomous factors, revealing epigenetic mechanisms regulating pDC and DC lineage commitment and differentiation, understanding the impact of microenvironmental signals on pDC and DC development and function, and determining how transcription factor networks affect functional outcomes (e.g. elucidation of specific gene targets regulated in differentiating pDCs and DCs). The pDC and DC lineages provide a diverse and malleable system for elucidating the regulatory events controlling hematopoiesis; in turn, this knowledge may reveal molecular targets to manipulate pDC and/or DC developmental or functional responses in clinical settings.
Acknowledgments
We thank Drs. Li Wu, Michel Gilliet, Wei Cao and Haiyan Li for helpful discussions and Drs. Wu and Gilliet for critical review of the manuscript. Work in the authors’ labs is supported by grants from NIH (AI059718, YJL; AI073587 and AR059010, SSW) and the NCI Core Grant P30CA16672 at M. D. Anderson Cancer Center.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. 1973;137:1142–1162. doi: 10.1084/jem.137.5.1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. II. Functional properties in vitro. J Exp Med. 1974;139:380–397. doi: 10.1084/jem.139.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steinman RM, Lustig DS, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. 3. Functional properties in vivo. J Exp Med. 1974;139:1431–1445. doi: 10.1084/jem.139.6.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinman RM, Adams JC, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. IV. Identification and distribution in mouse spleen. J Exp Med. 1975;141:804–820. [PMC free article] [PubMed] [Google Scholar]
- 6.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nat Rev Immunol. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 7.Liu YJ. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 2001;106:259–262. doi: 10.1016/s0092-8674(01)00456-1. [DOI] [PubMed] [Google Scholar]
- 8.Liu YJ. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 9.Shortman K, Naik SH. Steady-state and inflammatory dendritic cell development. Nature Rev Immunol. 2007;7:19–30. doi: 10.1038/nri1996. [DOI] [PubMed] [Google Scholar]
- 10.Naik SH. Demystifying the development of dendritic cell subtypes, a little. Immunol Cell Biol. 2008;86:439–452. doi: 10.1038/icb.2008.28. [DOI] [PubMed] [Google Scholar]
- 11.Vremec D, Pooley J, Hochrein H, Wu L, Shortman K. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J Immunol. 2000;164:2978–2986. doi: 10.4049/jimmunol.164.6.2978. [DOI] [PubMed] [Google Scholar]
- 12.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD8(−) dendritic cells cross- prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochrein H, Shortman K, Vremec D, Scott B, Hertzog P, O’Keeffe M. Differential production of IL-12, IFN-alpha, and IFN-gamma by mouse dendritic cell subsets. J Immunol. 2001;166:5448–5455. doi: 10.4049/jimmunol.166.9.5448. [DOI] [PubMed] [Google Scholar]
- 14.Edwards AD, et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 15.Zhong G, Reis e Sousa C, Germain RN. Antigen-unspecific B cells and lymphoid dendritic cells both show extensive surface expression of processed antigen-major histocompatibility complex class II complexes after soluble protein exposure in vivo or in vitro. J Exp Med. 1997;186:673–682. doi: 10.1084/jem.186.5.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Smedt T, et al. Regulation of dendritic cell numbers and maturation by lipopolysaccharide in vivo. J Exp Med. 1996;184:1413–1424. doi: 10.1084/jem.184.4.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heath WR, et al. Cross-presentation, dendritic cell subsets, and the generation of immunity to cellular antigens. Immunol Rev. 2004;199:9–26. doi: 10.1111/j.0105-2896.2004.00142.x. [DOI] [PubMed] [Google Scholar]
- 18.Henri S, et al. The dendritic cell populations of mouse lymph nodes. J Immunol. 2001;167:741–748. doi: 10.4049/jimmunol.167.2.741. [DOI] [PubMed] [Google Scholar]
- 19.Anjuere F, et al. Definition of dendritic cell subpopulations present in the spleen, Peyer’s patches, lymph nodes, and skin of the mouse. Blood. 1999;93:590–598. [PubMed] [Google Scholar]
- 20.Ginhoux F, et al. Blood-derived dermal langerin+ dendritic cells survey the skin in the steady state. J Exp Med. 2007;204:3133–3146. doi: 10.1084/jem.20071733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poulin LF, Henri S, de Bovis B, Devilard E, Kissenpfennig A, Malissen B. The dermis contains langerin+ dendritic cells that develop and function independently of epidermal Langerhans cells. J Exp Med. 2007;204:3119–3131. doi: 10.1084/jem.20071724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bursch LS, et al. Identification of a novel population of Langerin+ dendritic cells. J Exp Med. 2007;204:3147–3156. doi: 10.1084/jem.20071966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stingl G, Tamaki K, Katz SI. Origin and function of epidermal Langerhans cells. Immunol Rev. 1980;53:149–174. doi: 10.1111/j.1600-065x.1980.tb01043.x. [DOI] [PubMed] [Google Scholar]
- 24.Liu K, Nussenzweig MC. Origin and development of dendritic cells. Immunol Rev. 2010;234:45–54. doi: 10.1111/j.0105-2896.2009.00879.x. [DOI] [PubMed] [Google Scholar]
- 25.Merad M, Ginhoux F, Collin M. Origin, homeostasis and function of Langerhans cells and other langerin-expressing dendritic cells. Nat Rev Immunol. 2008;8:935–947. doi: 10.1038/nri2455. [DOI] [PubMed] [Google Scholar]
- 26.Ginhoux F, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med. 2009;206:3115–3130. doi: 10.1084/jem.20091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edelson BT, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207:823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bogunovic M, et al. Origin of the lamina propria dendritic cell network. Immunity. 2009;31:513–525. doi: 10.1016/j.immuni.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villadangos JA, Schnorrer P. Intrinsic and cooperative antigen-presenting functions of dendritic-cell subsets in vivo. Nat Rev Immunol. 2007;7:543–555. doi: 10.1038/nri2103. [DOI] [PubMed] [Google Scholar]
- 30.Pulendran B, Tang H, Denning TL. Division of labor, plasticity, and crosstalk between dendritic cell subsets. Curr Opin Immunol. 2008;20:61–67. doi: 10.1016/j.coi.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lanzavecchia A, Sallusto F. Regulation of T cell immunity by dendritic cells. Cell. 2001;106:263–266. doi: 10.1016/s0092-8674(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 32.Mellman I, Steinman RM. Dendritic cells: specialized and regulated antigen processing machines. Cell. 2001;106:255–258. doi: 10.1016/s0092-8674(01)00449-4. [DOI] [PubMed] [Google Scholar]
- 33.Merad M, Manz MG. Dendritic cell homeostasis. Blood. 2009;113:3418–3427. doi: 10.1182/blood-2008-12-180646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cella M, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 35.Siegal FP, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284:1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 36.Asselin-Paturel C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 37.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 38.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 39.Gregorio J, et al. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. 2010 doi: 10.1084/jem.20101102. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008;19:41–52. doi: 10.1016/j.cytogfr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lande R, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 42.Kadowaki N, Antonenko S, Lau JY, Liu YJ. Natural interferon alpha/beta-producing cells link innate and adaptive immunity. J Exp Med. 2000;192:219–226. doi: 10.1084/jem.192.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blasius AL, Cella M, Maldonado J, Takai T, Colonna M. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood. 2006;107:2474–2476. doi: 10.1182/blood-2005-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Keeffe M, et al. Mouse plasmacytoid cells: long-lived cells, heterogeneous in surface phenotype and function, that differentiate into CD8(+) dendritic cells only after microbial stimulus. J Exp Med. 2002;196:1307–1319. doi: 10.1084/jem.20021031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Okada T, Lian ZX, Naiki M, Ansari AA, Ikehara S, Gershwin ME. Murine thymic plasmacytoid dendritic cells. Eur J Immunol. 2003;33:1012–1019. doi: 10.1002/eji.200323616. [DOI] [PubMed] [Google Scholar]
- 46.Manz MG, Traver D, Miyamoto T, Weissman IL, Akashi K. Dendritic cell potentials of early lymphoid and myeloid progenitors. Blood. 2001;97:3333–3341. doi: 10.1182/blood.v97.11.3333. [DOI] [PubMed] [Google Scholar]
- 47.Merad M, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Nat Immunol. 2002;3:1135–1141. doi: 10.1038/ni852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu K, et al. In vivo analysis of dendritic cell development and homeostasis. Science. 2009;324:392–397. doi: 10.1126/science.1170540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- 50.Naik SH, et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat Immunol. 2007;8:1217–1226. doi: 10.1038/ni1522. [DOI] [PubMed] [Google Scholar]
- 51.Onai N, Obata-Onai A, Schmid MA, Ohteki T, Jarrossay D, Manz MG. Identification of clonogenic common Flt3(+)M-CSFR(+) plasmacytoid and conventional dendritic cell progenitors in mouse bone marrow. Nat Immunol. 2007;8:1207–1216. doi: 10.1038/ni1518. [DOI] [PubMed] [Google Scholar]
- 52.D’Amico A, Wu L. The early progenitors of mouse dendritic cells and plasmacytoid predendritic cells are within the bone marrow hemopoietic precursors expressing Flt3. J Exp Med. 2003;198:293–303. doi: 10.1084/jem.20030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fogg DK, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. 2006;311:83–87. doi: 10.1126/science.1117729. [DOI] [PubMed] [Google Scholar]
- 54.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–61. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inaba K, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-alpha cooperate in the generation of dendritic Langerhans cells. Nature. 1992;360:258–261. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 57.Kingston D, Schmid MA, Onai N, Obata-Onai A, Baumjohann D, Manz MG. The concerted action of GM-CSF and Flt3-ligand on in vivo dendritic cell homeostasis. Blood. 2009;114:835–843. doi: 10.1182/blood-2009-02-206318. [DOI] [PubMed] [Google Scholar]
- 58.Vremec D, Lieschke GJ, Dunn AR, Robb L, Metcalf D, Shortman K. The influence of granulocyte/macrophage colony-stimulating factor on dendritic cell levels in mouse lymphoid organs. Eur J Immunol. 1997;27:40–44. doi: 10.1002/eji.1830270107. [DOI] [PubMed] [Google Scholar]
- 59.King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med. 2010;207:953–961. doi: 10.1084/jem.20091844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Keeffe M, et al. Effects of administration of progenipoietin 1, Flt-3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood. 2002;99:2122–2130. doi: 10.1182/blood.v99.6.2122. [DOI] [PubMed] [Google Scholar]
- 61.Daro E, et al. Polyethylene glycol-modified GM-CSF expands CD11bhighCD11chigh but not CD11blowCD11chigh murine dendritic cells in vivo: a comparative analysis with Flt3 ligand. J Immunol. 2000;165:49–58. doi: 10.4049/jimmunol.165.1.49. [DOI] [PubMed] [Google Scholar]
- 62.Naik SH, et al. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat Immunol. 2006;7:663–671. doi: 10.1038/ni1340. [DOI] [PubMed] [Google Scholar]
- 63.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 64.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19:59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- 65.Cheers C, Haigh AM, Kelso A, Metcalf D, Stanley ER, Young AM. Production of colony-stimulating factors (CSFs) during infection: separate determinations of macrophage-, granulocyte-, granulocyte-macrophage-, and multi-CSFs. Infect Immun. 1988;56:247–251. doi: 10.1128/iai.56.1.247-251.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McKenna HJ, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. 2000;95:3489–3497. [PubMed] [Google Scholar]
- 67.Blom B, Ho S, Antonenko S, Liu YJ. Generation of interferon alpha-producing predendritic cell (Pre-DC)2 from human CD34(+) hematopoietic stem cells. J Exp Med. 2000;192:1785–1796. doi: 10.1084/jem.192.12.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gilliet M, et al. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J Exp Med. 2002;195:953–958. doi: 10.1084/jem.20020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karsunky H, Merad M, Cozzio A, Weissman IL, Manz MG. Flt3 ligand regulates dendritic cell development from Flt3+ lymphoid and myeloid-committed progenitors to Flt3+ dendritic cells in vivo. J Exp Med. 2003;198:305–313. doi: 10.1084/jem.20030323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maraskovsky E, et al. Dramatic increase in the numbers of functionally mature dendritic cells in Flt3 ligand-treated mice: multiple dendritic cell subpopulations identified. J Exp Med. 1996;184:1953–1962. doi: 10.1084/jem.184.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Manfra DJ, Chen SC, Jensen KK, Fine JS, Wiekowski MT, Lira SA. Conditional expression of murine Flt3 ligand leads to expansion of multiple dendritic cell subsets in peripheral blood and tissues of transgenic mice. J Immunol. 2003;170:2843–2852. doi: 10.4049/jimmunol.170.6.2843. [DOI] [PubMed] [Google Scholar]
- 72.Waskow C, et al. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat Immunol. 2008;944:676–683. doi: 10.1038/ni.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hochweller K, Miloud T, Striegler J, Naik S, Hammerling GJ, Garbi N. Homeostasis of dendritic cells in lymphoid organs is controlled by regulation of their precursors via a feedback loop. Blood. 2009;114:4411–4421. doi: 10.1182/blood-2008-11-188045. [DOI] [PubMed] [Google Scholar]
- 74.Fancke B, Suter M, Hochrein H, O’Keeffe M. M-CSF: a novel plasmacytoid and conventional dendritic cell poietin. Blood. 2008;111:150–159. doi: 10.1182/blood-2007-05-089292. [DOI] [PubMed] [Google Scholar]
- 75.Ginhoux F, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol. 2006;7:265–273. doi: 10.1038/ni1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Naik SH, et al. Cutting edge: generation of splenic CD8+ and CD8− dendritic cell equivalents in Fms-like tyrosine kinase 3 ligand bone marrow cultures. J Immunol. 2005;174:6592–6597. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- 77.Brasel K, De Smedt T, Smith JL, Maliszewski CR. Generation of murine dendritic cells from flt3-ligand-supplemented bone marrow cultures. Blood. 2000;96:3029–3039. [PubMed] [Google Scholar]
- 78.Brawand P, Fitzpatrick DR, Greenfield BW, Brasel K, Maliszewski CR, De Smedt T. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand-supplemented bone marrow cultures are immature APCs. J Immunol. 2002;169:6711–6719. doi: 10.4049/jimmunol.169.12.6711. [DOI] [PubMed] [Google Scholar]
- 79.Esashi E, Wang YH, Perng O, Qin XF, Liu YJ, Watowich SS. The signal transducer STAT5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28:509–520. doi: 10.1016/j.immuni.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wadleigh M, Stone RM. The role of myeloid growth factors in acute leukemia. J Natl Compr Canc Netw. 2009;7:84–91. doi: 10.6004/jnccn.2009.0007. [DOI] [PubMed] [Google Scholar]
- 81.Hahm B, Trifilo MJ, Zuniga EI, Oldstone MB. Viruses evade the immune system through type I interferon-mediated STAT2-dependent, but STAT1-independent, signaling. Immunity. 2005;22:247–257. doi: 10.1016/j.immuni.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 82.Franchini M, et al. Dendritic cells from mice neonatally vaccinated with modified vaccinia virus Ankara transfer resistance against herpes simplex virus type I to naive one-week-old mice. J Immunol. 2004;172:6304–6312. doi: 10.4049/jimmunol.172.10.6304. [DOI] [PubMed] [Google Scholar]
- 83.Biron CA. Interferons alpha and beta as immune regulators--a new look. Immunity. 2001;14:661–664. doi: 10.1016/s1074-7613(01)00154-6. [DOI] [PubMed] [Google Scholar]
- 84.Essers MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 85.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741–750. doi: 10.1016/j.immuni.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 86.Liu K, Waskow C, Liu X, Yao K, Hoh J, Nussenzweig M. Origin of dendritic cells in peripheral lymphoid organs of mice. Nat Immunol. 2007;8:578–583. doi: 10.1038/ni1462. [DOI] [PubMed] [Google Scholar]
- 87.Zenke M, Hieronymus T. Towards an understanding of the transcription factor network of dendritic cell development. Trends Immunol. 2006;27:140–145. doi: 10.1016/j.it.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 88.Sieweke MH, Graf T. A transcription factor party during blood cell differentiation. Curr Opin Genet Dev. 1998;8:545–551. doi: 10.1016/s0959-437x(98)80009-9. [DOI] [PubMed] [Google Scholar]
- 89.Orkin SH. Diversification of haematopoietic stem cells to specific lineages. Nat Rev Genet. 2000;1:57–64. doi: 10.1038/35049577. [DOI] [PubMed] [Google Scholar]
- 90.Shivdasani RA, Orkin SH. The transcriptional control of hematopoiesis. Blood. 1996;87:4025–4039. [PubMed] [Google Scholar]
- 91.Rothenberg EV, Scripture-Adams DD. Competition and collaboration: GATA-3, PU.1, and Notch signaling in early T-cell fate determination. Semin Immunol. 2008;20:236–246. doi: 10.1016/j.smim.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aliberti J, et al. Essential role for ICSBP in the in vivo development of murine CD8alpha + dendritic cells. Blood. 2003;101:305–310. doi: 10.1182/blood-2002-04-1088. [DOI] [PubMed] [Google Scholar]
- 93.Hacker C, et al. Transcriptional profiling identifies Id2 function in dendritic cell development. Nat Immunol. 2003;4:380–386. doi: 10.1038/ni903. [DOI] [PubMed] [Google Scholar]
- 94.Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cisse B, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carotta S, et al. The transcription factor PU.1 controls dendritic cell development and Flt3 cytokine receptor expression in a dose-dependent manner. Immunity. 2010;32:628–641. doi: 10.1016/j.immuni.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 97.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 98.Georgopoulos K, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. 1994;79:143–156. doi: 10.1016/0092-8674(94)90407-3. [DOI] [PubMed] [Google Scholar]
- 99.Wu L, Nichogiannopoulou A, Shortman K, Georgopoulos K. Cell-autonomous defects in dendritic cell populations of Ikaros mutant mice point to a developmental relationship with the lymphoid lineage. Immunity. 1997;7:483–492. doi: 10.1016/s1074-7613(00)80370-2. [DOI] [PubMed] [Google Scholar]
- 100.Anderson KL, Perkin H, Surh CD, Venturini S, Maki RA, Torbett BE. Transcription factor PU.1 is necessary for development of thymic and myeloid progenitor-derived dendritic cells. J Immunol. 2000;164:1855–1861. doi: 10.4049/jimmunol.164.4.1855. [DOI] [PubMed] [Google Scholar]
- 101.Guerriero A, Langmuir PB, Spain LM, Scott EW. PU.1 is required for myeloid- derived but not lymphoid-derived dendritic cells. Blood. 2000;95:879–885. [PubMed] [Google Scholar]
- 102.Rathinam C, et al. The transcriptional repressor Gfi1 controls STAT3-dependent dendritic cell development and function. Immunity. 2005;22:717–728. doi: 10.1016/j.immuni.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 103.Quong MW, Romanow WJ, Murre C. E protein function in lymphocyte development. Annu Rev Immunol. 2002;20:301–322. doi: 10.1146/annurev.immunol.20.092501.162048. [DOI] [PubMed] [Google Scholar]
- 104.Langlands K, Yin X, Anand G, Prochownik EV. Differential interactions of Id proteins with basic-helix-loop-helix transcription factors. J Biol Chem. 1997;272:19785–19793. doi: 10.1074/jbc.272.32.19785. [DOI] [PubMed] [Google Scholar]
- 105.Goldfarb AN, Lewandowska K, Shoham M. Determinants of helix-loop-helix dimerization affinity. Random mutational analysis of SCL/tal. J Biol Chem. 1996;271:2683–2688. doi: 10.1074/jbc.271.5.2683. [DOI] [PubMed] [Google Scholar]
- 106.Schiavoni G, et al. ICSBP is essential for the development of mouse type I interferon-producing cells and for the generation and activation of CD8alpha(+) dendritic cells. J Exp Med. 2002;196:1415–1425. doi: 10.1084/jem.20021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tsujimura H, Tamura T, Ozato K. Cutting edge: IFN consensus sequence binding protein/IFN regulatory factor 8 drives the development of type I IFN-producing plasmacytoid dendritic cells. J Immunol. 2003;170:1131–1135. doi: 10.4049/jimmunol.170.3.1131. [DOI] [PubMed] [Google Scholar]
- 108.Tamura T, et al. IFN regulatory factor-4 and -8 govern dendritic cell subset development and their functional diversity. J Immunol. 2005;174:2573–2581. doi: 10.4049/jimmunol.174.5.2573. [DOI] [PubMed] [Google Scholar]
- 109.Holtschke T, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 110.Turcotte K, Gauthier S, Tuite A, Mullick A, Malo D, Gros P. A mutation in the Icsbp1 gene causes susceptibility to infection and a chronic myeloid leukemia-like syndrome in BXH-2 mice. J Exp Med. 2005;201:881–890. doi: 10.1084/jem.20042170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tailor P, Tamura T, Morse HC, 3rd, Ozato K. The BXH2 mutation in IRF8 differentially impairs dendritic cell subset development in the mouse. Blood. 2008;111:1942–1945. doi: 10.1182/blood-2007-07-100750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giese NA, et al. Interferon (IFN) consensus sequence-binding protein, a transcription factor of the IFN regulatory factor family, regulates immune responses in vivo through control of interleukin 12 expression. J Exp Med. 1997;186:1535–1546. doi: 10.1084/jem.186.9.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Scharton-Kersten T, Contursi C, Masumi A, Sher A, Ozato K. Interferon consensus sequence binding protein-deficient mice display impaired resistance to intracellular infection due to a primary defect in interleukin 12 p40 induction. J Exp Med. 1997;186:1523–1534. doi: 10.1084/jem.186.9.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schroder K, et al. PU.1 and ICSBP control constitutive and IFN-gamma-regulated Tlr9 gene expression in mouse macrophages. J Leukoc Biol. 2007;81:1577–1590. doi: 10.1189/jlb.0107036. [DOI] [PubMed] [Google Scholar]
- 115.Tailor P, et al. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity. 2007;27:228–239. doi: 10.1016/j.immuni.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ichikawa E, et al. Defective development of splenic and epidermal CD4+ dendritic cells in mice deficient for IFN regulatory factor-2. Proc Natl Acad Sci USA. 2004;101:3909–3914. doi: 10.1073/pnas.0400610101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schotte R, Nagasawa M, Weijer K, Spits H, Blom B. The ETS transcription factor Spi-B is required for human plasmacytoid dendritic cell development. J Exp Med. 2004;200:1503–1509. doi: 10.1084/jem.20041231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Su GH, et al. Defective B cell receptor-mediated responses in mice lacking the Ets protein, Spi-B. EMBO J. 1997;16:7118–7129. doi: 10.1093/emboj/16.23.7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Iacobelli M, Wachsman W, McGuire KL. Repression of IL-2 promoter activity by the novel basic leucine zipper p21SNFT protein. J Immunol. 2000;165:860–868. doi: 10.4049/jimmunol.165.2.860. [DOI] [PubMed] [Google Scholar]
- 120.Levy DE, Darnell J, JE Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 121.Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 122.Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 123.Yang J, Stark GR. Roles of unphosphorylated STATs in signaling. Cell Res. 2008;18:443–451. doi: 10.1038/cr.2008.41. [DOI] [PubMed] [Google Scholar]
- 124.Dawson MA, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chou WC, Levy DE, Lee CK. STAT3 positively regulates an early step in B-cell development. Blood. 2006;108:3005–3011. doi: 10.1182/blood-2006-05-024430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Socolovsky M, Fallon AEJ, Wang S, Brugnara C, Lodish HF. Fetal anemia and apoptosis of red cell progenitors in Stat5a−/−5b−/− mice: a direct role for Stat5 in Bcl-XL induction. Cell. 1999;98:181–191. doi: 10.1016/s0092-8674(00)81013-2. [DOI] [PubMed] [Google Scholar]
- 127.Panopoulos AD, et al. STAT3 governs distinct pathways in emergency granulopoiesis and mature neutrophils. Blood. 2006;108:3682–3690. doi: 10.1182/blood-2006-02-003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinez GJ, Nurieva RI, Yang XO, Dong C. Regulation and function of proinflammatory TH17 cells. Ann N Y Acad Sci. 2008;1143:188–211. doi: 10.1196/annals.1443.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhang H, Nguyen-Jackson H, Panopoulos AD, Li HS, Murray PJ, Watowich SS. STAT3 controls myeloid progenitor growth during emergency granulopoiesis. Blood. 2010 doi: 10.1182/blood-2009-12-259630. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Laouar Y, Welte T, Fu XY, Flavell RA. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity. 2003;19:903–912. doi: 10.1016/s1074-7613(03)00332-7. [DOI] [PubMed] [Google Scholar]
- 131.Takeda K, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci USA. 1997;94:3801–3804. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Welte T, et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA. 2003;100:1879–1884. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bromberg JF, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 134.Onai N, Obata-Onai A, Tussiwand R, Lanzavecchia A, Manz MG. Activation of the Flt3 signal transduction cascade rescues and enhances type I interferon-producing and dendritic cell development. J Exp Med. 2006;203:227–238. doi: 10.1084/jem.20051645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hegde S, et al. Stat3 promotes the development of erythroleukemia by inducing Pu.1 expression and inhibiting erythroid differentiation. Oncogene. 2009;28:3349–3359. doi: 10.1038/onc.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Miyoshi K, et al. Structure of the mouse Stat 3/5 locus: evolution from Drosophila to zebrafish to mouse. Genomics. 2001;71:150–155. doi: 10.1006/geno.2000.6433. [DOI] [PubMed] [Google Scholar]
- 137.Moriggl R, et al. Stat5 tetramer formation is associated with leukemogenesis. Cancer Cell. 2005;7:87–99. doi: 10.1016/j.ccr.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 138.Cui Y, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24:8037–8047. doi: 10.1128/MCB.24.18.8037-8047.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yao Z, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kondo M, et al. Cell-fate conversion of lymphoid-committed progenitors by instructive actions of cytokines. Nature. 2000;407:383–386. doi: 10.1038/35030112. [DOI] [PubMed] [Google Scholar]
- 141.Iwasaki-Arai J, Iwasaki H, Miyamoto T, Watanabe S, Akashi K. Enforced granulocyte/macrophage colony-stimulating factor signals do not support lymphopoiesis, but instruct lymphoid to myelomonocytic lineage conversion. J Exp Med. 2003;197:1311–1322. doi: 10.1084/jem.20021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Laurence A, et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 143.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 144.Yang XO, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- 145.Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. 2009;325:217–218. doi: 10.1126/science.1171461. [DOI] [PubMed] [Google Scholar]
- 146.Georgescu C, et al. A gene regulatory network armature for T lymphocyte specification. Proc Natl Acad Sci USA. 2008;105:20100–20105. doi: 10.1073/pnas.0806501105. [DOI] [PMC free article] [PubMed] [Google Scholar]