

Abstract
In the CNS, the evolutionarily conserved Notch pathway regulates asymmetric cell fate specification to daughters of ganglion mother cells (GMCs). The E3 Ubiquitin ligase protein Neuralized (Neur) is thought to activate Notch-signaling by the endocytosis of Delta and the Delta-bound extracellular domain of Notch. The intracellular Notch then initiates Notch-signaling. Numb blocks N-signaling in one of the two daughters of a GMC, allowing that cell to adopt a different identity. Numb is asymmetrically localized in a GMC and is segregated to only one of the two daughter cells. In the typical GMC-1->RP2/sib lineage, we found that loss of Neur activity causes symmetric division of GMC-1 into two RP2s. We further found that Neur asymmetrically localizes in a late GMC-1 to the Numb domain and Neur mediates asymmetric division via two distinct, sequential mechanisms: by promoting the asymmetric localization of Numb in a GMC-1 via down-regulation of the transcription factor Pdm1, followed by enhancing the Notch-signaling via trans-potentiation of Notch in a cell committed to become a sib. In neur mutants the GMC-1 identity is not altered but Numb is non-asymmetrically localized due to an up-regulation of Pdm1. Thus, both its daughters inherit Numb, which prevents Notch from specifying a sib identity. Neur also enhances Notch since in neur; numb double mutants, both sibling cells often adopt a mixed fate as opposed to an RP2 fate observed in Notch;numb double mutants. Furthermore, over-expression of Neur can induce both cells to adopt a sib fate similar to gain of function Notch. Our results tie Numb and Notch-signaling through a single player, Neur, thus giving us a more complete picture of the events surrounding asymmetric division of precursor cells. We also show that Neur and Numb are interdependent for their asymmetric-localizations.
Keywords: Neuralized, asymmetric division, Neural precursor, Numb, Notch
Introduction
Loss of function mutations in the neurogenic gene, neuralized (neur), causes a hyperplastic nervous system at the expense of the ectoderm, similar to other neurogenic mutants such as Notch (N) and Delta (Dl). The neur gene encodes an intracellular peripheral membrane protein with two NEUR domains and a C-terminal RING domain (Lai and Rubin, 2001a, b; Lai et al., 2001; Price et al., 1993; Boulianne et al., 1993). Consistent with the presence of RING domain, it has been shown that Neur functions as an E3 Ubiquitin ligase (Lai et al., 2001; Yeh et al., 2001). Recent work indicates that Neur is also involved in N-signaling (Lai and Rubin, 2001a, b; Lai et al., 2001; Pavlopoulos et al., 2001). The N-signaling mediates a number of developmental cellular processes, most of which are involved in cell fate determination. During N-signaling, the extracellular domain of N (NEXTRA) interacts with the extracellular domain of Dl (or Serrate, another N ligand). This interaction leads to the proteolytic processing and release of NINTRA. NINTRA then translocates into the nucleus and mediates transcription of genes such as Enhancer of split, vestigial, etc. It appears that the activity of Neur is essential to the release of NINTRA. Current evidence suggests that Neur mediates endocytosis of the Delta-bound NEXTRA (Pavlopoulos et al., 2001). Such an activity of Neur can release the processed NINTRA from the membrane and potentiate N-signaling.
How does N-signaling regulate asymmetric cell identity specification? In the CNS of Drosophila embryo, the primary neuronal progenitor cells called neuroblasts (NBs) divide by asymmetric mitosis to “self-renew” and to produce a chain of ganglion mother cells (GMCs). Although a GMC is bipotential, it does not normally self-renew (Bhat and Apsel, 2004). Instead, it divides asymmetrically to generate two different post-mitotic neurons. Previous studies have shown that the N-signaling plays a crucial role not only in selecting a neural versus ectodermal fates during early neurogenesis, but also in the later asymmetric fate specification of daughter cells of GMCs (reviewed in Gaziova and Bhat, 2007). This later function of N-signaling has an interesting, antagonistic relationship to the function of cytoplasmic protein Numb. Numb localizes to the basal end of a GMC and during division, it segregates into one of its two daughter cells, where it inhibits the cleavage of NINTRA. This blocks the ability of Notch to specify a different fate (Buescher, et al., 1998; Wai et al., 1999). In the GMC-1->RP2/sib lineage, for example, the loss of function for Notch causes both the daughter cells of the GMC-1 to adopt an RP2 fate. This indicates that Notch specifies a sib fate. In the absence of Numb, both cells adopt the sib fate, thus, Numb is necessary to specify an RP2 fate. In the absence of both notch and numb, however, the two daughter cells adopt the RP2 fate indicating that Numb is necessary to specify an RP2 fate only when there is an intact N-signaling; Numb, thus, blocks Notch-signaling from specifying a sib fate to a cell.
In the CNS, while Neur plays a role in the selection of neural versus epidermal fates, it is not known if Neur plays any role in the terminal asymmetric divisions of GMCs or if Neur regulates N-signaling in this process. Therefore, we sought to examine the role of Neur in the asymmetric cell fate specification of GMCs in the CNS and its relation to Notch-signaling. We focused our efforts on the GMC-1->RP2/sib lineage, and to a lesser extent in another lineage, the GMC-1->aCC/pCC. The NB4-2→GMC-1->RP2/sib lineage is one of the very well studied lineages (see also Chu-LaGraff and Doe, 1993; Bhat and Schedl, 1994; Bhat, 1998; Buescher et al., 1998; Gaziova and Bhat, 2007). Many mutations and genes have been identified as required for the elaboration of this lineage. The asymmetric division of GMC-1 (also known as GMC4-2a) has been particularly proven useful to study asymmetric division of precursor cells and to determine the role of many molecules in this process (Buescher et al., 1993; Bhat and Schedl, 1994; Bhat et al., 1995; Mehta and Bhat, 2001; Bhat and Apsel, 2004).
Previous studies have shown that loss of function for not only Notch, but also for the other members of the Notch pathway such as mastermind (mam), causes GMC-1 of the RP2/sib lineage to undergo symmetric division into two RP2s instead of an asymmetric division into RP2 and sib cells (Wai et al., 1999; Yedvobnick et al., 2004). We sought to determine if the loss of function for neur would have a similar defect in the same lineage. In the CNS we found that Neur regulates asymmetric division by two distinct and sequential mechanisms: asymmetric distribution of Numb via down-regulation of Pdm, and trans-potentiation of Notch. Neur, which is asymmetrically localized to the same domain as Numb, primarily regulates asymmetric division through down-regulation of the POU protein Pdm1 and Numb localization. In neur mutants Numb is non-asymmetric in GMC-1 and both daughters inherit Numb, which then prevents the processing of Notch and the specification of sib identity. This symmetric division phenotype is also observed with the over-expression of the two POU genes, Pdm1 or Pdm2 (Mehta and Bhat, 2001; Bhat and Apsel, 2004). Consistent with this, Pdm1 is up-regulated in GMC-1 of neur mutants and down-regulated when Neur is over-expressed. In neur; numb double mutants, both sibling cells often adopt a mixed fate, which is different from the fate specification in Notch;numb double mutants where both daughter cells unambiguously adopt an RP2 fate. This result indicates that while Neur is also involved in Notch-signaling in order to specify the sib fate, its role appears to be not essential, but simply only to enhance the efficiency of Notch signaling. In agreement with this conclusion, over-expression of Neur, while induces both cells to adopt a sib fate similar to gain of function Notch, the penetrance of this phenotype is weak. Our results show that Neur down-regulates Pdm proteins to promote asymmetric localization of Numb. The localized Neur then segregates to the presumptive RP2 and enhances Notch signaling via a trans-activation mechanism. Finally, we show that while the localization of Numb is Neur-dependent, the localization of Neur is Numb-dependent, illustrating an inter-dependence of the two proteins to localize to the basal end of the precursor cell.
Materials and Methods
Fly strains, genetics
All flies and crosses are performed at 22 °C unless otherwise indicated. The following strains are used: for the analysis of neur, neurA101 (a P-element insertion at the 5′ end of the gene, Price at al., 1993) and neur1 (an EMS-induced point mutation) were used. A deficiency for neur Df(3R)BSC24 (cytology: 85B7-85D15; neur gene is located at 85C2-85C3), was also examined and the penetrance of the RP2 lineage defects was marginally higher in the deficiency homozygous embryos, suggesting that the point mutation is close to null as far as this lineage is concerned. The P-element insertion allele, neurA101, was also very strong hypomorph as far as this lineage is concerned with only a marginally lower penetrance of the defect compared to neur1 or the deficiency. To induce neur during development, we used a UAS-neur transgenic line (w; UAS-neur 7.5), which carries a full-length 7.5 kb neur transgene under the UAS promoter. UAS-neurΔRF transgenic line (w;UAS-neurΔRF) carries two copies of a truncated neur transgene with its RING domain missing, under the control of UAS promoter (source: Eric Lai). For inducing the UAS-neur and UAS-neurΔRF transgenes temporally, we used Heat-shock-GAL4 (GAL4 under the control of heat shock 70 promoter). Other mutant strains used were numb796, insc22, Delta, NotchTS1, and cyclin A [l(3) 03445)]. For wild type, Oregon-R flies were used. Various mutant combinations were generated using standard genetics. To exclude any maternal modifier effects of balancers (see Bhat et al., 2007; Gaziova and Bhat, 2009), homozygous mutant embryos were also tested by out-crossing the balancer-bearing mutants (mutant/balancer) to wild type and back crossing the non-balancer bearing mutant adults (mutant/+ x mutant/+) for embryo collection. Staging of embryos was done as described in Wieschaus and Nusslein-Volhard (Wieschaus and Nusslein-Volhard, 1986).
Immunohistochemistry
Standard immunostaining procedures were used with some modifications; modifications to the general fixation conditions and staining can be obtained by request. Embryos were fixed and stained with the following antibodies: Eve (rabbit, 1:2000 dilution; source: Manfred Frasch), Eve (mouse, 1:5; DSHB, University of Iowa), Zfh1 (mouse, 1:400: Zun Lai), 22C10 (mouse, 1:4; DSHB), LacZ (rabbit, 1:3000 or mouse, 1:400), BP102 (mouse, 1:10; DSHB) Fas II (mouse; 1:5; DSHB), Numb (rabbit, 1:100), Insc (rabbit, 1:50; Bill Chia), Neur (rabbit, 1:400; Eric Lai), Spectrin (mouse, 1:200; DSHB), and Pdm1 (mouse, 1:15; Steve Cohen). For confocal microscopy of embryos, Cy5 (1:400) and FITC (1:50)-conjugated secondary antibodies were used. For light microscopy, alkaline phosphatase or DAB-conjugated secondary antibodies were used. The relative density of Pdm1 and Eve staining signals (Fig. 4 and Fig. 5) was quantified by AlphaEaseFC (6.0, Alpha Innotech Corporation) program.
Figure 4. Expression of Pdm1 protein is up-regulated in neur mutants.
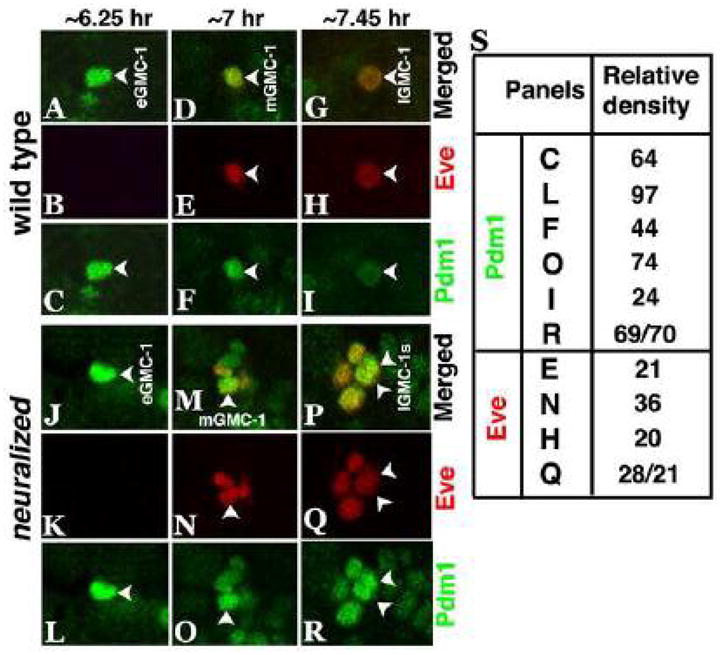
Wild type and neur mutant embryos are double stained with Eve (Red) and Pdm1 (Green) at different times during GMC-1 development. While in wild type (panels A–I), the level of Pdm1 protein goes down from an early GMC-1 to a late GMC-1, in the mutant, Pdm1 is at a much higher level in an early GMC-1 (panels J, L) as well as in a mid or a late GMC-1 (panels M, O, P, R). The level of Eve is the same in a mid GMC-1 or a late GMC-1; an early GMC-1 has not yet started to express Eve (panels B and K). A late GMC-1 is larger than a mid or an early GMC-1, and a mid GMC-1 is smaller than an early GMC-1. In panel S, quantification of Pdm1 and Eve in GMC-1 was done by measuring the relative densities of Pdm1 and Eve staining signals in GMC-1.
Figure 5. Over-expression of Neur in a GMC-1 down-regulates the levels of Pdm1.
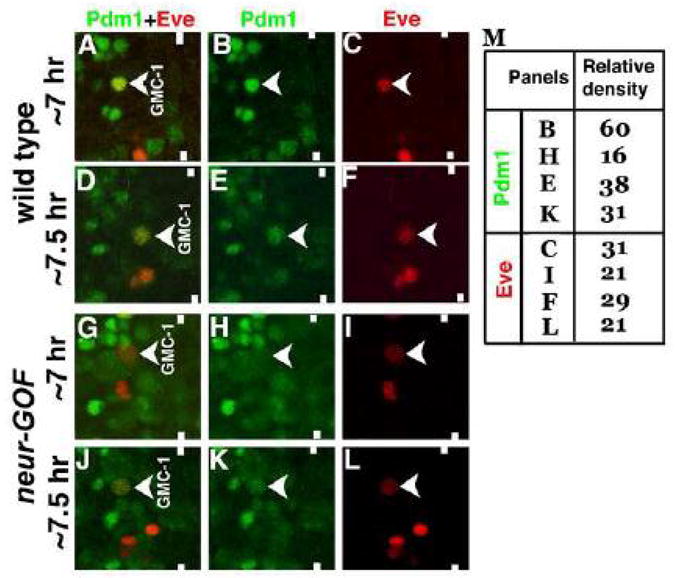
Embryos are double stained with Eve (Red) and Pdm1 (Green). Anterior is up, midline is marked by vertical lines. Pdm1 is expressed at high levels in a GMC-1 from a 7 hr old embryo (Panels A and B). The level drops as the development proceeds (Panels D and E). In neur-GOF embryos, with the induction of neur prior to the formation of GMC-1, the level of Pdm1 is drastically reduced as seen in a GMC-1 from a 7 hr old embryo (Panels G and H). The level of Pdm1 improves in a GMC-1 from a 7.5 hr old embryo (Panels J and K). In panel M, quantification of Pdm1 and Eve in GMC-1 was done by measuring the relative densities of Pdm1 and Eve staining signals in GMC-1.
Heat shock induction experiments
To determine the effect of over-expression of Neur during the formation of GMC-1, UAS-neur flies were crossed to Hs-GAL4 in cups and embryos (hereafter referred to as neur-gain of function or neur-GOF) were aged 5.5–6.0 hr at 22 °C, they were then subjected to heat shock for 30 min at 37 °C, and were fixed at different time points from immediately after the heat shock to allowing them to recover at 22 °C for one hour and two hours after the end of induction. These embryos were then fixed and stained for Eve. To determine the over-expression effect when the GMC-1 is dividing or has just divided, neur-GOF embryos were aged 7.25–7.5 hr at 22 °C, they were subjected to heat shock for 30 min at 37 °C, allowed to recover for about 30 min and were fixed and stained for Eve. However, we discovered that there was a developmental delay of about 1.5–2.0 hrs in most of these embryos, indicating that the actual age of the embryos when fixed were different than what it should be (we have observed this phenomenon with over-expression of pdm2, although the delay in this case was about 30 min). While we used this protocol for wild type, given the developmental delay with neur-GOF, we had to take a different approach to ascertain the GOF effects of neur.
To determine the effect of over-expression of Neur on the levels of Pdm1 in GMC-1, UAS-neur flies were crossed to Hs-GAL4 in cups and embryos, 4–6 hr (at 22 °C) old, were subjected to heat shock for 30 min at 37 °C. These embryos were allowed to age for another 4 hours and then fixed and stained for Eve and Pdm1. Because of the developmental delay in these embryos, the actual age of the embryos when fixed were between 6.5 hrs and 8.5 hrs old. We focused on embryos that were between 6.5–7.5 hrs of age for our analysis of GMC-1 (a GMC-1 becomes Pdm1-positive around 6.5 hours of development, but a GMC-1 is most likely formed between 5–5.5 hrs of development).
To determine the effect of over-expression of Neur on the asymmetric fate specification of GMC-1 of the RP2/sib lineage, several series of over-expression experiments were done. These were as follows: a) embryos were aged between 4–6 hrs, heat shocked for 30 min at 37 °C, then aged for 4 hours (with the developmental delay, this would correspond to 6.5–8.5 hrs), b) embryos were aged between 5–7 hrs (RT), heat shocked for 30 min at 37 °C, then aged for 3 hours (this would also correspond to 6.5–8.5 hrs), c) embryos were aged between 8–10 hrs (post GMC-1 division), heat shocked for 30 min at 37 °C, then aged for 5 hours (this corresponded to 12–14 hrs). These embryos were fixed and stained for Eve expression.
To determine the effect of over-expression of a truncated Neur missing the RING domain, similarly aged embryos (as for wild type described above) from the UAS-neurΔRF x Hs-GAL4 parents were heat shocked as above and stained for Eve and Pdm1, and Eve. These embryos did not show any developmental delays.
Results
Neuralized is required for the terminal asymmetric division of neural precursor cells
To determine the role of Neur in the asymmetric division of neural precursor cells of the CNS, we investigated the loss of function effects of neur gene on GMC-1 of the RP2/sib lineage. We stained neur loss of function mutant embryos with an antibody against Eve. The GMC-1->RP2/sib lineage is generated by NB4-2, formed as an S2 NB around 4.5 hrs of development. It generates its first GMC (GMC-1) by a self-renewing asymmetric division at around 6–6.5 hrs of development. This GMC-1 divides to generate an RP2 and a sib at around 7.45 hrs of development. The RP2 begins to project its axon ipsilaterally towards the intersegmental nerve bundle (ISN) by about 9.5 hrs of development and innervates muscle numbers 2, 9 and 11. There are several well-established ways to distinguish a GMC-1, an RP2, and a sib (Doe, 1992; Bhat and Schedl, 1994). Both nuclear division and cytokinesis of GMC-1 is asymmetric and thus, there is a size difference between a GMC-1 (7.5μM), an RP2 (~5μM), and a sib (~3μM). Similarly, the nuclear size of a GMC-1 is ~6.5μM, an RP2 is 4μM and a sib is 2.5μM. There is also a level difference in marker gene expression between an RP2 and a sib as well as a difference in the temporal dynamics of expression of these markers; the future RP2 cell has a stronger expression of markers like Even-skipped (Eve) compared to a future sib. The cell that assumes a sib identity undergoes a size reduction and further down-regulation of expression of RP2-specific marker genes. By ~14 hrs of development, expression of all those markers is completely lost from the sib. Finally, there is a subset of marker genes that only a mature RP2 expresses but not the sib or the GMC-1. Some of these markers include Mab 22C10 (MAP1B), which stains the membrane and the axonal projection of an RP2 (and a subset of other neurons such as aCC, in each hemisegment), allowing us to visualize axon morphology, and several transcription factors such as Cut, Zfh1, Pdm1 and Pdm2.
Loss of function mutations in neur causes severe hyperplastic nervous system due to formation of multiple NBs and therefore neurons of the same type. Thus, with Eve staining we observed a large number of RP2 neurons in a hemisegment in the mutant embryo (Fig. 1B) instead of one RP2 neuron per hemisegment in wild type (Fig. 1A). However, in neur1 mutants the RP2/sib lineage was not hyperplastic in 13% of the hemisegments ((n=240 hemisegments, from 12 mutant embryos), and 11% of the hemisegments had two RP2s with no sibs. In another neur allele, neurA101, the penetrance of this RP2 duplication phenotype was slightly lower (9%, n=240 hemisegments). Only a marginal increase in the penetrance was observed in embryos homozygous for a neur-deficiency (~12%, n=240 hemisegments. The duplication of RP2 in the absence of sib cells suggests that a symmetrical division of GMC-1 might have generated these RP2s. We further examined the hemisegments that had the neurogenic phenotype. Indeed, in most of such hemisegments, there were a lot more RP2s than sibs, suggesting that asymmetric division of a significant number of GMC-1s is affected in neur mutants. For example, in neur1 mutant embryos, we observed nearly 90% of the hemisegments (n=240) where the RP2s out numbered sibs indicating that the loss of asymmetric division occurred in nearly all hemisegments. The number was slightly less in neurtA101 (70% n=240) and about 90% in the deficiency that removes the neur gene. This suggests that the neur1 allele is close to null and we decided to use this allele extensively (neur refers to neur1 unless otherwise stated in the text). Given the severe neurogenic defect in all three mutat alleles, this is the best quantification that we were able to achieve. The duplicated RP2s were either of the same size or of different sizes (see Fig. 1B, midsection embryo), which is also the case in embryos mutant for Notch (Wai et al., 1999) or mastermind, which is downstream of Notch (Yedvobnick et al., 2004).
Figure 1. Loss of function for Neur causes symmetric division of neural precursor cells.
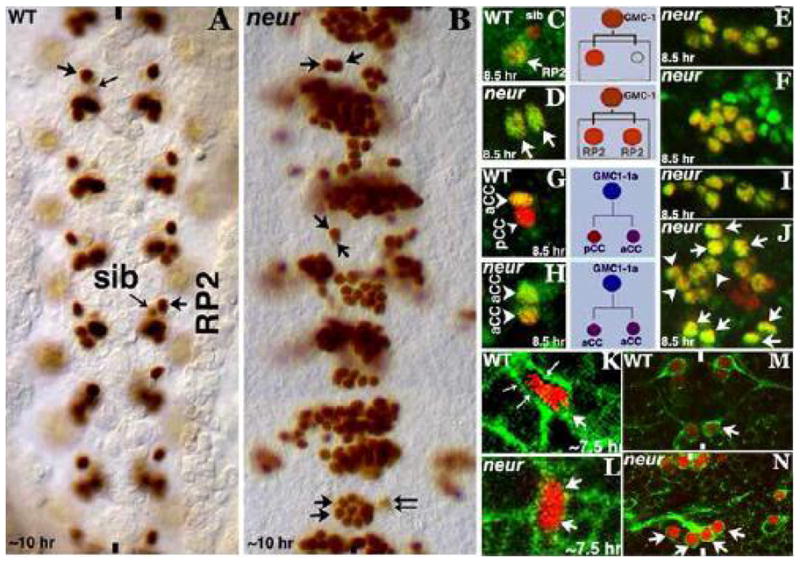
Embryos in panels A and B are stained for Eve, panels C-J are double stained for Eve (Red) and Zfhl (Green), panels in K and L are doubled stained for Eve (Red) and Spectrin (Green), panels M and N are stained for Eve (Red) and 22C10 (MAPIB; Green). Anterior end is up, midline is marked by vertical lines. An RP2 is indicated by an arrow, a sib by a small arrow, and an aCC by an arrowhead, and pCC by a small arrowhead. Two small-long arrows in panel K show the site of GMC-1 (of the RP2/sib lineage) cytokinesis. Panels A and B: Wild type and neur mutant, in the mutant (B), the GMC-1 in a non-neurogenic hemisegment has symmetrically divided into two RP2s (arrows). Note that in the middle segment, one of the duplicated RP2s is smaller than the other. While many hemisegments have multiple RP2s (arising from both symmetrical division and an earlier neurogenic defect, hemisegments with both RP2s and sibs can also be seen, bottom segment in panel B). Panel C: wild type, only a mature RP2 expresses Zfhl but not a sib. Panel D: Both the daughters of a GMC-1 in neur have Eve and Zfhl expression indicating their RP2 identity. Panels E and F: Even in those hemisegments where there is a neurogenic effect, only Eve and Zfhl positive RP2s are observed but not sib indicating symmetrical division of multiple GMCs. Panel G: Wild type, only aCC has both Eve and Zfhl, pCC has only Eve. Panel H: Both the daughters of GMC of the aCC/pCC have Eve and Zfhl although the transformed aCC has a lower expression of Zfhl compared to the bona fide aCC. Panels I and J: Many more aCCs are seen in the mutant embryo in those neurogenic hemisegments (arrowheads). Panel K: Wild type, a GMC-1 is unequally dividing into a larger RP2 and a smaller sib. Panel L: A GMC-1 in neur mutant is dividing equally into two RP2s. Panel M: Wild type, showing RP2s sending out their projection to the ISN bundle. Panel N: Duplicated RP2s with aberrant projections in the neur mutant.
Next, we examined neur mutant embryos with Eve and Zfh1, Zfh-1 is expressed at very low levels in a late GMC-1 just before its division, at high levels in an RP2 (Fig. 1C) and occasionally and transiently in a newly formed sib (Gaziova and Bhat, 2009). In neur1 mutant embryos, we observed Eve and Zfh1 positive RP2 neurons with no sibs in non-neurogenic hemisegments (Fig. 1D; 9% of the Hemisgemnets, n=144) or more of Eve and Zfh-1 positive RP2 neurons compared to only Eve positive sibs in as many as 80% of the neurogenic hemisegments (Fig. 1E and F; n=144). These results further confirm that a significant number of GMC-1s in neur mutants undergo symmetric division. This is also observed in another lineage, the GMC-1->aCC/pCC lineage. In this lineage, Eve is expressed in the GMC as well as in its two progeny, aCC and pCC. However, Zfh1 is expressed only in the aCC neuron (Fig. 1G). In both the neur mutants, we observed two Eve and Zfh1 positive aCC neurons but no pCCs in non-neurogenic hemisegments (Fig. 1H) and mostly aCCs in those neurogenic hemisegments (Fig. 1I and J). The asymmetric division of the GMC in aCC/pCC lineage was affected in most hemisegments.
The above symmetric division of GMCs was further verified by examining the GMC-1>RP2/sib lineage with Spectrin, which can be used to visualize dividing cells as it stains the cell cortex (Fig. 1K). We examined hemisegments that did not have any neurogenic phenotype in order to obtain an unequivocal answer. As shown in Figure 1L, examination of these hemisegments with Eve (to identify the GMC-1 and its progeny) and Spectrin antibody reveals a symmetrical division of GMC-1 to generate two RP2 neurons. Finally, the duplicated RP2 neurons in neur mutants were expressing MAPIB/22C10 on the membrane and axon projections, although the axon projections were misguided (Fig. 1N) as opposed to ipsilateral (Fig. 1M, wild type). These results show that loss of function for neur results in loss of asymmetric division of GMCs. That we did not observe a full penetrance of the symmetric division defects is unlikely due to a hypomorphic nature of the alleles used since the penetrance was similar in embryos homozygous for a deficiency that removes neur or in embryos that are transheterozygous for the two neur alleles or in trans with the neur deficiency. The incomplete penetrance could be due to a redundancy of the pathway with other E3 Ubiquitin ligases such as Mind-bomb-1 (Le Borgne et al., 2005; Lai et al., 2005).
Nuralized becomes asymmetrically localized to the basal pole prior to GMC-1 division
Given the symmetric division of GMC-1 into two RP2 neurons in neur mutants, we sought to determine if the localization of Neur is asymmetric in GMC-1. We examined the expression of Neur in the CNS in wild type embryos using an antibody against Neur. As shown in Figure 2, in GMC-1 of the RP2/sib lineage, initially Neur is not fully localized (panels A and B), but by ~7.5 hrs of development, it gets localized to the basal end (panels C, D and E). It then segregates to a future RP2 but not to a future sib, a pattern similar to the segregation of Numb. Neur is localized at the basal end in a subset of NBs as well (Fig. 2F), however, the parent NB of GMC-1, NB4-2, does not appear to express Neur, therefore that it is unlikely to be involved in the asymmetric division of NB4-2. Besides, if the duplication of RP2 in the mutant is due to a second GMC-1 generated by NB4-2 (or NB4-2 adopts a GMC-1 identity, i.e., NB4-2 symmetrically divides into two GMC-1s), we would have observed four cells, two RP2S and two sibs.
Figure 2. Neur is asymmetrically localized to the basal end in a GMC-1 prior to its division.
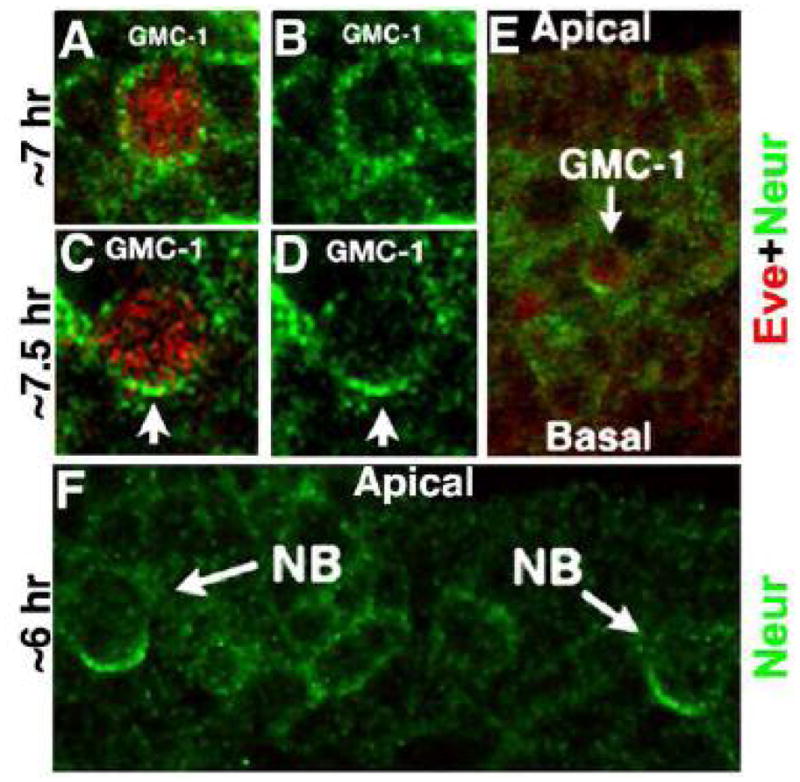
Embryos are double stained with Eve (Red) and Neur (Green) antibodies. Panels A–D are of the same magnification and a late GMC-1 (panels C, D) is larger than a mid GMC-1 (see Gaziova and Bhat, 2009). As shown in panels A and B, while Neur is less asymmetric and more uniform in a mid-stage GMC (a GMC-1 is normally born around 6–6.5 hrs of age), it is asymmetrically localized to the basal end of a late GMC-1 (panels C,D and E). Several NBs in a hemisegment also show a basally localized Neur (panel F), however, NB4-2 has no Neur expression. The GMC-1 development (timing) can be distinguished as an early, mid and late GMC-1 by looking at the development of the aCC/pCC lineage, appearance of cells of the EL lineage, and the migratory position of a GMC-1 within the nerve cord and the levels of expression of such proteins as Pdm1 and Pdm2. Moreover, a late GMC-1 is larger than a mid or an early GMC-1, and a mid GMC-1 is smaller than an early GMC-1 (see Gaziova and Bhat, 2009; see also Fig. 5).
Localization of Inscuteable is non-asymmetric in neur mutants
In a dividing GMC-1, Insc is asymmetrically localized to the apical pole (Buescher et al., 1998). We sought to determine if Insc localization is normal in neur embryos. We double stained embryos with Insc and Eve antibodies, Eve being the marker for GMC-1,. In wild type, Insc is on the apical side of a GMC-1 (Fig. 3A). In neur1 mutant embryos, we observed GMC-1s where the localization of Insc is non-asymmetric (Fig. 3B). Consistent with a non-asymmetric Insc localization in GMC-1, we also observed GMC-1s undergoing division with both newly forming cells inheriting Insc (Fig. 3C). These results indicate that localization of Insc to the apical pole is dependent on Neur/basally localized Neur. A similar pattern of mis-localization of Insc has been observed in GMC-1 of embryos with a brief over-expression of Pdm genes and such GMC-1s often undergo a symmetric division into two RP2s (Mehta and Bhat, 2001; Bhat and Apsel, 2004). We want to point out that GMC-1 is very sensitive to the varying levels of Pdm proteins, exhibiting different patterns of GMC-1 division with different levels of these proteins (see Yang et al., 1993; Bhat and Schedl, 1994; Bhat et al„ 1995; Mehta and Bhat, 2001; Bhat and Apsel, 2004; see also Discussion). The mis-localization of Insc in neur mutants is also different from numb mutants, where the localization of Insc is unaffected (data not shown).
Figure 3. Localization of Insc, Numb and Neur in GMC-1.
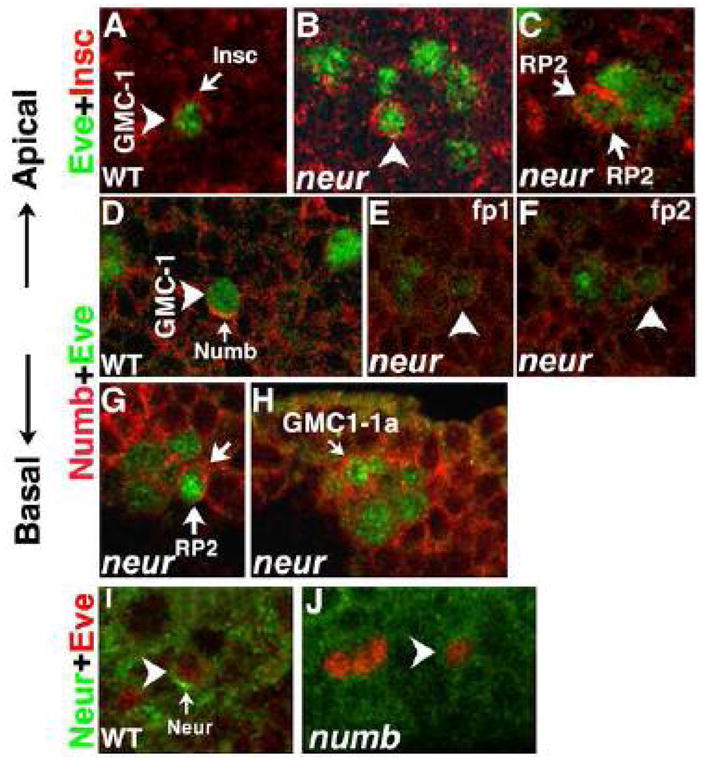
Panels A–C: Embryos are double stained with Eve (Green) and Insc (Red). Panel A: Asymmetric, apical localization of Insc in a hemisegment from a ~7.5-hr old wild type embryo is shown. Panel B: hemisegment from a ~7.5-hr old neur mutant embryo is shown with a GMC-1 that has non-asymmetric Insc. Panel C: hemisegment from a ~7.45-hr old neur mutant embryo is shown; the GMC-1 here is undergoing division and Insc is seen in both the newly forming daughter cells. Panels D–F: Embryos are double stained with Eve (Green) and Numb (Red). Panel D: Asymmetric, basal localization of Numb in a hemisegment from a ~7.5-hr old wild type embryo is shown. Panel E and F: Hemisegment from a ~7.5-hr old neur mutant embryo is shown (two different focal planes, fp1 and fp2 of the same hemisegment). Panels G: hemisegments from a ~8-hr old embryos are shown, with Numb in both the daughters from a GMC-1 division, GMC-1 in neur mutants (also in Notch and mam mutants) often divides to generate unequal sized cells but both assume an RP2 fate. In this panel, although one of the two cells appears smaller, both have Numb. The size difference could also be due to the focal plane of imaging. Panel H: The non-asymmetric localization of Numb in GMC-1-1a (of aCC/pCC lineage) is shown (in wild type, this GMC also has the basal localization of Numb, data not shown). Panels I and J: Embryos are double stained with Eve (Red) and Neur (Green). Panel I: Asymmetric, basal localization of Neur in a hemisegment from a ~7.5-hr old wild type embryo is shown. Panel J: Hemisegment from a ~7.5-hr old numb mutant embryo is shown, note the weak, non-asymmetric Neur in this cell. We have looked through hundreds of mutant embryos and we are confident that the localization of Insc and numb in neur and localization of Neur in numb mutants is non-asymmetric in GMC-1.
Localization of Numb in neur mutants and Neuralized in numb mutants is non-asymmetric
Because Neur co-localizes with Numb to the basal end of a GMC-1 (see Fig. 2C and Fig. 3I), we determined if Numb localization is disrupted in neur mutants. If the Numb localization is affected in neur mutants, loss of asymmetric division of GMC-1 in neur mutants could be due to mis-localization of Numb. We double stained neur embryos with Numb and Eve and examined the localization of Numb in GMC-1 (Fig. 3D–H). Unlike in wild type where Numb is localized to the basal end (Fig. 3D), in neur mutants, Numb is not localized, but is uniformly distributed along the rim of the entire cell (Fig. 3E and F). When such GMC-1s divide, both cells inherit Numb (Fig. 3G). This is also consistent with the above result that in neur mutants Insc is also non-asymmetric in GMC-1 in its localization. Since Numb will block Notch-signaling from specifying a sib fate, the daughter cells of those GMCs in which the distribution of Numb is non-asymmetric will inherit Numb and are expected to adopt an RP2 fate in neur mutants. This would account for the duplicated RP2 neurons in neur mutants. A similar non-asymmetric distribution of Numb can be also observed in GMC1-1a, the parent of aCC/pCC neurons (Fig. 3H), indicating that the distribution of Numb in neur mutants is affected in more than one lineage. Moreover, we found that nearly all the hemisegments (90%) that had a single GMC-1 had non-asymmetric Insc and Numb (N=80; for counting purposes we focused on only such hemisegments), which is consistent with the frequency of Eve-positive RP2 duplication phenotype observed in this allele.
We next examined if the localization of Neur is affected in numb mutants. Instead of a basal localization as Numb (Fig. 3I), the localization of Neur in numb mutants is affected the same way as localization of Numb is affected in neur mutants, with Neur distributed along the rim of the cell. These results show an interdependence of Neur and Numb proteins for their localization to the basal pole (see Discussion).
Neuralized is involved in the down-regulation of Pdm POU proteins
An up-regulation of POU proteins, Pdm1 and Pdm2, causes non-asymmetric localization of Insc; a non-asymmetric Insc causes non-asymmetric distribution of Numb. This results in a symmetric division of GMC-1 of the RP2/sib lineage into two RP2 neurons (Mehta and Bhat, 2001; Bhat and Apsel, 2004). Therefore, we determined whether or not loss of function for Neur causes an up-regulation of these POU proteins. As shown in Figure 4, we double stained wild type and neur mutant embryos with Pdm1 and Eve. In wild type, GMC-1 of the RP2/sib lineage expresses Pdm1 before the expression of Eve, and the expression of Pdm1 is quite high in a newly formed GMC-1. There is a down regulation of expression of Pdm1 in this cell as the development proceeds (Fig. 4A–L). A similar down-regulation of Eve was also observed in GMC-1 during development (Fig. 4A–L). However, in neur mutant embryos, the level of Pdm1 was not down-regulated, instead it was specifically high in a mid/late GMC-1 (Fig. 4P–R; compare these panels with Fig. 4D–I). The up-regulation of Pdm1 in the GMC-1 of neur mutant embryos was quantified by measuring the relative densities of Pdm 1 and Eve staining signals in the GMC-1 in wild type and neur embryos (Panel S). This quantification shows that the levels of Eve is significantly affected in neur embryos. For counting purposes, we focused on those hemisegments that had single GMC-1, and we found that nearly all those hemisegments with single GMC-1 had high levels of Pdm1 (N=44; note that many GMC-1s in those hemisegments that had multiple GMC-1s due to neurogenic phenotype also had high levels of Pdm1). Since we have previously shown that over-expression of Pdm mislocalizes Insc and Numb (Mehta and Bhat, 2001), the up-regulation of Pdm1 in GMC-1 is likely the reason for the mis-localization of Insc and Numb in neur mutants. It is also possible that a mis-localized Insc causes mis-localization of Numb since apical localization of Insc is necessary for the basal localization of Numb (Buescher et al., 1998).
We addressed this issue of regulation of Pdm1 level by Neur from another angle. Using a UAS-neur-transgenic line, we over-expressed neur in wild type embryos with GAL4 from an inducible heat shock 70 promoter linked GAL4 (Hs-GAL4), prior to formation of GMC-1 of the RP2/sib lineage from NB4-2. These neur-GOF embryos were then double stained with antibody against Pdm1 and Eve. Pdm1 is expressed at high levels in a 7 hr old GMC-1 (Fig. 5A, B), and the level of Pdm1 begins to go down in a GMC-1 by 7.5 hrs of age (Fig. 5D and E). In neur-GOF embryos, a 7 hr old GMC-1 had almost no or very little of Pdm1 (Fig. 5G, H). The level of Pdm1 slightly improves in a 7.5 hr old neur-GOF embryo (Fig. 5J and H). Furthermore, the level of Eve is also low in such GMC-1s (Fig. 5I and L). The down-regulation of Pdm1 in the GMC-1 of neur-GOF embryos was quantified by measuring the relative densities of Pdm1 and Eve staining signals in the GMC-1 of 7 hrs and 7.5 hrs old wild type and neur-GOF embryos (Panel M). This quantification shows that the levels of Eve are also affected in neur-GOF embryos. Finally, the effect of neur-GOF on Pdm1 expression in GMC-1 is quite strong in terms of the penetrance with ~60% of the GMC-1s show the defect (N=65 hemisegments). It has been previously shown that loss of function for Pdm1 and or Pdm2 causes loss of GMC-1 identity and loss of Eve expression (Bhat and Schedl, 1994; Bhat et al., 1995; Yeo et al., 1995). This loss/reduction of Eve in GMC-1 in neur-GOF embryos is likely due to the negative effect of over-expression of Neur on Pdm1. The down-regulation of Pdm1 protein in GMC-1 in gain of function neur is consistent with the finding that loss of function for neur causes an up-regulation of Pdm1 protein.
Neuralized also mediates asymmetric cell fate specification post GMC division
The first clue that Neur might also function during the processing of Notch comes from the finding that the symmetrical division of GMC-1 into two RP2s has similarity to the symmetrical division in Notch mutants. That is, in wild type, the asymmetric division of GMC-1 is not only asymmetric in fates but also in sizes—a sib is smaller than an RP2 and this occurs during the cytokinesis itself. In Notch mutants, one of the two RP2s from the symmetrical division of a GMC-1 is usually smaller than the other (Fig. 6A; see also Wai et al., 1999; Buescher et al., 1998). In neur mutants we generally find hemisegments with two RP2s from a GMC-1 that are of equal sizes; however, we also find two RP2s of unequal sizes (see Fig. 1B). The precise percentage of penetrance of these two phenotypes was difficult to determine due to the neurogenic defect. However, by focusing only on those hemisegments where the RP2s are just two (with no accompanying sibs), we found a frequency of about 70% (equal sizes) and 30% unequal sizes (n=110 hemisegments).
Figure 6. Neur also mediates asymmetric cell fate specification by activating Notch.
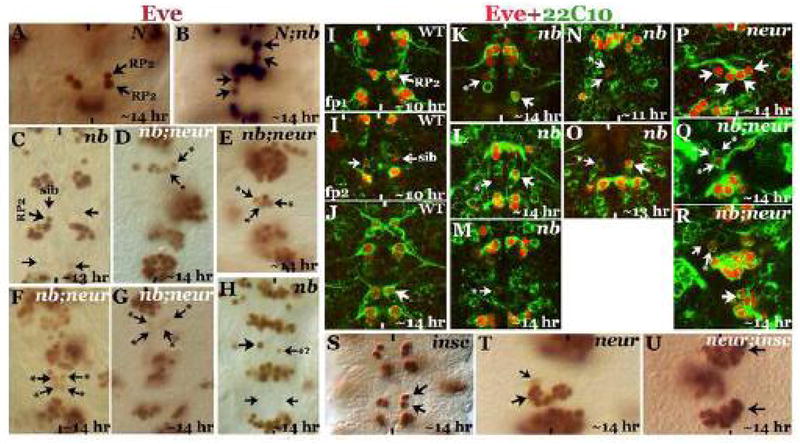
Embryos in panels A–H and S–U are stained with Eve; embryos in panels I-R are doubled stained with Eve and 22C10. Anterior is up, midline is marked by vertical lines. Larger arrow indicates anRP2, smaller arrow indicates a sib; smaller arrow with a star indicates mixed RP2-sib identity. Panel A: NotchTS mutant embryo showing two RP2s from the symmetrical division of GMC-1. Panel B: Notch; numb double mutant embryo with Notch phenotype epistatic to the numb phenotype. Panel C: numb embryo with both progeny of GMC-1 adopting a sib identity with a barely detectable Eve expression. Panels D–G: neur;numb double mutant embryos; many of the daughter cells of GMC-1 have mixed RP2-sib identity with reduced expression of Eve. Panel H: An older numb mutant embryo with a rarely seen phenotype of a smaller cell with weak Eve expression, as if this cell has a mixed RP2-sib identity. Panels I–I’: Two different focal planes (fp1 and fp2) of the same 10-hr old wild type embryo showing an RP2 with its projection (Panel I) and a sib with its weak 22C10 expression although no axon projection can be observed from it. Panel J: ~14 hour old wild type embryo with an RP2 with its projection; the sib cell has no Eve by this time and no 22C10 expressing cell is observed in the location where a sib normally resides. Panels K–O: numb mutant embryos. In Panels K and L, two examples of a larger cell with weak Eve and 22C10 expression are shown, in panel M, a smaller cell with weak Eve and 22C10 is shown. In panel N (younger 11-hr old embryo), a pair of cells, one larger than the other but both with weak Eve and 22C10 expression, is shown; the smaller cell is a normal sib. In panel O, an example of a larger cell with strong 22C10 expression is shown. These examples indicate a mixed identity for the cells of the RP2/sib lineage in numb mutants (see text). Panel P: A neur mutant embryo with strong Eve and 22C10 expression in the duplicated pairs of RP2 neurons. Panels Q and R: numb; neur double mutant embryos showing larger cells and smaller cells with weak Eve and 22C10 expression. Panels S–U: In panel S, duplication of the RP2 in insc mutants is shown; in panel T, hemisegments of a neur mutant embryo with both RP2s and sibs are shown. In panel U, neur; insc double mutant is shown with many more RP2s and no sibs, indicating that insc is epistatic to neur and enhances the neur symmetric division defect.
To explore if Neur is also involved at the later stage during Notch-processing, we sought to determine the relationship between neur and numb. Numb blocks the intracellular processing of Notch to prevent Notch from specifying a sib fate. Thus, while in Notch mutants, a GMC-1 divides to give rise to two RP2s (Fig. 6A), in numb mutants GMC-1 divides to give rise to two sib cells (Fig. 6C and H). The specification of a sib fate in numb mutants is Notch-dependent, which is indicated by the fact that in N;numb double mutants both the daughter cells adopt an RP2 fate and that the Notch-phenotype is epistatic to the numb-phenotype (Fig. 6B). This means that in the absence of Numb activity, Notch processing occurs in both daughter cells of a GMC-1 and the intracellular domain of Notch specifies a sib fate to both the daughter cells. In order to specify an RP2 fate, Numb is needed only if there is an intact Notch.
It has been suggested that Neur is non-autonomously involved in the endocytosis of the extracellular domain of Notch-Delta complex, thus Neur is part of the Notch-signaling. It is not clear if this Neur-mediated process is required for the processing of Notch or whether it simply removes the Delta-bound extracellular domain of Notch after the processing of Notch. We decided to test if Neur indeed functions as a part of the Notch-signaling during the specification of sib identity by examining embryos that are double mutant for numb and neur. In the absence of Numb, Notch processing should normally occur in both daughters of GMC-1; if Neur is involved in Notch processing/Notch-signaling during the specification of sib fate, in the double mutant both daughters should adopt an RP2 fate. Examination of the double mutant embryos indicated an ambiguous result. We observed hemisegments where the two daughter cells appear to be RP2s but with reduced levels of Eve (Fig. 6D-G, arrow with a star). Approximately 70% of the cells in affected hemisegments had this reduced Eve-phenotype, 30% had normal Eve (n=total number of cells counted in affected hemisegments, 72). In some hemisegments, the levels of Eve in such cells were very low, indicating that these may be even sib cells (Fig. 6G; 18% of the affected cells, n=72). However, we did not observe hemisegments where the progeny cells adopted a complete sib fate by losing all of Eve expression. This raises the possibility that the progeny cells adopt a mixed identity in the double mutant.
The above result prompted us to take a closer look at the sib transformation in numb embryos. Numb is maternally deposited to developing embryos (Lear et al, 1999). For example, in embryos homozygous for a deletion that removes the numb gene has one or two hemisegments with loss of asymmetric division defect (our unpublished results). However, in embryos homozygous for one of the alleles of numb, numb796, the loss of asymmetric division in the RP2/sib lineage can be see between 1–14 hemisegments, with an average of 4–5 hemisegments. This allele is a loss of function allele since maternal and zygotic null for numb has a fully penetrant defect (Lear et al., 1999). The numb gene in this allele has not been sequenced, and we think that the Numb protein in this allele somehow interferes with the maternal Numb to create a strong loss of function effect.
When we carefully examined numb796 embryos with Eve staining, we observed hemisegments with a single cell with weak Eve expression and of the same size as a sib cell, even in embryos that were as old as 14–15 hours (Fig. 6H, arrow with a star; ~2% among the hemisegments affected showed this phenotype). It is possible that such cells with weak Eve expression in numb mutants and the cells with weak Eve expression in numb;neur double mutants have a mixed identity, perhaps due to a partial processing of Notch in this allele of numb.
We therefore determined if such cells have an axonal projection, since a normal sib reportedly has no axonal projection. First, we examined if a sib in a 10-hr wild type embryo has projections by staining with Mab 22C10, against MAPIB. As shown in Figure 6I, an RP2 has already sent out its axon ipsilaterally towards the ISN. However, a sib in the same hemisegment has no visible axon projection but it has a weak 22C10 expression (Fig. 6I′). This expression disappears by 14 hours of age (Fig. 6J) indicating that the 22C10 expression is transient in a sib cell. We next examined the expression in embryos mutant for numb. Several results are of interest. We observed hemisegments with a large cell of the size of an RP2, but with weak Eve and weak 22C10 expression (Fig. 6K, L, arrow-with-a-star). Hemisegments with smaller or small cell of the size of a sib with weak Eve and 22C10 expression (Fig. 6M) were also observed. Interestingly, in 11-hr old embryos we observed hemisegments with a smaller than normal RP2/sib pairs with weak Eve and 22C10 expression (Fig. 6N) as if both cells are adopting a sib fate. Rarely, we observed hemisegments that have cells of the size of an RP2 with weak Eve but strong 22C10 expression (Fig. 6O). This cell on the other hand, had no visible 22C10 positive projection. These results suggest that in numb mutants there are hemisegments where a cell in the RP2/sib lineage has a mixed or a partially transformed identity.
We next examined the RP2/sib fates in numb;neur double mutant embryos. As shown in Figure 6, panels Q and R, we observed a numb-like phenotype with hemisegments showing partial transformation phenotypes: cells of RP2 size but week Eve expression (panels Q and R) and with or without an axonal projection. A complete transformation into sib fate as in numb mutants was not evident in these numb;neur double mutant embryos. These results suggest the following. If there is none or some Numb activity, there is processing of Notch, and a variety of transformation phenotypes, from complete to partial transformation into sib, are observed. In embryos that lack both Neur activity and Numb activity, a complete sib transformation was not observed, indicating that Neur is needed for Notch processing and a full sib-transformation. However, some processing of Notch does occur without having Neur activity, to the extent that it is sufficient to induce a partial transformation. These results suggest that Neur function is necessary in order to efficiently process or potentiate, thereby enhancing the Notch activity.
Ectopic expression of Neuralized can induce sib fate to both daughters of GMC-1
Given the above results, we next determined if over-expression of neur could induce both the daughters of GMC-1 to adopt a sib fate. We expressed UAS-neur at high levels from a heat shock-GAL4 driver at different time points during the development of a GMC-1. Over-expression of Neur prior to the formation of GMC-1 causes a down-regulation of Pdm1 (Fig. 5) and non or mis-specification of GMC-1 identity as indicated by the loss of Eve expression (Fig. 7, panels D, E). However, over-expression of Neur prior to the division of GMC-1 causes a loss of Eve expression from both daughters of GMC-1, consistent with their specification as sib cells (Fig. 7F). This phenotype is similar to the phenotype induced with the over-expression of the intracellular domain of Notch or the phenotype observed in numb mutants (Wai et al., 1999). However, the penetrance of this transformation phenotype was low with about 15% of the hemisegments (n=200) showing the two-sibs phenotype. This specification of a sib fate to both daughter cells can be either due to mis-localization of Numb and both daughter cells inheriting Numb protein, or that Numb localization is normal, however, because of high levels of Neur, even the cell that has Numb is unable to block Notch processing, resulting in adopting a sib fate. Since the over-expression of Neur was done just prior to the division of GMC-1, it seems more likely that Numb localization is not disrupted but gain of function for Neur overcomes the Numb-mediated blocking of Notch signaling and induce a sib phenotype to daughters of a GMC-1. Moreover, if the localization of Numb is disrupted, it is more likely that the penetrance of the phenotype is much higher. Thus, this result is consistent with the conclusion that Neur is involved in potentiating Notch, although Neur is not indispensable to Notch potentiation. Finally, while the source of Neur for the trans-effect on Notch-signaling in wild type is most likely from the “RP2” cell, it appears that the surrounding cells can also be the source for Neur in the absence of an RP2 cell. This is indicated by the result that while the GMC-1 in embryos mutant for cyclin A adopts an RP2 fate (Fig. 8B), the same GMC-1 in a cyclin A; numb double mutant adopts a sib fate (Fig. 8D; see also Wai et al., 1999). If the Neur function for the specification of a sib cell came exclusively from an RP2 cell, cyclin A; numb double mutants would have an RP2 cell, not a sib cell.
Figure 7. Ectopic expression of neur induces both daughter cells of a GMC-1 to adopt a sib fate.
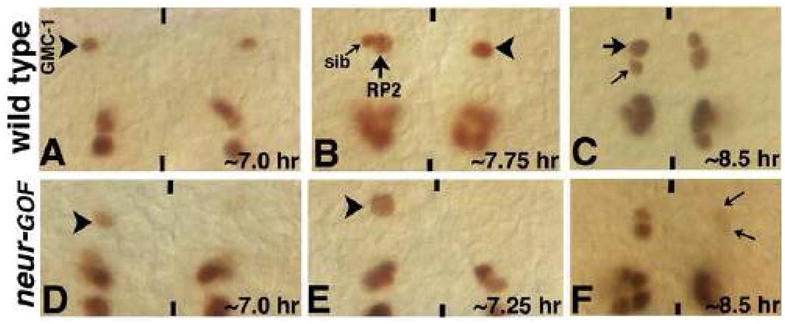
Wild type (Panels A–C) and neur-GOF embryos (panels D–F) were subjected to heat shock at different time points during the development of the GMC-1 lineage (see text for details). In panels D and E, neur was induced prior to the formation of GMC-1, whereas in panel F, neur was induced prior to GMC-1 division, note the weak Eve expression in the RP2/sib pairs indicating their transformation to sib fate. Black arrowhead indicates a normal GMC-1, white arrowhead indicates either a GMC-1 with very weak Eve expression (panel D) or the location of a missing/eve-negative GMC-1 (panel E); large arrow indicates an RP2, small arrow indicates a sib.
Figure 8. The source of Neur for the endocytosis of Delta and the Delta-bound extracellular domain of Notch is outside of the lineage.
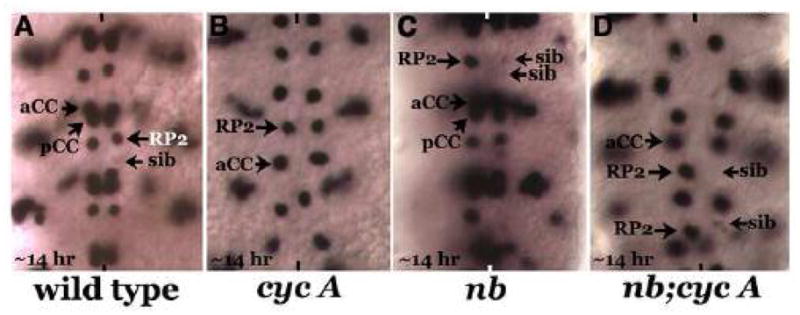
Embryos are stained with Eve, anterior end is up, and midline is marked by vertical lines. Panel A: Wild type, rarely a sib is visible in a 14 hr old embryo with weak Eve expression. Panel B: cyclin a mutant embryo, the GMC-1 fails to divide, and it adopts an RP2 identity. Panel C: a numb mutant embryo, both daughter cells of a GMC-1 adopts a sib fate. The penetrance of this phenotype is partial (see text). Panel D: numb;cyc a double mutant embryo, the numb phenotype in terms of the fate specification, is epistatic to the cyc a phenotype (cell division defect of cyc a is unaffected in the double mutant and this is expected since Numb is not required for cell division).
Discussion
Our results in this paper, summarized in Figure 9, tie the localization of Numb and the signaling-processing of Notch through a single upstream player, Neur. This gives us a more complete picture of the events that surround asymmetric division of neural precursor cells. We have shown that the E3 Ubiquitin ligase protein Neur regulates asymmetric division of Numb and Notch-sensitive neural precursor cells in the CNS via two distinct, sequential mechanisms: first, by promoting the asymmetric localization of Insc and Numb in GMCs and second, via non-cell autonomously potentiating or enhancing the activation of Notch signaling in the Numb-negative daughter cell. While Neur is known to activate Notch-signaling by the endocytosis of Delta and the Delta-bound extracellular domain of Notch, an earlier role for it in asymmetric division via Insc and Numb localization has not been discovered. In fact, our results show that this is the primary role for Neur in generating asymmetry in the CNS. That Neur plays a secondary role or a role which is not absolute in the potentiation or enhancement of Notch signaling is indicated by our finding that in neur; numb double mutants, both sibling cells often but not always adopt a mixed fate as opposed to an RP2 fate seen in Notch;numb double mutants. If the role of Neur in Notch potentiation in this lineage is an absolute one, we would have seen the same result in neur; numb as a N; numb mutants. Furthermore, over-expression of Neur can induce both cells to adopt a sib fate similar to gain of function Notch, however, the penetrance of this effect weak.
Figure 9. Summary of the role of Neur is the GMC-1->RP2/sib lineage development.
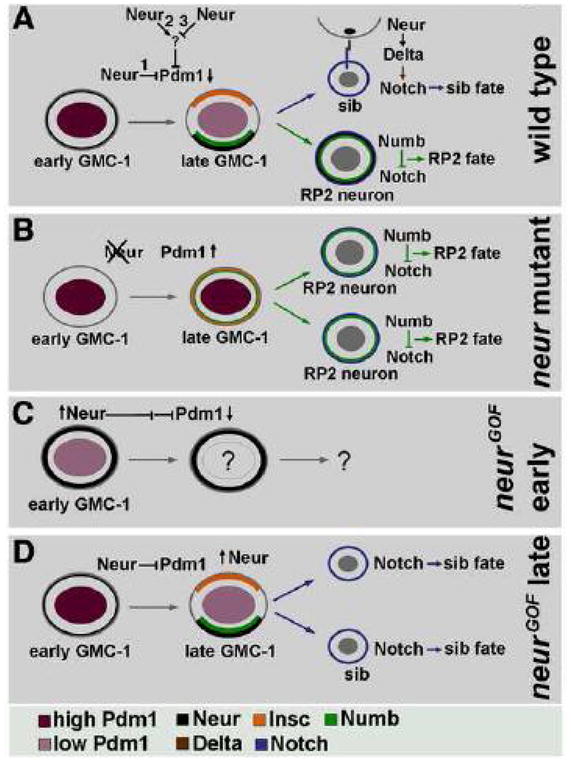
Panel A: In wild type, the level of Pdm1 is very high in a newly formed GMC-1, which drops in a late GMC-1. This drop corresponds to the expression of Neur in a GMC-1, which is mostly non-asymmetric at this point in a GMC-1. We do not know if the regulation of Pdm1 by Neur is via a direct mechanism (possibility 1), or via other indirect ways (possibilities 2 and 3). In a late GMC-1, prior to its division, Neur becomes localized to the basal Numb-domain, presumably segregating to a future RP2 similar to Numb. Neur also plays a role in endocytosing Delta bound extracellular domain of Notch, thus releasing the intracellular Notch to induce a sib fate. The source of this Neur appears to be from surrounding cells and not necessarily from the RP2 (see Fig. 8). Panel B: In neur mutants, absence of Neur activity leads to a high level of Pdm1 in a late GMC-1, resulting in non-asymmetric localization of Insc and Numb; thus both the progeny cells of GMC-1 adopt an RP2 fate due to the presence of Numb in them. Panel C: When Neur is ectopically expressed at high levels prior to the formation of a GMC-1, no to very little of Pdm1 can be seen in this cell. Lack of Pdm1 causes loss of GMC-1 identity and no RP2/sib cells are formed. Panel D: when neur is induced at a later time point prior to the division of GMC-1, both the daughter cells adopt a sib fate, presumably by forcing the release of the intracellular Notch from the progeny cells; this must overcome the inhibitory activity of Numb in this Numb-positive cell, which is otherwise destined to become an RP2.
Previous studies had shown that the RP2-sib binary fate decision is regulated by unequal segregation of the Notch regulator Numb. Here, the simplest interpretation of the results would suggest that Neur is required for sib fate specification via Notch. However, our results indicate that the requirement of Neur for sib-specification to a daughter cell of a GMC-1 via regulating Notch is preceded by its requirement in GMC-1 for Numb localization, where Neur itself is expressed and becomes asymmetrically localized to the basal Numb-domain. Thus, the loss of sib identity in neur mutants appears to be mainly due to the non-asymmetric localization of Insc and Numb in GMC-1. Moreover, the levels of Pdm1 are responsive to both loss of function neur (Pdm1 level is up-regulated) and gain of function neur (the Pdm1 level is down-regulated), which are more likely a consequence of Neur function within GMC-1. This regulation of Insc and Numb localization appears to be via regulation of Pdm1 levels inside GMC-1, whereas regulating Notch processing is later and the source of Neur is from outside. By regulating asymmetric localization of Numb, Neur ensures that one of the two daughters is free of Numb, thus, later on the activation of Notch-signaling in that cell can occur (Fig. 9). The source of Neur for this Notch processing appears to be from outside of the lineage since a division-arrested GMC-1 in numb;cyc A double mutant can still adopt a sib fate (Fig. 8). Thus, the two roles of Neur in this lineage are distinct and separable. But then is it possible Notch has a role in the asymmetric localization of Numb and this activity of notch is regulated by Neur? it certainly is possible but then one would have to disregard the presence of asymmetrically localized Neur in a GMC-1 as anything but of no consequence to the asymmetric division of GMC-1. We must also point out that the identity of GMC-1 per se in neur is not altered, if it did, we would have seen two neurons of some other identities, not RP2s (or sibs).
A previous study in the sensory system of the PNS indicated that Neur protein localizes asymmetrically in the pI cell of SOP. It then segregates to pIIb, where it is thought to enhance the endocytosis of Dl to promote N activation in the pIIa cell (Pavlopoulos et al., 2001; Le Borgne and Schweisguth, 2003; Yeh et al., 2000). This represents a trans-differentiation mechanism to specify different cell fates. Our results confirm the findings in SOP lineage but at the same time extends the data on SOP lineage in that this trans-determination process is a potentiation step to mediate a more efficient Notch-signaling-processing, but it is not necessarily a deterministic one. What is new and different from the SOP lineage is that Neur controls not only the asymmetric localization of Numb during mitosis, but also controls the localization of Insc, an apical cue that controls spindle orientation and participates in numb basal localization. In neur mutant cells, Insc is no longer asymmetric indicating that Neur is somehow needed to localize Insc, the fact that Neur is somehow needed for Insc localization is also consistent with the finding that genetically insc is epistatic to neur, therefore that it is downstream of neur.
Finally, while insc is epistatic to neur in the RP2 lineage defect in insc; neur double mutants, as for the neurogenic phenotype, neur is epistatic. This is not surprising since epistasis relationships are lineage/cell-type/tissue specific, depending upon whether or not the two genes in question are expressed in the same lineage and if the two single mutants give the same (or opposing) phenotype. Insc has no role during the neural versus ectodermal fate decisions and loss of function for insc does not cause a neurogenic phenotype, hence, we do expect the neurogenic phenotype of neur mutants to be present (epistatic) in the double mutant.
Neuralized regulates asymmetric division of GMCs
It is clear from our results that Neur regulates asymmetric division of GMCs in the CNS. We have examined this in at least two different GMCs, GMC of the RP2/sib lineage (GMC-1 or GMC4-2a of NB4-2) and GMC of the aCC/pCC lineage (GMC-1 or GMC1-1a of NB1-1). In neur, these GMCs symmetrically divide to generate two of the same cells, RP2 neurons in the case of GMC-1 and aCC neurons in the case of GMC1-1a. We think that many more GMC lineages are affected by loss of function for neur. Being a neurogenic protein, Neur is also involved in selecting neural versus ectodermal fates for the neuroectodermal cells. Due to its neurogenic property, the mutant will generate extra copies of many of the NBs in the nerve cord, which in turn, will generate more of the GMCs and neurons. Several lines of evidence indicate that symmetric division of a GMC indeed occurs at high frequency in the CNS in neur mutants. For example, GMC-1 normally generates an RP2 and a sib, RP2 is larger than the sib and the two have distinct gene expression profiles and patterns. This is also the case for aCC/pCC pairs—they also have distinct gene expression profiles. We used these specific criteria to separate the ones that are generated by the symmetric division from those generated due to a neurogenic effect of neur mutation.
Several additional evidences indicate a role for Neur in generating asymmetry. These include the asymmetric localization of Neur in GMCs, non-asymmetric localization of Numb in GMC-1 in neur mutants, non-asymmetric localization of Neur in numb mutants, genetic interaction results and effect on downstream players such as Pdm and Numb. All these results point to a specific role for Neur in regulating asymmetric mitosis of precursor cells.
How does Neuralized regulate Insc and Numb localization?
Our results show that Neur itself is asymmetrically localized in GMC-1 to the Numb-domain and opposite to that of the Insc-domain (Neur is also localized to the basal end of several NBs, the significance of which is not known). In neur mutants, both Insc and Numb are not localized but found uniformly distributed along the cell cortex. This suggests that Neur is upstream of Insc and Numb localization but not their expression per se. The levels of Numb or Insc are also not affected in neur mutants indicating that Neur does not participate in Numb degradation (via ubiquitination, or otherwise). We do not have as yet any evidence that Neur has a direct role in the localization of Numb. Do these results therefore mean Neur basically regulates the identity or the fate (i.e. gene expression program) of the GMC-1 prior to its division and therefore that Neur has only one function, which is potentiating Notch signaling? We have examined the GMC-1 in neur mutants with several different GMC-1 markers (Eve, Pdm1, Zfh-1, Spectrin, etc) and with the exception of a higher than normal Pdm1 in a late GMC-1, none of these markers were affected. A higher than normal levels of Pdm1 does not change the identity of a GMC-1. Indeed, several studies have shown that high levels of Pdm1 or its sister protein Pdm2 will induce a GMC-1 to undergo symmetric division to produce two GMC-1s and then two RP2s and two sibs (Yang et al., 1993; Bhat and Schedl, 1994; Bhat et al., 2005). In order for a GMC-1 to change its identity, many of its genes should be turned off and a new set of genes has to be initiated. Such a drastic change does not occur in GMC-1 of neur mutants. Similarly, an identity change should result in this GMC-1 in neur mutants to produce different sets of neurons, which it does not. Instead, it produces two RP2s. Given these results and that Neur is necessary for the normal localization of Numb, whether this is directly mediated or indirectly mediated, our conclusion that Neur regulates asymmetric division at two different levels during the lineage development is based on firm grounds.
The main question is how might Neur regulate Insc and Numb localization. A clue to this question comes from some of our previous studies (Bhat and Apsel, 2004). We showed that over-expression of Pdm POU transcription factors (Pdm1 or Pdm2) in GMC-1 causes non-localization of Insc and Numb and their segregation to both daughter cells of GMC-1; these cells then adopt an RP2 fate, with Numb blocking the N-signaling from specifying a sib fate. Pdm1 was up-regulated in GMC-1 in neur mutants and down-regulated with over-expression of Neur. This shows that the localization of Insc and Numb is altered in neur mutants indirectly via the upregulation of Pdm protein. At the moment, it is not clear how an up-regulation of Pdm alters Insc or Numb localization. A most likely possibility is that Pdm proteins, being transcription factors, their over-expression may cause changes in the expression of genes that are needed for the proper localization of Insc and Numb but without altering the cell-identity itself (since this cell still produces RP2 neurons and not some other neurons). These conclusions are all consistent with the overall expression pattern and mutant effects of pdm genes: Pdm proteins are down-regulated in GMC-1 prior to its division (Bhat and Schedl, 1994; Bhat et al., 1995), loss of function for Pdm causes loss of GMC-1 identity (Bhat and Schedl, 1994; Bhat et al., 1995; Yeo et al., 1995).
The gain of function for these pdm genes indicates that the GMC-1 division is quite sensitive to varying levels and timings of expression of these POU proteins. For example, a high level of pdm gene expression in a GMC-1 from pdm transgenes causes a symmetric division of GMC-1 into two GMC-1s and then each of these GMC-1s generates an RP2 and a sib (Yang et al., 1993; Bhat et al., 1995). On the other hand, we can also observe a symmetric division of GMC-1 into two RP2s in these embryos (Mehta and Bhat, 2001; Bhat and Apsel, 2004). In this case, the cells from the GMC-1 express Zfh1; a GMC-1 does not continually express (a late GMC-1 about to divide does express Zfh1 at a very low level, see Gaziova and Bhat, 2009), a sib transiently expresses Zhf-1 (Gaziova and Bhat, 2009), and an RP2 stably expresses Zfh-1 (Gaziova and Bhat, 2009). Moreover, both these cells inherit Insc and Numb (Mehta and Bhat, 2001). No more cells are produced from these two cells, and each of these cells generates a projection that of an RP2. When these genes are over-expressed for a prolonged period of time, a GMC-1 divides multiple times producing a GMC-1 and a differentiated progeny: we observe first two unequal sized cells, only one of the two (the smaller cell) expresses markers such as Zfh1. Later on, we sequentially see three cells, and then five cells, etc, all in a tight cluster; from these clusters, as many as 5 RP2s are formed (Bhat and Apsel, 2004), Indeed, with this prolonged over-expression of pdm genes for 90 min from a heat shock promoter causes hemisegments with all the above types of divisions depending upon the time of over-expression (see Bhat and Apsel, 2004). On the other hand, it is not clear the sensitivity range of GMC-1 to varying concentrations in terms of the kind of division pattern generated. One clue to this comes from an earlier study (Bhat and Schedl, 1994), that GMC-1 in embryos carrying a duplication chromosome for the chromosomal region containing the two POU genes undergo a single self-renewing asymmetric division of GMC-1. This suggests that when the copy numbers for these genes are doubled, presumably results in producing twice the amount of these proteins (from their own promoters), causes a single self-renewing division. Having said that, we have also found that in neur mutants a GMC-1 rarely divides symmetrically into two GMC-1s and then each produces a sib and an RP2, or a GMC-1 dividing more than once with self-renewing asymmetric division as in pdm-GOF situations (data not shown).
Based on these results with gain of function for pdm genes, a loss of function for pdm genes should suppress the neur defects. However, this experiment is not possible to do since loss of function for the pdm genes causes loss of GMC-1 identity (GMC-1 becomes some other GMC) and therefore GMC-1 is undetectable with GMC-1 markers.
While we do not know the exact mechanism as to how the level of Pdm1 is up-regulated in GMC-1 of neur mutants or down-regulated when Neur is over-expressed in GMC-1, one possibility is that Neur in involved in the degradation of Pdm1 in GMC-1. This scenario is most likely since Neur has the RING domain, one of the signature domains for E3 Ubiquitin-ligase proteins involved in protein degradation. Neur has also been shown to ubiquitinate proteins in vitro (Lai et al., 2001). One indication that Neur might be involved in the degradation of Pdm1 is our result that while ectopic or over-expression of full length neur from a transgene down-regulated Pdm1 and resulted in the same phenotype as loss of function for pdm genes, a similar ectopic or over-expression of a neur transgene missing the RING domain (Hs-neurΔRF) did not result in a down-regulation of Pdm1 or resulted in any phenotypes. Pdm1 appears to be specifically affected in GMC-1 of the RP2/sib lineage and not in other cells where Pdm proteins are present. Even if the up-regulation of Pdm proteins in neur mutants is via an indirect mechanism, say via factor X or Y, our results define a major role for Neur in regulating asymmetric division prior to the Notch-potentiation role of Neur: regulating Numb localization via down-regulating (directly or indirectly) Pdm proteins.
Neuralized enhances the efficiency of Notch-signaling during the specification of sib fate
Results from the analysis of neur, neur; numb double mutant embryos and neur gain-of-function embryos show that Neur functions to increase the efficiency of Notch-signaling but not essential for it. None of the previous studies have made this important distinction. Previous results have indicated that Neur activates Notch-signaling via endocytosis of Delta and the Delta bound extracellular domain of Notch. However, in neur null mutants (embryos homozygous for a deficiency that removes neur completely), sib specification still occurs in ~10% of the hemisegments. While this may arguably be due to a partial redundancy for neur, there is another line of evidence that suggests a role for Neur in enhancing the efficiency of Notch signaling. That is, while in Notch;numb double mutants both daughter cells of a GMC-1 adopt an RP2 fate (note that for the specification of an RP2 fate Numb is needed only when there is an intact Notch-signaling), in neur; numb double mutants the daughters often adopt a mixed identity. This result indicates that Notch is still able to specify some features of a sib identity (i.e., reduced levels of Eve expression) in the absence of Neur activity. If Neur is absolutely needed for Notch signaling, the double mutant results would have been exactly the same as Notch;numb double mutants where both daughters adopt an unambiguous RP2 fate.
On the other hand, the results from Neur over-expression experiments indicate that when present at high levels Neur is able to overcome the Numb block and induce both the progeny of GMC-1 to adopt a sib fate (Fig. 7F). This phenotype is strikingly similar to the phenotype observed with the over-expression of the intracellular domain of Notch or the phenotype in numb mutants (Wai et al., 1999). These results suggest that over-expression of Neur leads to processing of Notch in the cell that has Numb. We also want to point out that the source of Neur for the trans-effect on Notch-signaling need not be only from the “RP2” cell, but may also be from the neighboring cells. This is indicated by the previous result that while the GMC-1 in embryos mutant for cyclin A adopts an RP2 fate, the same GMC-1 in cyclin A; numb double mutants adopts a sib fate (Wai et al., 1999; see Fig. 8).
Inter-dependence between Neuralized and Numb for their basal localization
Our results show that the asymmetric basal localization of Numb in neur mutants and Neur in numb mutants is affected. This shows the inter-dependence of localization of these two proteins. We have examined whether there is any initial localization of Numb or Neur in the two mutants to determine if the localization of the one protein falls apart in the absence of localization of the other. However, no such initial localization was observed for either of the two proteins. It is possible that both Neur and Numb control the same pathway(s) that directly or indirectly mediates localization of the other. Perhaps Neur and Numb interact physically with each other in the cytoplasm prior to any localization and it is this Neur-Numb complex that gets localized to the basal pole; in the absence of either of the two proteins, no such complex is formed, and no localization occurs. We have not yet tested this model due to lack of appropriate reagents. On the other hand, loss of Numb-localization in neur could be due to loss of Insc localization; loss of Neur localization in numb mutants could be more direct where Neur is downstream of Numb and Numb mediates directly or indirectly the localization of Neur. The function of Neur in GMC-1, however, appears to be required for the down-regulation of Pdm and allow localization of such proteins as Insc. Thus, Neur is both upstream and downstream of Numb in GMC-1. Another important distinction between Neur and Numb is that while non-asymmetric localization of Numb in GMC-1 will lead to both daughters of GMC-1 inheriting Numb and adopting RP2 fates, a non-asymmetric localization of Neur and inheritance of Neur by both daughters will not make them adopt an RP2 fate, but a sib fate.
In numb mutants, the localization of Neur is affected in such a way that both daughters inherit Neur. Does this have a consequence? Our results argue that unlike Numb there is no consequence to the non-asymmetric localization and segregation of Neur to both daughters, For instance, in wild type the sib cell does not inherit Neur, thus, the potentiation of Notch in this cell by Neur occurs in a cell non-autonomous mechanism (removing the extracellular domain of Notch bound by Delta) and there is no role for Neur in the sib itself. Thus, in numb mutants although both daughters inherit Neur, they still adopt a sib fate.
Research highlights.
In the CNS, Notch regulates asymmetric cell fate specification to daughters of precursor cells. The E3 Ubiquitin ligase protein Neuralized (Neu) is thought to activate Notch-signaling by the endocytosis of Delta and the Delta-bound extracellular domain of Notch. The intracellular Notch then initiates Notch-signaling. We have known for years that the asymmetrically localized and inherited Numb blocks N-signaling in one of the two daughters of a GMC to allow that cell to adopt a different identity. What we have found is that loss of Neu activity causes symmetric division of precursor cells. Neu is asymmetrically localized prior to the precursor cell division to the Numb domain and most importantly, it regulates asymmetric division via two distinct, sequential mechanisms: by asymmetric distribution of Numb via down-regulation of Pdm 1 (a transcription factor), followed by the trans-potentiation of Notch. Our results tie Numb localization and Notch processing-signaling through a single player, Neu, thus giving us a more complete picture of the events that surround asymmetric division of precursor cells. Finally, this work also shows that there is interdependence between Neu and Numb for their asymmetric localization to the basal end.
Acknowledgments
We thank the following researchers for providing antibodies: Drs. Manfred Frasch (Eve), Zun Lai (Zfh-1), Eric Lai (Neuralized), Bill Chia (Insc), Steve Cohen (Pdm1), Iowa Hybridoma Center (22C10, Spectrin). We thank Dr. Eric Lai for sending us the UAS-neur and UAS-neurΔRING lines, and the Bloomington stock center for the neur mutants, deficiencies, and other lines used in this study, and members of the Bhat lab for comments on the manuscript. This work is supported by a grant from the National Institutes of General Medicine, NIH, to KB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bhat KM. frizzled and frizzled 2 play a partially redundant role in wingless signaling and have similar requirements to wingless in neurogenesis. Cell. 1998;95:1027–36. doi: 10.1016/s0092-8674(00)81726-2. [DOI] [PubMed] [Google Scholar]
- Bhat KM. Segment polarity genes in neuroblast formation and identity specification during Drosophila neurogenesis. Bioessays. 1999;21:472–485. doi: 10.1002/(SICI)1521-1878(199906)21:6<472::AID-BIES4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Bhat KM, Schedl P. The Drosophila miti-mere gene, a member of the POU family, is required for the specification of the RP2/sibling lineage during neurogenesis. Development. 1994;120:1483–1501. doi: 10.1242/dev.120.6.1483. [DOI] [PubMed] [Google Scholar]
- Bhat KM, Apsel N. A mechanism for the self-renewing asymmetric division of neural precursor cells in the Drosophila CNS. Development. 2004;131:1123–1134. doi: 10.1242/dev.01014. [DOI] [PubMed] [Google Scholar]
- Bhat KM, Poole SJ, Schedl P. The miti-mere and pdm1 genes collaborate during specification of the RP2/sib lineage in Drosophila neurogenesis. Mol Cell Biol. 1995;15:4052–4063. doi: 10.1128/mcb.15.8.4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KM, Gaziova I, Krishnan S. Regulation of Axon Guidance by Slit and Netrin Signaling in the Drosophila Ventral Nerve Cord. Genetics. 2007;176:2235–2246. doi: 10.1534/genetics.107.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher M, Yeo SL, Udolph G, Zavortink M, Yang X, Tear G, Chia W. Binary sibling neuronal cell fate decisions in the Drosophila embryonic central nervous system are nonstochastic and require inscuteable-mediated asymmetry of ganglion mother cells. Genes Dev. 1998;12:1858–1870. doi: 10.1101/gad.12.12.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulianne GL, de la Concho A, Campos-Ortega JA, Jan LY, Jan YN. The Drosophila neurogenic gene neuralized encodes a novel protein and is expressed in precursors of larval and adult neurons. EMBO J. 1991;10:2975–2983. doi: 10.1002/j.1460-2075.1991.tb07848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu La Graff Q, Doe CQ. Neuroblast specification and formation regulated by wingless in Drosophila CNS. Science. 1993;261:1594–1597. doi: 10.1126/science.8372355. [DOI] [PubMed] [Google Scholar]
- Gaziova I, Bhat KM. Generating asymmetry: with and without self-renewal. Prog Mol Subcell Biol. 2007;45:143–178. doi: 10.1007/978-3-540-69161-7_7. [DOI] [PubMed] [Google Scholar]
- Gaziova I, Bhat KM. Ancestry - independent fate specification and plasticity of a developmental timing of a typical Drosophila neuronal lineage. Development. 2009;136:263–274. doi: 10.1242/dev.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Deblandre GA, Kintner C, Rubin GM. Drosophila Neuralized is a ubiquitin ligase that promotes internalization and degradation of Delta. Developmental Cell. 2001;1:783–794. doi: 10.1016/s1534-5807(01)00092-2. [DOI] [PubMed] [Google Scholar]
- Lai Z, Fortini ME, Rubin GM. The embryonic expression pattern of zfh1 and zfh2, two Drosophila genes encoding novel zinc-finger homeodomain proteins. Mech Dev. 1991;34:123–134. doi: 10.1016/0925-4773(91)90049-c. [DOI] [PubMed] [Google Scholar]
- Lai EC, Roegiers F, Qin X, Jan YN, Rubin GM. The ubiquitin ligase Drosophila Mind bomb promotes Notch signaling by regulating the localization and activity of Serrat e and Delta. Development. 2005;132:2319–2332. doi: 10.1242/dev.01825. [DOI] [PubMed] [Google Scholar]
- Lai EC, Rubin GM. ‘Neuralized is essential for a subset of Notch pathway-dependent cell fate decisions during Drosophila eye development. Proc Natl Acad Sci USA. 2001a;98:5637–5642. doi: 10.1073/pnas.101135498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Rubin GM. neuralized functions cell-autonomously to regulate a subset of Notch-dependent processes during Drosophila Development. Dev Biol. 2001b;231:217–233. doi: 10.1006/dbio.2000.0124. [DOI] [PubMed] [Google Scholar]
- Lear BC, Skeath JB, Patel NH. Neural cell fate in rca1 and cycA mutants: the roles of intrinsic and extrinsic factors in asymmetric division in the Drosophila central nervous system. Mech Dev. 1999;88:207–219. doi: 10.1016/s0925-4773(99)00190-2. [DOI] [PubMed] [Google Scholar]
- Le Borgne R, Schweisguth F. Unequal segregation of neutralized biases notch activation during asymmetric cell division. Dev Cell. 2003;5:139–148. doi: 10.1016/s1534-5807(03)00187-4. [DOI] [PubMed] [Google Scholar]
- Le Borgne R, Remaud S, Hamel S, Schweisguth F. Two distinct E3 ubiquitin ligases have complementary functions in the regulation of delta and serrate signaling in Drosophila. PLoS Biol. 2005;3:e96. doi: 10.1371/journal.pbio.0030096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta B, Bhat KM. Slit signaling promotes the terminal asymmetric division of neural precursor cells in the Drosophila CNS. Development. 2001;128:3161–3168. doi: 10.1242/dev.128.16.3161. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E, Kluding H. Mutations affecting the pattern of the larval cuticle in Drosophila melanogaster. I. Zygotic loci on the second chromosome. Roux Arch Dev Biol. 1984;193:267–282. doi: 10.1007/BF00848156. [DOI] [PubMed] [Google Scholar]
- Pavlopoulos E, Pitsouli C, Klueg KM, Muskavitch MA, Moschonas NK, Delidakis C. Neutralized encodes a peripheral membrane protein involved in delta signaling and endocytosis. Dev Cell. 2001;6:807–816. doi: 10.1016/s1534-5807(01)00093-4. [DOI] [PubMed] [Google Scholar]
- Price BD, Chang Z, Smith R, Bockheim S, Laughon A. The Drosophila neuralized gene encodes a C3HC4 zinc finger. EMBO J. 1993;12:2411–2418. doi: 10.1002/j.1460-2075.1993.tb05895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Shepherd S, Ackerman L, Jan LY, Jan YN. numb, a gene required in determination of cell fate during sensory organ formation in Drosophila embryos. Cell. 1989;58:349–360. doi: 10.1016/0092-8674(89)90849-0. [DOI] [PubMed] [Google Scholar]
- Wai PB, Truong B, Bhat KM. Cell division genes promote asymmetric interaction between Numb and Notch in the Drosophila CNS. Development. 1999;126:2759–2770. doi: 10.1242/dev.126.12.2759. [DOI] [PubMed] [Google Scholar]
- Yeh E, Zhou L, Rudzik N, Boulianne GL. ‘euralized functions cell autonomously to regulate drosophila sense organ development. EMBO J. 2000;19:4827–4837. doi: 10.1093/emboj/19.17.4827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh E, Dermer M, Commisso C, Zhou L, McGlade CJ, Boulianne GL. Neuralized functions as an E3 ubiquitin ligase during Drosophila development. Curr Biol. 2001;11:1675–1679. doi: 10.1016/s0960-9822(01)00527-9. [DOI] [PubMed] [Google Scholar]
- Yedvobnick B, Kumar A, Choudhury P, Opraseuth J, Mortimer N, Bhat KM. The asymmetric division function of Mastermind is separable and distinct from its neurogenic function during drosophila neurogenesis. Genetics. 2004;166:1281–1289. doi: 10.1534/genetics.166.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo SL, Lloyd A, Kozak K, Dinh A, Dick T, Yang X, Sakonju S, Chia W. On the functional overlap between two Drosophila POU homeo domain genes and the cell fate specification of a CNS neural precursor. Genes Dev. 1995;9:1223–1236. doi: 10.1101/gad.9.10.1223. [DOI] [PubMed] [Google Scholar]