

Abstract

The synthesis and properties of a full set of four benzo-expanded ribonucleosides (xRNA), analogous to A, G, C, and U RNA monomers, are described. The nucleosides are efficient fluorophores with emission maxima of 369–411 nm. The compounds are expected to be useful as RNA pathway probes, and as components of an unnatural ribopolymer.
Ribonucleosides and ribonucleotides with unnatural nucleobases have found widespread use as chemical tools to study the functional roles1a,1b, hydrogen bonding requirements1c, structure/activity relationships1d, and mechanistic details1e of RNAs in biological systems. In particular, the use of fluorescent ribonucleotides as biological probes and sensors has received much attention2. In an effort to minimally perturb the recognition of RNAs by natural biomolecules, most fluorescent ribonucleotides are designed to retain some of the original canonical Watson-Crick base-pairing recognition groups found in natural RNA3.
Previously, Leonard et al. described the synthesis of fluorescent ribonucleoside analogs of adenosine (1, Figure 1) and guanosine (2) with a benzene ring incorporated within the purine scaffold that expanded their size by 2.4 Å4,5. Among other features, these analogs retained their canonical Watson-Crick base-pairing groups. Nucleoside triphosphate derivatives of 1 and 2 have been prepared enzymatically6; however, technological limitations at the time did not allow for the synthesis of oligoribonucleotides using these novel RNA monomers. Furthermore, analogous benzo-expanded analogs of cytidine (3) and uridine (4) have never been reported. One example of a size-expanded uridine analog has been reported7; however, the glyosidic bond is at the same N1-position as natural pyrimidine ribonucleosides and is therefore not structurally analogous to 1 and 2 in a size-expanded RNA genetic set.
Figure 1.
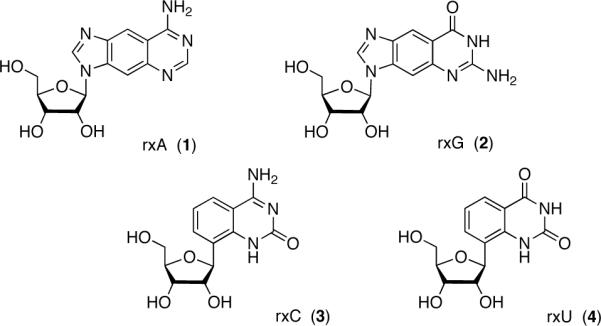
The xRNA genetic set.
Our laboratory has previously reported the design and synthesis of a set of four size-expanded DNA analogs (xDNA)8,9. After incorporation into oligonucleotides using automated oligonucleotide synthesis, these compounds were studied for their unusual biophysical and biochemical properties in DNA10,11. Herein we describe the synthesis of the complete set of size-expanded RNA analogs (xRNA, Figure 1) and report their photophysical properties. This information will provide a foundation on which to design experiments to use xRNA monomers and polymers in the study of ribonucleotide and RNA pathways in biological systems.
The syntheses of the expanded purine nucleosides 1 (rxA) and 2 (rxG) were similar to the syntheses of their xDNA counterparts, dxA and dxG8,9, and to previous approaches to the compounds as reported by Leonard and co-workers4,5. The protected xA nucleobase (5, Scheme 1) was coupled to 1,2,3,5-tetra-O-acetyl-β-D-ribose (rib) under Vorbrüggen conditions12. This method for forming N-nucleosides is a less hazardous alternative to Leonard's approach of using mercuric cyanide as a coupling agent4. Although both methods are known to form the desired β-anomers of the nucleosides, they also generate a mixture of N3- and undesired N1-regioisomers. After chromatographic purification of the N-ribosylated products, an overall yield of 61% was obtained in a ratio of 60:40 (6a:6b). The correct regioisomer (6a) was identified by 2D-NMR (Suppl. Figure S1). To obtain additional amounts of the desired N3-regioisomer, 6b was converted to 6a via a transisomerization reaction involving catalytic amounts of p-toluenesulfonic acid13. Leonard did not report the overall yield and regioisomeric ratios of their N- ribosylation reaction, so it is difficult to compare the prior approach to the effectiveness of the present Vorbrüggen approach for generating the rxA precursors. A tandem O-deacetylation/nucleophilic aromatic substitution reaction of 6a with ammonia generated 1 in good yield. Using these improved methods, compound 1 can be synthesized in 5.1% overall yield over 8 steps, or 6.0% overall yield in 9 steps (including the transisomerization of 6b).
Scheme 1.
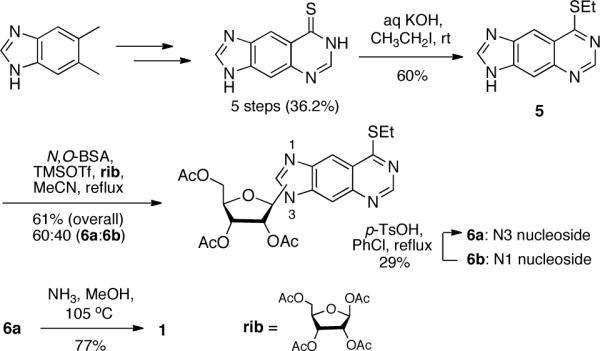
Synthesis of rxA triol (1).
The synthesis of rxG, as shown in Scheme 2, has been previously reported by us as an intermediate in dxG synthesis by reaction of 7 with rib under Vorbrüggen conditions9. However, the N3- and N1-regioisomeric mixture (8a and 8b, respectively) could only be separated in later steps. To improve rxG synthesis, we found an eluant mixture for column chromatography that successfully separated 8a and 8b. The correct regioisomer (8a) was identified using 2D-NMR (Suppl. Figure S1). As was done in rxA synthesis, we were also able to generate more of the desired 8a regioisomer by transisomerization of 8b. Mild deacylation by aminolysis of 8a generated 2 in 87% yield. Compound 2 was prepared in 5.0% overall yield over 7 steps, or 6.3% overall yield in 8 steps (including the transisomerization of 8b).
Scheme 2.
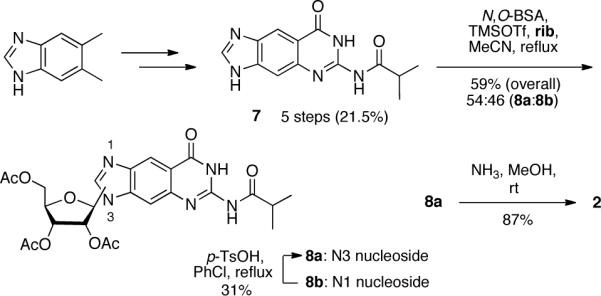
Synthesis of rxG triol (2).
The novel expanded pyrimidines 3 (rxC) and 4 (rxU) were modeled after their xDNA counterparts, dxC and dxT8,9. A major structural difference between the xDNA and xRNA pyrimidine sets is the presence and absence of a methyl group on the C6 position of their nucleobases, respectively. This difference required a substantial change of synthetic approach. Our strategy for the synthesis of xRNA C-nucleosides involved metallation of an aryl halide nucleobase followed by nucleophilic addition to 2,3,5-tri-O-benzyl-β-D-ribono-1,4,-lactone (rlac) and subsequent reduction at C-1', as was done previously in our laboratory14. For the synthesis of rxU, we envisioned the use of 8-bromo-2,4-dimethoxyquinazoline (12, Scheme 3) as aryl halide. After reduction at C-1', the nucleoside would serve as a common intermediate between rxU and rxC syntheses.
Scheme 3.
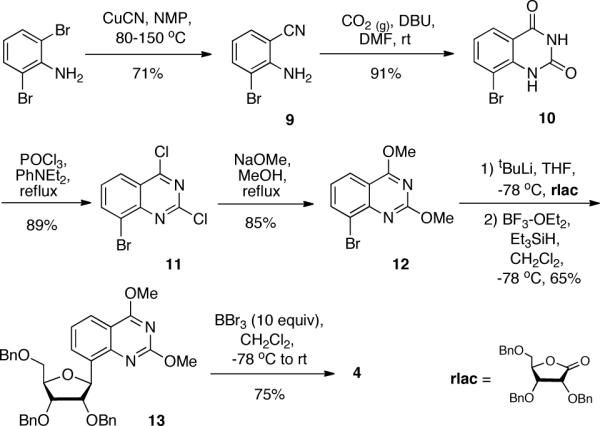
Synthesis of rxU triol (4).
To the best of our knowledge, the synthesis of the brominated heterocycle intermediate 12 has not been previously reported. We investigated various synthetic approaches to generate a sizeable amount of 12. In the end, we found that this could be best accomplished beginning with the synthesis of 2-amino-3-bromobenzonitrile, 9. Jiang and Lai previously reported the synthesis of 2-amino-3-chlorobenzonitrile from 2,6-dichloroaniline using copper (I) cyanide in NMP in 31% yield15. Replacement of the starting material with commercially available 2,6-dibromoaniline and improving reaction conditions afforded 9 in 71% yield. Cyclization of 9 using carbon dioxide gas in the presence of DBU produced 10 in 91% yield, as was reported for the cyclization of various 2-aminobenzonitriles16. Following this, compounds 11 and 12 were synthesized in excellent yield employing methods previously used by our laboratory8.
Various metallating agents were screened in the C-ribosylation reaction of 12 with the ribonolactone. We found that 1.1 equivalents of tert-butyllithium best generated the activated species for the nucleophilic addition reaction. Following reduction at C-1' using BF3·OEt2 and triethylsilane, a single product, 13, was formed in 65% yield. gHMBC and ROESY studies on 13 revealed that the xU nucleobase was attached to the sugar in the correct C8-position and that the nucleoside was the desired β-anomer (Suppl. Figure S2).
At this point, we were interested in using 13 as the branching point between rxU and rxC syntheses. It is known that boron tribromide is an effective debenzylating reagent17. BBr3 also serves as a demethylating agent for aryl methyl ethers18. We found that by controlling the amount of BBr3 and reaction conditions on 13, we could generate two different and useful products. Using 10 equivalents of BBr3 beginning at −78 °C and warming to room temperature while stirring overnight generated 4 in 75% yield. On the other hand, using milder conditions (3 equivalents of BBr3 kept at −78 °C with stirring for 3 hours) generated the 2,4-dimethoxyquinazoline triol nucleoside, 14 (Scheme 4), in 79% yield. To obtain the desired rxC precursor, 15, we performed an aromatic nucleophilic substitution reaction on 14 using ammonia in methanol. We found that only one mono-amino substituted product was formed in very good yield. 1H-NMR data showed the presence of a methoxy and amino group in the compound. ROESY analysis of this product (15) revealed that the methoxy and amino groups were in the desired 2- and 4-positions, respectively (Suppl. Figure 2). Demethylation of 15 using sodium iodide in acetic acid generated 3 in excellent yield. Compound 3 was synthesized in 19.2% overall yield over 8 steps and compound 4 was prepared in 22.6% yield in 6 steps.
Scheme 4.
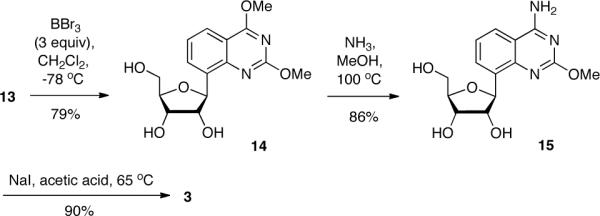
Synthesis of rxC triol (3).
To evaluate the utility of the xRNA nucleosides to be employed as fluorescence probes, we measured the photophysical properties of the entire genetic set (1–4) in methanol (Table 1) and water at pH 7.0 (Suppl. Table S1). Previously, we reported the photophysical properties of the xDNA monomers in methanol11. In addition, partial photophysical data for rxA and rxG triols has been reported by Leonard4,5,6a.
Table 1.
Photophysical data for xRNA triols in water.
| xRNA triol | λmax abs.a (nm) | λmax ex. (nm) | λmax em. (nm) | e260 (L/mol·cm) | emax (L/mol·cm) | Fb | brightnessc (L/mol·cm) | t (ns) |
|---|---|---|---|---|---|---|---|---|
| rxA | 331 | 331 | 373 | 18,500 | 10,500 | 0.44 | 4,600 | 3.6 |
| rxG | 326 | 337 | 411 | 4,900 | 4,500 | 0.40 | 1,800 | 7.3 |
| rxC | 322 | 322 | 379 | 5,300 | 4,500 | 0.48 | 2,200 | 4.2 |
| rxU | 311 | 311 | 369 | 1,000 | 3,900 | 0.27 | 1,050 | 2.9 |
long l maximum
error ≤ ± 0.03
brightness calculated as emax × F.
Our experiments reveal that all four nucleosides have high quantum yields (Table 1). The data show that rxA is the brightest of the four, owing to its higher molar absorptivity. In water, the data for rxA are in good agreement with that of Leonard4. In methanol, the fluorescence spectra of rxA (Fig. 2A) and that of dxA are similar, with the exception that the emission of dxA is red-shifted by 20 nm11.
Figure 2.

Fluorescence spectra of rxA (A), rxG (B), rxC (C), and rxU (D) triols in methanol. Excitation spectra were monitored at the emission maxima.
The rxG nucleoside emits furthest to the red (411 nm) of the set. Our data for rxG in water seem to be in general agreement with previous limited data for rxG triol in aqueous buffer5,6a. We also find that rxG in methanol (Fig. 2B) has very similar photophysical properties to dxG in the same solvent11. Interestingly, the emission and excitation wavelengths of rxG are all blue-shifted (by ca. 20 nm) in water as compared to data taken in methanol.
We also measured the photophysical properties of the previously unknown xRNA pyrimidines (rxC and rxU). In comparison to the xDNA pyrimidines11, there is a small blue-shifting of absorption, excitation, and emission maxima by ~10 nm (in each case) in the xRNA pyrimidine nucleosides. This difference is likely due to the absence of the C6 methyl group in rxC and rxU. We find that the two have virtually the same properties in methanol (Fig. 2C and 2D) or water.
In summary, we have synthesized a complete four-letter benzo-expanded RNA genetic set, in good yield and purity. We have also demonstrated the photophysical properties of the xRNA nucleosides, which we find to be efficient fluorophores. Future work will be directed toward the use of these compounds as nucleotide probes and in unnatural RNA polymers.
Supplementary Material
Acknowledgment
This work was supported by the National Institutes of Health (GM063587). We also acknowledge the NIH for support of A.H. as a Ruth L. Kirschstein NRSA predoctoral fellow (GM084680). We thank Dr. Urvashi Sahni (Stanford University) for technical assistance and Dr. Stephen R. Lynch (Stanford University) for assistance with NMR experiments.
Footnotes
Supporting Information Available Experimental procedures, NMR data, fluorescence experiment details, UV-Vis and fluorescence spectra, and fluorescence lifetime data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).(a) Lu J, Li N-S, Koo SC, Piccirilli JA. J. Org. Chem. 2009;74:8021. doi: 10.1021/jo9016919. [DOI] [PubMed] [Google Scholar]; (b) Cho HD, Oyelere AK, Strobel SA, Weiner AM. RNA. 2003;9:970. doi: 10.1261/rna.2110903. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Somoza A, Chelliserrykattil J, Kool ET. Angew. Chem. Int. Ed. 2006;45:4994. doi: 10.1002/anie.200601311. [DOI] [PubMed] [Google Scholar]; (d) Pokharel S, Jayalath P, Maydanovych O, Goodman RA, Wang SC, Tantillo DJ, Beal PA. J. Am. Chem. Soc. 2009;131:11882. doi: 10.1021/ja9034076. [DOI] [PubMed] [Google Scholar]; (e) Araki L, Morita K, Yamaguchi M, Zhao Z-Y, Wilson TJ, Lilley DMJ, Harusawa S. J. Org. Chem. 2009;74:2350. doi: 10.1021/jo802556s. [DOI] [PubMed] [Google Scholar]
- (2).(a) Stengel G, Urban M, Purse BW, Kuchta RD. Anal. Chem. 2010;82:1082. doi: 10.1021/ac902456n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Srivatsan SG, Tor Y. Nat. Protoc. 2007;2:1547. doi: 10.1038/nprot.2007.222. [DOI] [PubMed] [Google Scholar]; (c) Kimoto M, Mitsui T, Harada Y, Sato A, Yokoyama S, Hirao I. Nucleic Acids Res. 2007;35:5360. doi: 10.1093/nar/gkm508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Zhang C-M, Liu C, Christian T, Gamper H, Rozenski J, Pan D, Randolph JB, Wickstrom E, Cooperman BS, Hou Y-M. RNA. 2008;14:2245. doi: 10.1261/rna.1158508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Leonard NJ, Sprecker MA, Morrice AG. J. Am. Chem. Soc. 1976;98:3987. doi: 10.1021/ja00429a040. [DOI] [PubMed] [Google Scholar]
- (5).Keyser GE, Leonard NJ. J. Org. Chem. 1979;44:2989. [Google Scholar]
- (6).(a) Leonard NJ, Keyser GE. Proc. Natl. Acad. Sci. USA. 1979;76:4262. doi: 10.1073/pnas.76.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leonard NJ, Scopes DIC, Van Der Lijn P, Barrio JR. Biochem. 1978;17:3677. doi: 10.1021/bi00611a001. [DOI] [PubMed] [Google Scholar]
- (7).Xie Y, Maxson T, Tor Y. J. Am. Chem. Soc. 2010;132:11896. doi: 10.1021/ja105244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu H, Gao J, Maynard L, Saito YD, Kool ET. J. Am. Chem. Soc. 2004;126:1102. doi: 10.1021/ja038384r. [DOI] [PubMed] [Google Scholar]
- (9).Liu H, Gao J, Kool ET. J. Org. Chem. 2005;70:639. doi: 10.1021/jo048357z. [DOI] [PubMed] [Google Scholar]
- (10).Gao J, Liu H, Kool ET. J. Am. Chem. Soc. 2004;126:11831. [Google Scholar]
- (11).Krueger AT, Kool ET. J. Am. Chem. Soc. 2008;130:3989. doi: 10.1021/ja0782347. [DOI] [PubMed] [Google Scholar]
- (12).Vorbrüggen H, Hölfe G. Chem. Ber. 1981;114:1256. [Google Scholar]
- (13).Boryski J, Golankiewicz B. Nucleos. Nucleot. 1989;8:529. [Google Scholar]
- (14).Silverman AP, Kool ET. J. Am. Chem. Soc. 2007;129:10626. doi: 10.1021/ja072791b. [DOI] [PubMed] [Google Scholar]
- (15).Jiang J, Lai Y-H. J. Am. Chem. Soc. 2003;125:14296. doi: 10.1021/ja0382260. [DOI] [PubMed] [Google Scholar]
- (16).Mizuno T, Ishino Y. Tetrahedron. 2002;58:3155. [Google Scholar]
- (17).Kundu NG, Hertzberg RP, Hannon SJ. Tetrahedront Lett. 1980;21:1109. [Google Scholar]
- (18).Sakamoto T, Kondo Y, Sato S, Yamanaka H. Tetrahedron Lett. 1994;35:2919. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.