

Abstract
A facile protocol to prepare highly effective and durable in-line enzyme bioreactors inside capillary electrophoresis (CE) columns was developed. To demonstrate the methodology, L-glutamic dehydrogenase (GLDH) was selected as the model enzyme. GLDH was first immobilized onto 38 nm dia. gold nanoparticles (GNPs), and the functionalized GNPs were then assembled on the inner wall at the inlet end of the CE capillary treated with polyethyleneimine (PEI), producing an in-line GLDH bioreactor. Compared with a GLDH bioreactor prepared by immobilizing GLDH directly on PEI-treated capillary, the GNP-mediated bioreactor showed a higher enzymatic activity and a much better stability. The in-capillary enzyme bioreactor was proven very useful for screening of GLDH inhibitors deploying the GLDH-catalyzed α-ketoglutaric acid reaction. The screening assay was preliminarily validated by using a known GLDH inhibitor, perphenazine. A Z′ factor value of 0.95 (n = 10) was obtained, indicating the screening results were highly reliable. Screening of GLDH inhibitors present in medicinal plant extracts by the proposed method was demonstrated. Inhibition percentage was found to be 53% for radix scutellariae, 45% for radix codonopsis, 37% for radix paeoniae alba, and 0% for the other 22 extracts tested at a concentration of 0.6 mg extract /mL.
Keywords: Enzyme bioreactor, capillary electrophoresis, gold nanoparticles, inhibitor screening, medicinal plant extracts
Enzyme inhibitor screening is an effective approach to identify drug leads because many enzymes are involved in regulatory cellular processes and, thus become therapeutic drug targets [1–3]. High throughput screening (HTS) is the most commonly used method for screening of enzyme inhibitors [4–6]. It is based on colorimetric or fluorometric measurements carried out on multiwell microplates. It works perfectly with large libraries of pure compounds, but not applicable sometimes to assay complicate samples such as extracts of medicinal plants that may contain many UV-absorbing or fluorescent compounds [7–8]. Over the past years, search for enzyme inhibitors present in medicinal plants has been one of the major interests in drug discovery and development [9–11]. In fact, many new drugs developed between 1981 and 2002, particularly for treating cancer and infectious diseases originated from natural sources [12]. To study these complex sample matrices, screening assays based on liquid chromatography (LC), mass spectrometry (MS), and capillary electrophoresis (CE) were developed [13–15]. In most of these methods, an in-line enzyme bioreactor prepared by immobilizing the enzyme onto a solid support that was retained inside the LC or CE column was used. A test solution containing enzyme substrate was injected and incubated in the enzyme bioreactor for certain time. The product resulting from the enzymatic reaction was then separated from other co-exiting components including un-reacted substrate and quantified. If an enzyme inhibitor was present in the test solution, the amount of product was decreased. Obviously, having an effective and durable enzyme bioreactor is essential for the success of such a screening method.
To prepare enzyme bioreactors, immobilization techniques including physical adsorption, ionic binding, covalent binding, and sol-gel entrapment were deployed [16]. Wainer and co-workers described the immobilization of enzymes on silica particles that were then packed into LC columns for studying enzyme activity and screening of inhibitors [17]. Enzyme bioreactors prepared by entrapping enzymes in sol-gel derived monolithic capillary columns were reported for protein analysis by capillary electrophoresis [18] and for inhibitor screening by MS [19]. Using glutaraldehyde as a coupling reagent, enzymes were covalently immobilized onto amine-functionalized magnetic nanoparticles, to prepare an αglucosidase bioreactor used for inhibitor screening by CE [20]. In addition, Zhang et al. described a procedure to covalently immobilize trypsin onto a monolithic stationary phase that was activated by glutaraldehyde [21]. The trypsin bioreactor was used in μHPLC-MS/MS proteomic analysis with a digestion speed about 6600 times faster than that of digestion in free solutions. Compared with all other immobilization techniques, ionic binding is certainly the most convenient and fastest technique to immobilize enzymes. In an immobilization procedure via ionic binding, the solid support is first coated with a polyelectrolyte to obtain a suitably charged carrier surface (either positively or negatively depending on the predominant charge on the enzyme). The solid support with a charged surface is then soaked with an enzyme solution. Due to the electrostatic interactions, enzyme molecules bind to the surface of the solid support. The coating-and-binding process can be repeated several times, forming a layer-by-layer assembly of enzyme molecules on the carrier surface. Kang and co-workers described a bioreactor of angiotensin-converting enzyme using hexadimethrine bromide as the polyelectrolyte [22] and a layer-by-layer assembly bioreactor of acetylcholinesterase using polydiallyldimethylammonium chloride as the polyelectrolyte inside CE capillary columns that were used for enzyme inhibitor screening [23].
We describe here a new strategy for preparing CE in-column enzyme bioreactors based on ionic binding technique. The innovative aspect of the proposed procedure is that the enzyme is first immobilized on gold nanoparticles (GNPs) and the functionalized GNPs (instead of enzyme molecules) are then assembled onto the polyelectrolyte-modified carrier surface, forming a GNP-mediated enzyme bioreactor. The rationale for this experimental design is that enzyme loading in the bioreactor mediated by GNPs is expected to be much greater than that prepared from a free enzyme solution because enzyme molecules can be enriched on GNPs with a vast surface-to-mass ratio. In addition, a much better stability of the bioreactor can be expected because of the fact that enzyme-GNP conjugates are much more stable than enzyme-polyelectrolyte conjugates. It is well documented that thiol groups (-SH) present in protein molecules bind to GNPs strongly, which contributes to the high stability of protein-GNP conjugates [24]. Figure 1 illustrates an enzyme bioreactor prepared by the proposed approach. In this work, L-glutamic dehydrogenase (GLDH) was selected as the test enzyme. The prepared CE in-column GLDH bioreactor was evaluated in terms of enzyme activity, bioreactor stability, and its usefulness for screening of GLDH inhibitors present in medicinal plants by CE.
Figure 1.
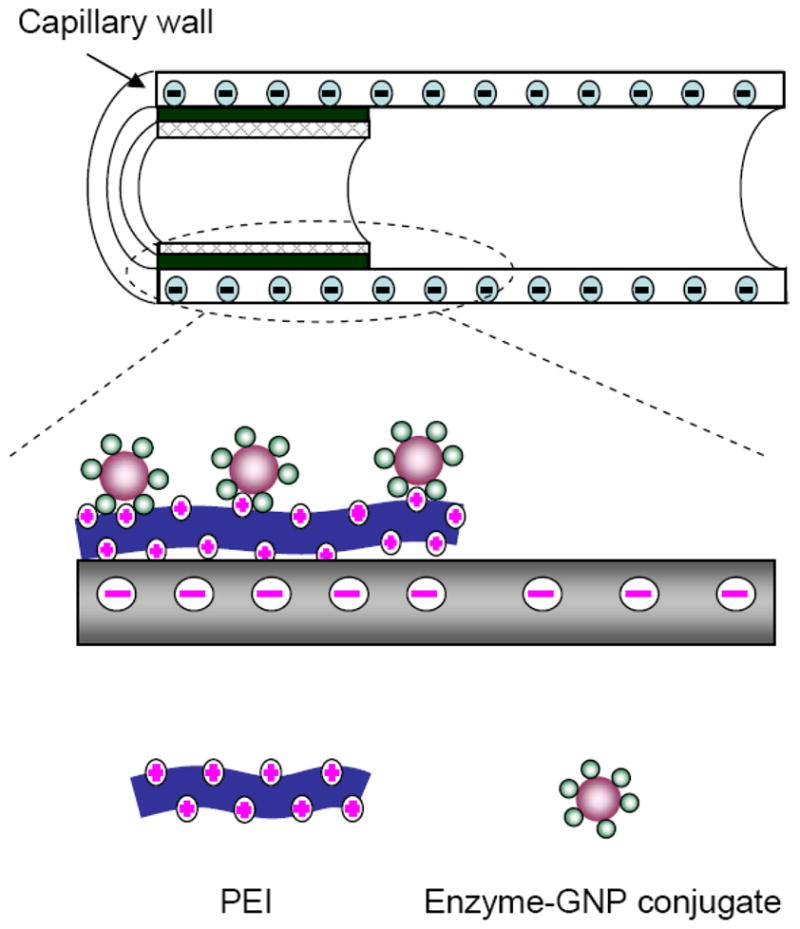
Schematic of a gold nanoparticles-mediated enzyme bioreactor prepared in a CE column.
Materials and methods
Chemicals and solutions
L-Glutamic dehydrogenase (GLDH), nicotinamide adenine dinucleotide (β-NAD+), β-nicotinamide adenine dinucleotide, reduced disodium salt (β-NADH), poly(ethyleneimine) (PEI, Mw= 750,000, 50% (w/v) solution), α-ketoglutaric acid, and perphenazine were purchased from Sigma–Aldrich Chemicals (St. Louis, MO, USA). All medicinal plants tested were collected from local markets. The GLDH solution was prepared by dissolving 0.0020 g GLDH in 200 μL 30% glycerol solution and kept at −20 °C before use. The PEI solution was freshly prepared by diluting the 50% (w/v) PEI solution with water to obtain a concentration of 4% (w/v) PEI.
CE System
All CE experiments were performed using an HP3D capillary electrophoresis system (Hewlett Packard, Waldbonm, Germany). UV detection was at a fixed wavelength of 254 nm. Data were processed with a HP Chemstation.
Preparation of GLDH-functionalized GNPs
GNPs (38 nm dia.) were prepared as described previously [25]. Briefly, 100 mL of a 0.01% (w/v) HAuCl4 solution was heated to boiling, and then 1.0 mL of a 1% trisodium citrate solution was added with stirring. The solution was refluxed for 30 min. After cooling to room temperature, the solution was filtered through a 0.45 μm nylon membrane. The pH of the GNP solution was adjusted to pH 9.0 with K2CO3 and kept at 4 °C. The size and monodispersity of GNPs prepared was determined by using a JEOL 100CX transmission electron microscope. To prepare GLDH-functionalized GNPs, 95 μL of the GNP solution was mixed with 5 μL of the above prepared 10 mg /mL GLDH solution. The mixture was vortexed for 1 min and then let stand alone at 4 °C for 1 hr.
Preparation of a GLDH bioreactor in a CE column
A piece of 50 μm ID × 38 cm (29.5 cm to detection window) fused-silica capillary was used. The capillary was treated with a 1 M NaOH solution for 1 hr and then flushed with by water. A ~0.5 cm section of the capillary at the inlet end was filled with the PEI solution above prepared. After 10 min, the PEI solution was washed out with water. The GLDH-functionalized GNP solution was introduced and filled the PEI modified section of the capillary. Negatively charged GLDH-GNP conjugates bound to the positively charged PEI coating on capillary wall. After 2 hrs, the capillary was flushed with water and ready for use in CE experiments.
Preparation of the extracts of medicinal plants
Air-dried medicinal plants were ground to fine powders. Isopropanol-H2O (1:1) solution was added to the powder in an Erlenmeyer flask (40 mL isopropanol solution / 10 g powder). The mixture was sonicated for 2.5 hrs at a frequency of 100 kHz and 60 °C by using an ultrasonic cleaning bath. The mixture was filtered. The filtrate collected was evaporated to nearly complete dryness. The residue was dried in a vacuum drier at 30 °C and 0.07 MPa, obtaining the extract. Solutions of extracts were prepared in 50% isopropanol at 6 mg /mL. The solution was appropriately diluted with the CE running buffer before assay.
Screening of GLDH inhibitors by CE
The screening assay was based on the following enzymatic reaction
For enzyme activity studies, a substrate solution was injected into the CE capillary with an enzyme bioreactor at the inlet end by pressure at 40 mbar for 5 s. A voltage of −28 kV was applied to separate β-NAD+ from β-NADH and potentially any other existing compounds in the sample solution. After the separation, β-NAD+ was quantified by UV absorbance at 254 nm. Enzyme activity was assessed based on the amount of β-NAD+ produced from the enzymatic reaction.
For inhibition studies, the CE running buffer containing a known inhibitor (or extract of a medicinal plant) was injected into the capillary and left in place for 1 min. A inhibitor (or the extract) -containing substrate solution was then injected. The CE separation was started and β-NAD+ was quantified as described above. The inhibition percentage was calculated according to the peak area of β-NAD+ compared with the reference electropherogram obtained in the absence of the inhibitor (or extract).
Results and discussion
CE Quantification of β-NAD+
β-NAD+ is a coenzyme found in all living cells. β-NAD+ and its reduced form, β-NADH are involved as a reactant /product pair in many enzymatic reactions. For example, in the GLDH catalyzed reaction of α-ketoglutaric acid selected in this work for screening of GLDH inhibitors, β-NADH is converted to β-NAD+. Therefore, quantification of β-NAD+ in the reaction mixture serves well the purpose of monitoring these enzymatic reactions if β-NAD+ doesn’t occur in the sample matrix (i.e. the herbal extracts in this work). CE was proven to be a very effective technique to separate these two compounds. Thus, a relatively short CE column (29.5 cm effective length) was used to achieve a fast quantification. Because phosphate ions affect GLDH activity while borate ions don’t [26], a borate buffer solution (25 mM at pH 9.0) was chosen as the CE running buffer. Under the experimental conditions selected, β-NAD+ and β-NADH were separated within 3 min (Figure 2). For quantification, seven-point calibration curves were prepared by analyzing β-NAD+ standard solutions at varying concentrations from 0.0100 to 0.400 mM. β-NAD+ was quantified using the CE peak area. Linear regression resulted in the following regression equation:
Figure 2.
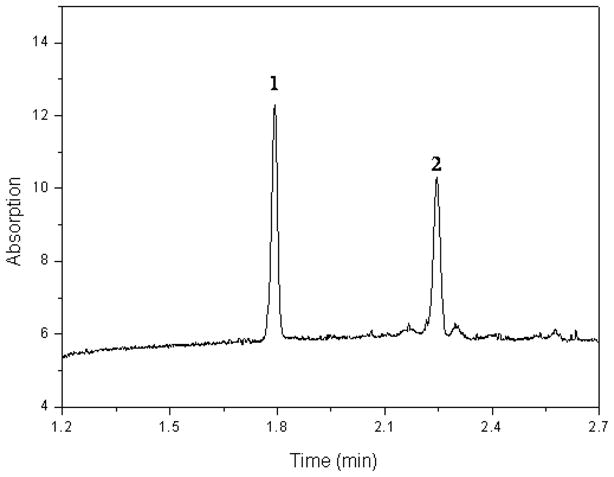
CE separation of β-NAD+ and β-NADH. Experimental conditions: CE column, 50 mm ID x 29.5 cm effective length fused silica capillary; injection, 40 mbar for 5 s; running buffer, 25 mM borate buffer (pH 9.0); voltage applied, -28 kV; column temperature, 25 °C; UV detection at 254 nm. Peak identifications: 1, β-NAD+; and 2, β-NADH (0.10 mM each).
Where Y was the peak area, X was β-NAD+ concentration in mM, and r2 was the correlation coefficient. The detection limit was calculated to be 0.002 mM for β-NAD+ (S/N = 3). To determine the assay reproducibility, a 0.050 mM β-NAD+ standard solution was analyzed 10 times. The assay reproducibility was found to be 1.4% (RSD, n = 10). The reproducibility of β-NAD+ migration time (RSD, n = 10) was 0.65%.
Performance of the in-line GLDH bioreactor
The GLDH bioreactor was first evaluated in terms of GLDH activity of the bioreactor. In these studies, a substrate solution containing 16 mM α-ketoglutaric acid, 0.050 mM β-NADH and 80 mM ammonium acetate was injected into the CE capillary with an in-line GLDH bioreactor at the inlet end and the solution was left in the bioreactor for some time (from 0 to 3 min) before a CE voltage was applied to separate and quantify β-NAD+ produced from the enzymatic reaction. The results indicated that the enzymatic reaction was more than 75% complete with zero incubation time (i.e. the CE separation was started immediately after injecting the substrate solution). Figure 3 shows the relationship between β-NAD+ amount produced and the incubation time. Within 1.0 min the enzymatic reaction was more than 97% complete. The reaction velocity was unexpectedly high considering that the enzyme quantity loaded in the bioreactor was extremely small. The high reaction velocity was likely due to the vast GNP surface area for enzyme immobilization, and thus a high level of contact between the immobilized enzyme and the substrate.
Figure 3.

Enzyme activity tests: incubation time versus the amount of β-NAD+ produced from the enzymatic reaction. The substrate solution injected contained 80 mM ammonium acetate, 16 mM α-ketoglutaric acid, and 0.050 mM β-NADH in CE running buffer. CE experimental conditions were as in Fig. 2.
The value of Michaelis-Menten constant (Km), a most important parameter of an enzymatic reaction, was also determined for the GLDH bioreactor by using Lineweaver & Burk’s plotting method:
| (1) |
where v and Vmax are the initial and maximal velocity of the reaction, and [S] is the substrate concentration. Six β-NADH solutions at different concentrations ranging from 0.020 to 0.100 mM were analyzed. Each of the solutions was analyzed three times. The peak area of β-NAD+ was used to represent the initial reaction rate. A double-reciprocal plot defined by Eq. (1) was constructed. Linear regression analysis of the data produced the following equation: 1/v = 0.373 (1/[S]) + 24.85 (r2 = 0.9961). From this equation, Km was calculated to be 0.015 mM that was similar to that obtained from the GLDH reaction in free solutions (0.02 mM [27]). The results suggested that no significant change in the substrate-GLDH binding property was caused by enzyme immobilization.
Stability of the in-line GLDH bioreactor was assessed. A substrate mixture solution was repetitively injected for analysis up to 110 times on the same CE capillary with an in-line GLDH bioreactor. Figure 4 shows the results of β-NAD+ peak area versus the number of assays. As can bee seen, the efficacy of the GLDH bioreactor (measured by β-NAD+ peak area) tended to decrease as the number of assays increased. However, the decrease was relatively insignificant (less than 15% after an extensive use for 110 times). In addition, the bioreactor could be re-generated easily by dipping the inlet end of the capillary into the GLDH-functionalized GNP solution for 10 s and flushing the capillary with water 2 hrs after. For comparison, a GLDH bioreactor was prepared by immobilizing GLDH (instead of GLDH-functionalized GNPs) onto the inner wall of capillary treated with PEI. It was found that the enzymatic activity decreased very quickly. The bioreactor lost about 40% of its initial activity after only 5 assays. The improved stability of the GNP-mediated bioreactor likely resulted from the fact that GLDH-GNP conjugate was much more stable than GLDH-PEI conjugate.
Figure 4.

Stability of the in-line GLDH bioreactor: enzyme efficacy versus the number of assays. Experimental conditions were as in Fig. 3.
The above described studies showed that the in-line GLDH bioreactor had not only a high enzymatic activity, but also a good stability. In addition, the CE quantification allowed accurate, reliable, and quick monitoring of the GLDH reaction. These results combined suggested that the CE-based method was well suited for screening of GLDH inhibitors. To evaluate its applicability for GLDH inhibitor screening, inhibition studies using a known GLDH inhibitor, perphenazine, were performed. In these tests, a perphenazine solution prepared in the CE running buffer was injected and left in place for 1 min. The substrate solution containing perphenazine at 0.125 mM was then injected. It was noted that without the inhibitor in the bioreactor a significant amount of the substrate was converted to the product before the enzyme was inhibited due to a very fast enzymatic reaction rate. The inhibition percentage was calculated using the following equation:
| (2) |
where x and blank are the peak areas of β-NAD+ measured with or without the inhibitor, respectively. The quality of screening assays can be assessed by using Z’ factor calculated as following [28]:
| (3) |
where μs and μc are the means of the signal (i.e. β-NAD+ peak area in this work) obtained from the standard (in the absence of inhibition) and control assays (100% inhibition by a known inhibitor), respectively, whereas σs and σc are the standard deviations of the data. A Z’ factor value between 0.5 and 1 indicates that the quality of the screening data is excellent, i.e. the inhibitor screening assay is accurate and reliable. In this work, perphenazine solutions at 0.06 and 0.12 mM were assayed. The Z′ factor value was calculated as 0.95 (n=10). The result indicated that the accuracy of the present CE-based screening method was excellent for the screening of GLDH inhibitors.
Screening of GLDH Inhibitors in Chinese medicinal plants
Isopropanol extracts were prepared from 25 Chinese medicinal plants. In addition, perphenazine (a known GLDH inhibitor) was also included into the chemical library for the purpose of assay validation (as a positive control). Triplicate assays were made for each isopropanol extract. The averaged peak areas from the three measurements were used to calculate the inhibition percentage by Equation (2). Fig. 5 shows several electropherograms from the screening assays. Fig. 5a was from a blank solution (i.e. in the absence of inhibition). The injected substrate solution contained α-ketoglutaric acid, ammonium acetate, and β-NADH. Fig. 5b and Fig. 5c were from assaying radix scutellariae and radix codonopsis extracts (both at 0.6 mg /mL), respectively. The injected sample solutions were the CE running buffer containing the extract at 0.6 mg /mL for pre-incubation and then the substrate solution containing the extract. As can be seen, the peak areas of β-NAD+, the product of the enzymatic reaction were reduced clearly compared with that in Fig. 5a from a blank solution, indicating clearly GLDH inhibition: 53% inhibition from Radix Scutellariae extract and 45% from radix codonopsis extract. It’s worth noting that several endogenous compounds present in the extract were separated from β-NAD+ and β-NADH as shown in Fig 5b. This might eliminate potential interference with the quantification and, therefore, false screening results. As can be seen in Table 1, perphenazine (at 0.125 mM) and 3 extracts at 0.6 mg /mL were found to be positive for GLDH inhibition. The other extracts didn’t show any inhibitory effects. They served well as negative controls for the screening assay whereas perphenazine served as a positive control. Extract of radix scutellariae showed a 53% inhibition on GLDH activity. Radix scutellariae is widely used for the treatment of inflammation, fever, hepatitis and allergic diseases and hypertension, etc. in Chinese traditional medicine.28 Studies have shown that radix scutellariae lowers blood pressure and has sedative effects on the central nervous system [30–31].
Figure 5.

Electropherograms obtained from the screening assays: a) blank solution (i.e. the substrate solution); b) radix Scutellariae extract at 0.6 mg /mL; c) radix codonopsis extract at 0.6 mg /mL; and d) perphenazine (a known GLDH inhibitor) at 0.125 mM. Assay conditions were as in Fig. 3. Peak identifications: 1) β-NAD+; and 2) β-NADH.
Table 1.
Results of GLDH inhibitor screening in medicinal plant extracts*
| Medicinal Plant | Inhibition %** | Medicinal Plant | Inhibition %** |
|---|---|---|---|
| Perphenazine (a known GLDH inhibitor) | 65 | Exocarpium citri rubrum | 0 |
| Radix astagali | 0 | Wrinkled gianthyssop | 0 |
| Fructus crataegi | 0 | Herba verbenae | 0 |
| Poria | 0 | Radix scutellariae | 53 |
| Radix achyranthis bidentatae | 0 | Radix scrophulariae | 0 |
| Fructus hordei germinatus | 0 | Semen lablab album | 0 |
| Radix paeoniae alba | 37 | Pericarpium citri reticulatae | 0 |
| Fructus piperis | 0 | Leaf of henon bamboo | 0 |
| Rhizoma chuanxiong | 0 | Fructus amomi rotundus | 0 |
| Rhizoma anemarrhenae | 0 | Radix et rhizoma rhei | 0 |
| Radix ophiopogonis | 0 | Semen arecae | 0 |
| Rhizoma pinelliae | 0 | Radix codonopsis | 45 |
| Rhizoma atractylodis macrocephalae | 0 | Bulbus fritillariae cirrhosae | 0 |
Concentration of extract was 0.6 mg /mL; and perphenazine concentration was 0.125 mM.
Mean of triplicate assays.
Conclusion
GLDH could be easily immobilized on 38 nm dia. GNPs. The functionalized GNPs were assembled on the inner wall at the inlet end of a CE capillary modified by PEI, producing an in-line GLDH bioreactor. The GNP-mediated GLDH bioreactor showed a high enzymatic activity and a very good stability. The value of Michaelis-Menten constant (Km) was 0.015 mM that was very similar to that obtained from the GLDH reaction in free solutions. The result suggested that no significant changes in the substrate-GLDH binding property were caused by enzyme immobilization. Use of the highly effective and durable in-line GLDH bioreactor in combination with the proposed CE quantification of β-NAD+ allowed accurate and reliable GLDH inhibitor screening. The proposed screening method was applied to analyzing extracts of 25 medicinal plants. Inhibition percentage was found to be 53% for radix scutellariae, 45% for radix codonopsis, 37% for radix paeoniae alba, and 0% for the other 22 extracts tested. The screening method was proved to be accurate, easy to carry out, and well suited for assaying complex samples such as medicinal plant extracts.
Acknowledgments
Financial support from the National Natural Science Foundations of China (No. 20875019 to SZ), the Guangxi Science Foundation of China (No. 0832004 to SZ), and US National Institutes of Health (SC1 GM089557 to YML) is gratefully acknowledged.
Abbreviations used
- GNPs
gold nanoparticles
- CE
capillary electrophoresis
- GLDH
glutamic dehydrogenase
- β-NAD+
nicotiamide adenine dinucleotide
- PEI
polyethyleneimine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johnson TO, Ermolieff J, Jirousek MR. Protein tyrosine phosphatase 1B inhibitors for diabetes. Nat Rev Drug Discov. 2002;9:696–709. doi: 10.1038/nrd895. [DOI] [PubMed] [Google Scholar]
- 2.Noble MEM, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. doi: 10.1126/science.1095920. [DOI] [PubMed] [Google Scholar]
- 3.Stravopodis DJ, Margaritis LH, Voutsinas GE. Drug-mediated targeted disruption of multiple protein activities through functional inhibition of the Hsp90 chaperone complex. Curr Med Chem. 2007;14:3122–3138. doi: 10.2174/092986707782793925. [DOI] [PubMed] [Google Scholar]
- 4.von Ahsen O, Bomer U. High-throughput screening for kinase inhibitors. ChemBiochem. 2005;6:481–490. doi: 10.1002/cbic.200400211. [DOI] [PubMed] [Google Scholar]
- 5.Ma H, Horiuchi KY. Chemical microarray: a new tool for drug screening and discovery. Drug Discov Today. 2006;11:661–668. doi: 10.1016/j.drudis.2006.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kool J, Lingeman H, Niessen W, Irth H. High throughput screening methodologies classified for major drug target classes according to target signaling pathways. Comb Chem High Throughput Screen. 2010;13:548–61. doi: 10.2174/138620710791515815. [DOI] [PubMed] [Google Scholar]
- 7.Grepin C, Pernelle C. High-throughput screening. Drug Discov Today. 2000;5:212–214. doi: 10.1016/s1359-6446(00)01491-4. [DOI] [PubMed] [Google Scholar]
- 8.Gao H, Leary JA. Multiplex inhibitor screening and kinetic constant determinations for yeast hexokinase using mass spectrometry based assays. J Am Soc Mass Spectrom. 2003;14:173–181. doi: 10.1016/S1044-0305(02)00867-X. [DOI] [PubMed] [Google Scholar]
- 9.Uehara Y. Natural product origins of Hsp90 inhibitors. Curr Cancer Drug Targets. 2003;3:325–30. doi: 10.2174/1568009033481796. [DOI] [PubMed] [Google Scholar]
- 10.Lam KS. New aspects of natural products in drug discovery. Trends Microbiol. 2007;15:279–289. doi: 10.1016/j.tim.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Shiao MS, Chiu JJ, Chang BW, Wang J, Jen WP, Wu YJ, Chen YL. In search of antioxidants and anti-atherosclerotic agents from herbal medicines. Biofactors. 2008;34:147–57. doi: 10.1002/biof.5520340206. [DOI] [PubMed] [Google Scholar]
- 12.Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the last 25 years. J Nat Prod. 2003;66:1022–1037. doi: 10.1021/np030096l. [DOI] [PubMed] [Google Scholar]
- 13.Glatz Z. Determination of enzymatic activity by capillary electrophoresis. J Chromatogr B. 2006;841:23–37. doi: 10.1016/j.jchromb.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 14.Greis KD. Mass spectrometry for enzyme assays and inhibitor screening: an emerging application in pharmaceutical research. Mass Spectrom Rev. 2007;26:324–39. doi: 10.1002/mas.20127. [DOI] [PubMed] [Google Scholar]
- 15.Hoffmann T, Martin MM. CE-ESI-MS/MS as a rapid screening tool for the comparison of protein-ligand interactions. Electrophoresis. 2010;31:1248–55. doi: 10.1002/elps.200900585. [DOI] [PubMed] [Google Scholar]
- 16.Hanefeld U, Gardossi L, Magner E. Understanding enzyme immobilization. Chem Soc Rev. 2009;38:453–468. doi: 10.1039/b711564b. [DOI] [PubMed] [Google Scholar]
- 17.Bartolini M, Andrisano V, Wainer IW. Development and characterization of an immobilized enzyme reactor based on glyceraldehyde-3-phosphate dehydrogenase for on-line enzymatic studies. J Chromatogr A. 2003;14:331–40. doi: 10.1016/s0021-9673(02)01809-5. [DOI] [PubMed] [Google Scholar]
- 18.Sakai-Kato K, Kato M, Toyo’oka T. On-line trypsin-encapsulated enzyme reactor by the sol-gel method integrated into capillary electrophoresis. Anal Chem. 2002;74:2943–2949. doi: 10.1021/ac0200421. [DOI] [PubMed] [Google Scholar]
- 19.Hodgson RJ, Besanger TR, Brook MA, Brennan JD. Inhibitor screening using immobilized enzyme reactor chromatography/mass spectrometry. Anal Chem. 2005;77:7512–7519. doi: 10.1021/ac050761q. [DOI] [PubMed] [Google Scholar]
- 20.Hu F, Deng C, Zhang X. Development of high performance liquid chromatography with immobilized enzyme onto magnetic nanospheres for screening enzyme inhibitor. J Chromatogr B. 2008;871:67–71. doi: 10.1016/j.jchromb.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 21.Ma J, Liang Z, Qiao X, Deng Q, Tao D, Zhang L, Zhang Y. Organic-inorganic hybrid silica monolith based immobilized trypsin reactor with high enzymatic activity. Anal Chem. 2008;80:2949–2956. doi: 10.1021/ac702343a. [DOI] [PubMed] [Google Scholar]
- 22.Tang ZM, Kang JW. Enzyme inhibitor screening by capillary electrophoresis with an on-column immobilized enzyme microreactor created by an ionic binding technique. Anal Chem. 2006;78:2514–2520. doi: 10.1021/ac052030w. [DOI] [PubMed] [Google Scholar]
- 23.Tang ZM, Wang TD, Kang JW. Screening of acetylcholinesterase inhibitors in natural extracts by CE with electrophoretically mediated microanalysis technique. Electrophoresis. 2007;28:2981–2987. doi: 10.1002/elps.200600327. [DOI] [PubMed] [Google Scholar]
- 24.Thobhani S, Attree S, Boyd R, Kumarswami N, Noble J, Szymanski M, Porter RA. Bioconjugation and characterisation of gold colloid-labelled proteins. J Immunol Methods. 2010;356:60–69. doi: 10.1016/j.jim.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Zhao S, Niu T, Song Y, Liu YM. Gold nanoparticle-enhanced chemiluminescence detection for CE. Electrophoresis. 2009;30:1059–1065. doi: 10.1002/elps.200800283. [DOI] [PubMed] [Google Scholar]
- 26.Prisco GD. Effect of pH and ionic strength on the catalytic and allosteric properties of native and chemically modified ox liver mitochondrial glutamate dehydrogenase. Arch Biochem Biophys. 1975;171:604–612. doi: 10.1016/0003-9861(75)90070-3. [DOI] [PubMed] [Google Scholar]
- 27.McCarthy AD, Tipton AF. Ox glutamate dehydrogenase. Comparison of the kinetic properties of native and proteolysed preparations. Biochem J. 1985;230:95–99. doi: 10.1042/bj2300095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 29.Wang YS, Deng WL, Que CS. Pharmacology and application of Traditional Chinese Medicine. Human Health Press; Beijing: 1998. pp. 972–982.pp. 1004–1029. [Google Scholar]
- 30.Chen CS, Chen NJ, Lin LW, Hsieh CC, Chen GW, Hsieh MT. Effects of Scutellariae Radix on gene expression in HEK 293 cells using cDNA microarray. J Ethnopharmacol. 2006;105:346–351. doi: 10.1016/j.jep.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Kang K, Oh YK, Choue R, Kang SJ. Scutellariae radix extracts suppress ethanol-induced caspase-11 expression and cell death in N(2)a cells. Brain Res Mol Brain Res. 2005;142:139–145. doi: 10.1016/j.molbrainres.2005.09.006. [DOI] [PubMed] [Google Scholar]