

Abstract
Two mouse models, the CochG88E/G88E or “knock-in” and the Coch−/− or “knock-out” (Coch null), have been developed to study the human late-onset, progressive, sensorineural hearing loss and vestibular dysfunction known as DFNA9. This disorder results from missense and in-frame deletion mutations in COCH (coagulation factor C homology), encoding cochlin, the most abundantly detected protein in the inner ear. We have performed hearing and vestibular analyses by auditory brainstem response (ABR) and vestibular-evoked potential (VsEP) testing of the Coch−/− and CochG88E/G88E mouse models. Both Coch−/− and CochG88E/G88E mice show substantially elevated ABRs at 21 months of age, but only at the highest frequency tested for the former and all frequencies for the latter. At 21 months, 9 of 11 Coch−/− mice and 4 of 8 CochG88E/G88E mice have absent ABRs. Interestingly Coch−/+ mice do not show hearing deficits, in contrast to CochG88E/+, which demonstrate elevated ABR thresholds similar to homozyotes. These results corroborate the DFNA9 autosomal dominant mode of inheritance, in addition to the observation that haploinsufficiency of Coch does not result in impaired hearing. Vestibular evoked potential (VsEP) thresholds were analyzed using a two factor ANOVA (Age X Genotype). Elevated VsEP thresholds are detected in Coch−/− mice at 13 and 21 months, the two ages tested, and as early as seven months in the CochG88E/G88E mice. These results indicate that in both mouse models, vestibular function is compromised before cochlear function. Analysis and comparison of hearing and vestibular function in these two DFNA9 mouse models, where deficits occur at such an advanced age, provide insight into the pathology of DFNA9 and age-related hearing loss and vestibular dysfunction as well as an opportunity to investigate potential interventional therapies.
Keywords: Coch, cochlin, DFNA9 mouse models, ABR, VsEP
1. Introduction
The COCH (coagulation factor C homology) gene, encoding the abundant inner ear protein cochlin, has been shown to be mutated in the human autosomal dominant, late-onset and progressive nonsyndromic hearing loss and vestibular disorder, DFNA9. The true incidence of COCH mutations is unknown, as systematic genetic testing for this and other late-onset forms of hearing loss is not currently performed. However, to date, 11 different missense mutations, and one in-frame deletion have been reported in patients from four continents (Fig. 1, Table I). In addition, potential roles of COCH in presbycusis and balance disorders have been implicated (de Kok et al., 1999; Fransen et al., 1999). Cochlin has also been shown to play an important role in autoimmune sensorineural hearing loss via presence of auto-antibodies against cochlin (Boulassel et al., 2001; Tebo et al., 2006) as well as by T-cell mediated mechanisms (Baek et al., 2006).
Figure 1.

Schematic representation of the deduced amino acid structure of human COCH, encoding the protein cochlin, shows a predicted signal peptide (SP), followed by a domain initially designated as FCH (factor C-homology), also known as the LCCL domain, (Limulus factor C, cochlin, lung gestational protein), followed by an intervening domain, and two von Willebrand factor A-like domains (vWFA1 and vWFA2) separated by an intervening domain. Twelve mutations, (all missense except for one in-frame deletion) causing DFNA9 deafness and vestibular disorder, are indicated. The positions of all cysteine residues are shown as “C”.
Table 1.
COCH mutations in DFNA9
| Origin | Exon | Nucleotide change |
Amino acid change |
Protein domain |
Reference |
|---|---|---|---|---|---|
| Belgium & The Netherlands |
4 | C207T | P51S | FCH/LCCL | (Fransen et al., 1999) (de Kok et al., 1999) |
| United States | 4 | T253G | V66G | FCH/LCCL | (Robertson et al., 1998) |
| The Netherlands | 5 | G315T | G87W | FCH/LCCL | (Collin et al., 2006) |
| United States & The Netherlands |
5 | G319A | G88E | FCH/LCCL | (Robertson et al., 1998) (Kemperman et al., 2005) |
| Hungary | 5 | 366_368delGTA | V104del | FCH/LCCL | (Nagy et al., 2004) |
| The Netherlands | 5 | T382C | I109T | FCH/LCCL | (Pauw et al., 2007) |
| Australia | 5 | T382A | I109N | FCH/LCCL | (Kamarinos et al., 2001) |
| United States & Korea |
5 | T405C | W117R | FCH/LCCL | (Robertson et al., 1998) (Baek et al., 2010) |
| Japan | 5 | G411A | A119T | FCH/LCCL | (Usami et al., 2003) |
| China | 12 | T1591C | M512T | vWFA2 | (Yuan et al., 2008) |
| United States | 12 | G1681T | C542F | vWFA2 | (Street et al., 2005) |
| China | 12 | G1681A | C542Y | vWFA2 | (Yuan et al., 2008) |
To understand better the functions of cochlin and the etiology of DFNA9, we developed and previously reported a knock-in mouse model, CochG88E/G88E (Robertson et al., 2008). The Coch−/− mouse model was also previously reported and testing of hearing by auditory brainstem response (ABR) of these mice was performed only up to 5 months of age, showing no hearing impairment at this time-point (Makishima et al., 2005). Because DFNA9 is a late-onset disorder, we obtained the Coch−/− mice and carried out ABR testing at progressively more advanced ages. We also tested vestibular function, not previously performed on the Coch−/− mouse, by measurement of the vestibular evoked potentials (VsEPs) at several ages. Histological evaluation of these mice was carried out. In addition, we performed ABR and VsEP testing in the CochG88E/G88E mice at progressively younger ages than in our previous study, in order to determine the age of onset for hearing and vestibular deficits. We present in this report comprehensive testing and comparison of the two mouse models and human DFNA9, providing genotype/phenotype correlations and insight into pathogenic mechanisms of COCH mutations.
2. Materials and methods
2.1. ABR and VsEP measurements
2.1.1 Animals and animal preparation
The use of animals for these studies was approved by the Institutional Animal Care and Use Committee at East Carolina University and at Harvard Medical School. Both the Coch−/− and CochG88E/G88E mouse models were back-crossed for 12 generations with the CBA/CaJ strain, which has been demonstrated to have no hearing loss well into advanced ages (Zheng et al., 1999). Because these two mouse models are now in the CBA/CaJ background, the analyses are not confounded by background, age-related hearing loss. In addition, our data are based on comparison of the three genotypes of mice from the same cohorts, to control for any potential genetic background effects.
Auditory brainstem responses (ABRs) were measured for Coch−/− (n = 11), Coch−/+ (n = 12) and Coch+/+ (n = 9) littermates at 13 months of age and for Coch−/− (n = 10), Coch−/+ (n = 7) and Coch+/+ (n = 8) littermates at 21 months.
Vestibular evoked potentials (VsEPs) were measured for Coch−/− (n = 5), Coch−/+ (n = 5) and Coch+/+ (n = 5) littermates at 13.5 months of age and for Coch−/− (n = 10), Coch−/+ (n = 7) and Coch+/+ littermates (n = 8) at 21 months. VsEPs were also measured for CochG88E/G88E mice at 5 months (n=7) and at 7 months (n=12).
Mice were anesthetized with a ketamine (18 mg/ml) and xylazine (2 mg/ml) solution (5-7 μl per gram body weight injected intraperitoneally). Core body temperature was maintained at 37.0 ± 0.1°C using a homeothermic heating pad system (FHC, Inc., Bowdoin, ME).
2.1.2. ABR stimulus and stimulus coupling
For ABR testing, tone burst stimuli were generated and controlled using Tucker Davis Technologies (TDT, Gainesville, FL) System III (RX6, PA5 components). Tone bursts at 8, 16, 32 and 41.2 kHz had 1.0 ms rise-fall times with 1.0 ms plateau (3 ms total duration) and alternating stimulus polarity. Stimuli for ABR testing were calibrated using a Bruel & Kjaar ¼″ microphone and Nexus amplifier. Stimuli were calibrated in dB peSPL and were presented via high frequency transducers (TDT ED1 driver, EC1 speakers) coupled at the left ear via PE tubing. Auditory stimuli were presented at a rate of 17 stimuli/sec.
2.1.3. Vestibular stimulus and stimulus coupling
VsEP recordings were based on methods for mice (Jones et al., 1999a; Jones et al., 2004; Jones et al., 2002) and are briefly described below. Linear acceleration pulses, 2 ms duration, were generated and controlled with TDT System III processors and presented to the cranium via a non-invasive spring clip that encircled the head anterior to the pinna and secured the head to a voltage-controlled mechanical shaker. Stimuli were presented along the naso-occipital axis using two stimulus polarities, normal (+Gx axis) and inverted (−Gx axis). Stimuli were presented at a rate of 17 pulses/sec. Stimulus amplitude ranged from +6 dB to −18 dB re: 1.0g/ms (where 1g = 9.8 m/s2) adjusted in 3 dB steps.
2.1.4 VsEP and ABR recording parameters
Stainless steel wire was placed subcutaneously at the nuchal crest to serve as the noninverting electrode. Needle electrodes were placed posterior to the left pinna and at the ventral neck for inverting and ground electrodes, respectively. Traditional signal averaging was used to resolve responses in electrophysiological recordings. Ongoing electroencephalographic activity was amplified (equivalent to 200,000X), filtered (300 to 3000Hz) and digitized (24 kHz sampling rate). 512 primary responses were averaged for each VsEP or ABR response waveform. All responses were replicated. VsEP intensity series was collected beginning at the maximum stimulus intensity (i.e., +6 dB re: 1.0g/ms) with and without acoustic masking, then descending in 3 dB steps to −18 dB re: 1.0g/ms. A broad band forward masker (50 to 50,000 Hz, 94 dB SPL) was presented during VsEP measurements to verify absence of cochlear responses (Jones et al., 1999b). ABR intensity series was collected with a descending series of stimulus intensities (5 dB steps) beginning at approximately 100 dB peSPL. Thresholds were obtained from the VsEP (measured in dB re: 1.0g/ms) and ABR (in dB peSPL) intensity series. Descriptive statistics were calculated for each measure for each genotype.
2.2 Tissue Processing
Mouse tissues were obtained according to guidelines and protocols approved by the Harvard Medical School Standing Committee on Animals (Boston, MA). For histologic evaluation, osmium tetroxide staining and embedding in araldite resins (Polysciences, Warrenton, PA) was performed, as this technique yields better morphology of tissues than paraffin embedding. Mice were perfused intracardially with 2.5% glutaraldehyde and 1.5% paraformaldehyde in a 65 mM phosphate buffer. Both petrous temporal bones were extracted and the round and oval windows were opened to allow intralabyrinthine perfusion of fixative. After overnight postfixation (in 120 mM) EDTA for 1 week and osmication (1% OsO4 in dH20) for 1 hour, cochleas were dehydrated in ethanols and propylene oxide and then embedded in araldite resins and sectioned at 40 μM with a carbide steel knife. For cytocochleogram analysis in the cochlea, cell loss was assessed semi-quantitatively in every section through each ear. In this analysis, the fractional survival of the following cell types was estimated: inner and outer hair cells, fibrocytes in the spiral ligament and spiral limbus, the stria vascularis and neuronal cell bodies in the spiral ganglion. Cochlear position was converted to frequency according to a map for the mouse (Muller et al., 2005).
3. Results and discussion
3.1 Analysis of hearing and vestibular function in the Coch−/Coch− mouse model and further evaluation of CochG88E/G88E mice
We have undertaken the testing of auditory and vestibular functions in Coch−/− and CochG88E/G88E mouse models. Vestibular evoked potential (VsEP) measurements are a reflection of vestibular function, more specifically of gravity receptor or otolithic organs, the saccule and utricle. We showed previously that VsEP thresholds were elevated in CochG88E/G88E mice assessed at 11 months of age, compared to Coch+/+ littermates (Robertson et al., 2008). We have now obtained data regarding vestibular dysfunction at the earliest age of onset in this mouse model. Further testing has shown that vestibular dysfunction is present as early as 7 months in these mice (n=12), but appears to be normal at 5 months (n=7) (Fig. 2A). We have also performed vestibular testing of the Coch−/− mice which was previously uncharacterized. VsEP thresholds for the Coch−/− mice were elevated at 13 and 21 months of age, the two ages tested (Fig. 2B). It is possible that vestibular function in this mouse model shows deficits at younger ages.
Figure 2.
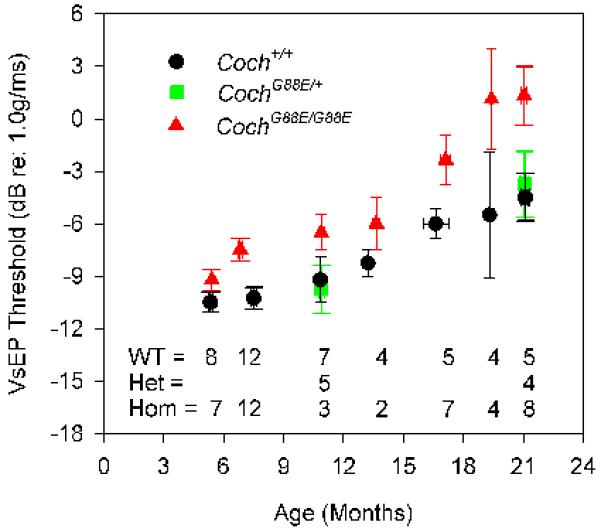
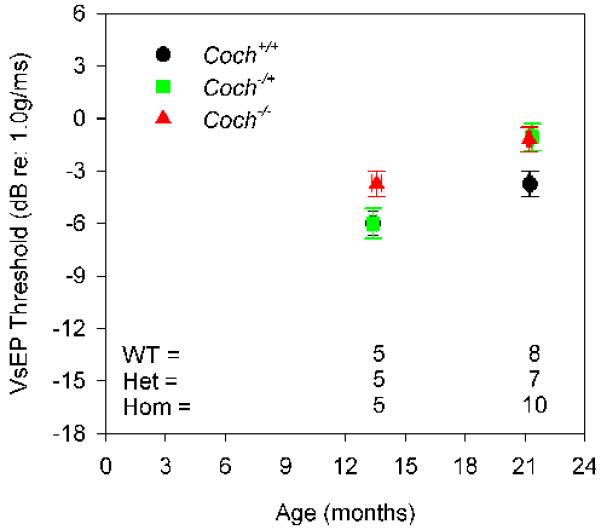
Average VsEP thresholds for (A) CochG88E/G88E and (B) Coch−/− mouse models. In the CochG88E/G88E mouse, vestibular dysfunction is present as early as 7 months, but appears to be normal at 5 months. VsEP thresholds of the Coch−/− mice were elevated at 13 months (earliest age tested) and at 21 months of age. VsEP thresholds suggest that vestibular dysfunction precedes hearing loss for both mouse models. Error bars represent standard error. [Color figure can be viewed in the online issue, which is available at http://www.sciencedirect.com/science/journal/03785955.]
In both mouse models, vestibular function is compromised at earlier ages than hearing function as shown by VsEP and ABR data. Although a wide range of ages for onset of hearing loss and vestibular dysfunction have been reported in DFNA9 mutation carriers, it is recognized to begin at “mid-life”. There have been several more thorough reports for ages of onset and progression of hearing and vestibular problems in DFNA9. A study of a Dutch family bearing the G88E COCH mutation showed deterioration of hearing from age 46-49 and onward, whereas deterioration of vestibular function started from ~46 years of age (Kemperman et al., 2005). A larger and more comprehensive study (n=74) of individuals in the Netherlands bearing the P51S mutation (in the same domain of COCH as the G88E mutation) revealed that vestibular deficits began at ~34 years of age, preceding onset of hearing impairment at ~43 years of age (Bischoff et al., 2005).
Hearing status of both mouse models was evaluated by auditory brainstem response (ABR) testing. In a previous report (Makishima et al., 2005), Coch−/− mice did not show ABR threshold elevation at 5 months, the oldest age tested. However, because of the late age of onset of dysfunction in humans with DFNA9, we performed testing of more advanced ages for the Coch−/− mice. At 13.5 months of age, Coch−/− mice (n=6) showed no significant elevation of ABR thresholds in comparison to the Coch+/+ (n=5) and Coch−/+ (n=6) littermates (Fig. 3A). At 21 months, Coch−/− mice (n= 11) show elevated ABR thresholds or absent ABRs at the highest frequency tested (Fig. 3A) as compared to Coch+/+ (n=9) and Coch−/+ (n=12) littermates. In fact, 9 of 11 Coch−/− mice, had absent ABRs at 41.2 kHz. In the CochG88E/G88E mouse model, ABR thresholds were elevated for all frequencies at 21 months of age, with 4 of 9 showing absent ABR. The earliest sign of elevation in these mice is at about 19 months (data not shown).
Figure 3.
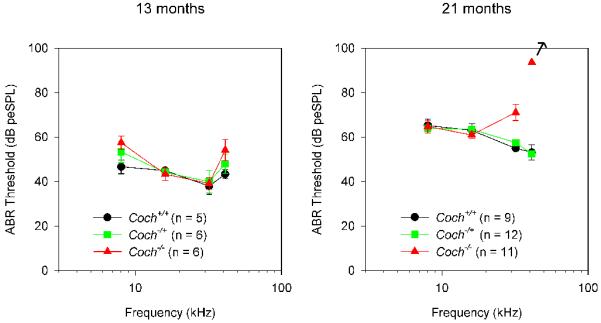

Average ABR thresholds at two ages for (A) Coch−/− and (B) CochG88E/G88E mouse models. At 13 months of age, the Coch−/− mice (n=6) showed no significant elevation of ABR thresholds in comparison to the Coch+/+ (n=5) and Coch−/+ (n=6) littermates. At 21 months, Coch−/− mice (n= 11) showed elevated ABR thresholds or absent ABRs at the highest frequency tested as compared to Coch+/+ (n=9) and Coch−/+ (n=12) littermates, with 9 of 11 Coch−/− mice showing absent ABRs at 41.2 kHz. In the CochG88E/G88E mouse model, ABR thresholds were elevated for all frequencies at 21 months of age, with 4 of 8 showing absent ABR. In contrast to Coch−/+ mice, CochG88E/+ mice show ABR threshold elevations similar to those of the CochG88E/G88E mice. Error bars represent standard error. [Color figure can be viewed in the online issue, which is available at http://www.sciencedirect.com/science/journal/03785955.]
Coch−/− mice show elevated thresholds or absent ABRs only at high frequencies (Fig. 3A), whereas CochG88E/G88E and CochG88E/+ mice show elevated or absent ABRs at all frequencies tested (Fig. 3B). Individuals with DFNA9 display elevated ABR thresholds first at higher frequencies then progressing to all frequencies. The observation of elevated ABR thresholds in all frequencies in CochG88E/G88E mice at 21 months of age may be a reflection of the late symptoms of DFNA9 where all frequencies are affected, suggesting that CochG88E/G88E mice show a more advanced degree of hearing loss than Coch−/− mice at the same age. These findings may also suggest that pathology of CochG88E/G88E manifests differently from that of Coch−/−.
Interestingly, Coch−/+ mice did not show elevation of ABR thresholds (as did Coch−/−), in contrast to CochG88E/+ mice, which show ABR threshold elevations similar to those of CochG88E/G88E. This observation is in agreement with the human DFNA9 pathology, as individuals heterozygous for the G88E mutation and the other DFNA9 mutations are all affected, reflecting the autosomal dominant mode of inheritance. Data on the two mouse models corroborate the hypothesis that DFNA9 mutations are likely to have a dominant negative effect, resulting in the observed pathology. It should be noted that the Coch−/+ mice did show vestibular malfunction (but only at the latest age tested), even though their hearing was not compromised. One possible explanation for the difference in the hearing vs. the vestibular phenotype of the Coch−/+ mice may be based largely on the fact that the age of onset of the vestibular malfunction is much earlier than that of cochlear deficits, reflecting the higher sensitivity of the vestibular system to changes in cochlin level. It should also be noted that the expressivity of vestibular malfunction is highly variable in DFNA9 individuals, as may also be the case with the mouse models tested. It is possible that Coch haploinsufficiency may also eventually affect hearing if testing were to be carried out to more advanced ages, since the onset of hearing deficits is much later that that of vestibular anomalies. We do see that the complete lack of cochlin expression does affect both hearing and vestibular status in Coch−/− mice, but likely by a different mechanism than that of Coch missense mutations. Our functional data on the knock-in mouse model, which is a more accurate recapitulation of the genetics of the human COCH mutations, still support the dominant negative effect and gain of function of these mutations.
This same observation of gain of function of Coch mutations is also supported by biochemical studies in a recent report (Yao et al., 2010). Transfection of mammalian cells with Coch missense mutation constructs resulted in secretion and misfolding, leading to dimerization of mutant cochlins, whereas transfections with the wild-type Coch did not. However, in the presence of mutant cochlin, the wild-type cochlin is “recruited” to form dimmers and eventually stable oligomers with the mutant, as in the case of the oligomerization of mutant cochlins alone.
3.2 Histopathology
Coch−/− mice at 21 months of age were evaluated histologically by osmium tetroxide staining and araldite embedding of temporal bones. Examination of Coch−/− mice in comparison to Coch+/+ mice, did not reveal any significant histological changes, despite the fact that these mice had shown significant ABR abnormalities, with absent ABRs at the highest frequencies tested, as well as vestibular deficits. Representative midmodiolar sections through the 32 kHz region of the cochlear duct are shown in Figure 4. Comparison of the wild-type and Coch−/− mutant ears shows no hair cell loss, and no obvious changes in morphology of accessory structures of the cochlear duct, including spiral ligament, stria vascularis, limbus or tectorial membrane (Figs. 4 A,B). Spiral ganglion cells and their myelinated axons are darkly stained by osmium in these thick sections: they are better seen at high power, where the cell density in the Coch−/− mutant appears normal (Figs. 4 C,D). The only cochlear histopathology in these aged ears was a scattered loss of outer hair cells (OHCs) in the basal and apical ends of the cochlea; however as quantified in Figure 5, this hair cell loss was similar in Coch−/− and wild-type ears. The semi-quantitative analysis also revealed no histopathology in the saccular or utricular sensory epithelia or ganglion.
Figure 4.

Photomicrographs comparing cochlear morphology in aged ears (21 months) from Coch−/− (A,C) and wild-type littermates (B,D). Low-power views of the cochlear duct (A,B) are from the mid-basal turn at roughly the 32 kHz location. High-power views of the spiral ganglion (C,D) are from the same sections shown in the left column. Arrows in C and D indicate spiral ganglion cells for which the nucleus and nucleolus are in focus. Scale bars in A and C apply to B and D, respectively. [Color figure can be viewed in the online issue, which is available at http://www.sciencedirect.com/science/journal/03785955.]
Figure 5.

Cytocochleograms showing scattered OHC loss in two 21-month Coch−/− ears and two age-matched wild-type littermates.
In a recent study, mouse cochleae injected with mutant cochlin oligomers (from media of transfected cells) showed loss of fibrocyte types I-IV in the spiral ligament (but not limbus), and thinning of the stria vascularis (Yao et al., 2010). These observations demonstrate cytotoxicity of mutant cochlin, but not in the exact same structures of the cochlea as in DFNA9. An ABR threshold increase of ~40 db was also observed in these mice at one week, although some had recovered at 4 weeks. These results represent consequences from a one-time delivery of a mass of mutant cochlin, whereas the mouse models represent long-term and gradual exposure to mutant cochlin.
In addition, in this same study (Yao et al., 2010) where dimerization and oligomerization of cochlins were detected on Western blots, no microscopically visible aggregates were observed. This is in contrast to the polyglutamine neurodegenerative disorder, Huntington’s disease, where expression of huntingtin in mammalian cells leads to immediate and rapid formation of highly insoluble microscopic visible aggregates. The formation of aggregates in DFNA9 is likely to have much slower kinetics, as suggested by the late-onset and progressive nature of the disorder.
Our previous study also showed a lack of deposits in CochG88E/G88E mice (Robertson et al., 2008). In this case, it is possible that these microfibrillar deposits have not yet formed to the degree of visibility and are at a lower level than detectable even at an advanced age in the mouse. It is also possible that a longer duration of time, (end-stage of human life-span), rather than the much shorter life-span of the mouse is necessary for the formation of the deposits. Coch−/− mice also did not show any fibrillar deposits characteristic of DFNA9. This observation is not surprising because the Coch−/− mouse model does not provide an exact parallel to DFNA9 pathology caused by missense mutations and their possible deleterious gain of function.
Lack of detection of any gross morphological changes in the Coch−/− mouse is a similar finding to that of the CochG88E/G88E mouse model (Robertson et al., 2008), and similar to other examples of mutant mouse models with elevated ABR thresholds that do not show detectable histological abnormalities (Maison et al., 2006; Maison et al., 2010). Our studies suggest that structural anomalies may not be detectable at a visible level, despite presence of overt symptoms, suggesting that pathology may be due to events occurring on a molecular level. In order to pursue this possibility, studies are underway through use of expression and antibody microarrays of both the Coch−/− and CochG88E/G88E mice as compared to age-matched wild-type mice, to look for differences in mRNA expression as well as protein content. Furthermore, these studies will also potentially reveal proteins that may interact with cochlin and may be involved in the same functional pathways.
3.3 Late-onset hearing loss mouse models
Many mouse models of age-related hearing loss (ARHL) exist (recently reviewed in Noben-Trauth et al., 2009). In general, the only commonality among all models is that the animals are born hearing but lose hearing as they age. In some cases, the onset and progression of hearing loss is early and fast, such as in strains with a knockout of Kcnq4 (Kharkovets et al., 2006) or Col9a1 (Suzuki et al., 2005), which begin to show hearing loss by one month of age. In other ARHL strains, measurable hearing loss does not begin for several months, such as strains with a Polg mutation (Kujoth et al., 2005), or a Cdh23, Pcdh15 double heterozygote (Zheng et al., 2005). The onset of hearing loss in the CochG88E/G88E and Coch−/− mouse models appears to be the latest onset reported in the literature to date. Therefore, the two Coch mutant strains are uniquely positioned as models for a later onset hearing loss, more similar in time scale to that in humans (latter decades of life). In addition, none of the 20+ ARHL strains reviewed in Noben-Trauth et al., (2009) report any indication of vestibular dysfunction, whereas the two Coch models present a combination of hearing loss accompanied by vestibular dysfunction, which may also provide similarities with humans where problems of balance are also seen in advanced ages.
Acknowledgements
The authors would like to thank T. Lever, K. Mills, and J. Pierce for assistance with data collection and tissue preparation. This research was supported by the National Institutes of Health (R01 DC006443 to S.M.J., R01 DC03402 to C.C.M., and R01 DC00188 and P30 DC05209 to M.C.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baek MJ, Park HM, Johnson JM, Altuntas CZ, Jane-Wit D, Jaini R, Solares CA, Thomas DM, Ball EJ, Robertson NG, Morton CC, Hughes GB, Tuohy VK. Increased frequencies of cochlin-specific T cells in patients with autoimmune sensorineural hearing loss. J Immunol. 2006;177:4203–10. doi: 10.4049/jimmunol.177.6.4203. [DOI] [PubMed] [Google Scholar]
- Baek JI, Cho HJ, Choi SJ, Kim LS, Zhao C, Sagong BR, Kim UK, Jeong SW. The Trp117Arg mutation of the COCH gene causes deafness in Koreans. Clin Genet. 2010;77:399–403. doi: 10.1111/j.1399-0004.2009.01362.x. [DOI] [PubMed] [Google Scholar]
- Bischoff AM, Huygen PL, Kemperman MH, Pennings RJ, Bom SJ, Verhagen WI, Admiraal RJ, Kremer H, Cremers CW. Vestibular deterioration precedes hearing deterioration in the P51S COCH mutation (DFNA9): an analysis in 74 mutation carriers. Otol Neurotol. 2005;26:918–25. doi: 10.1097/01.mao.0000185048.84641.e3. [DOI] [PubMed] [Google Scholar]
- Boulassel MR, Tomasi JP, Deggouj N, Gersdorff M. COCH5B2 is a target antigen of anti-inner ear antibodies in autoimmune inner ear diseases. Otol Neurotol. 2001;22:614–8. doi: 10.1097/00129492-200109000-00009. [DOI] [PubMed] [Google Scholar]
- Collin RW, Pauw RJ, Schoots J, Huygen PL, Hoefsloot LH, Cremers CW, Kremer H. Identification of a novel COCH mutation, G87W, causing autosomal dominant hearing impairment (DFNA9) Am J Med Genet A. 2006;140:1791–4. doi: 10.1002/ajmg.a.31354. [DOI] [PubMed] [Google Scholar]
- de Kok YJM, Bom SJH, Brunt TM, Kemperman MH, van Beusekom E, van der Velde-Visser SD, Robertson NG, Morton CC, Huygen PLM, Verhagen WIM, Brunner HG, Cremers CWRJ, Cremers FPM. A Pro51Ser mutation in the COCH gene is associated with late onset autosomal dominant progressive sensorineural hearing loss with vestibular defects. Hum Mol Genet. 1999;8:361–6. doi: 10.1093/hmg/8.2.361. [DOI] [PubMed] [Google Scholar]
- Fransen E, Verstreken M, Verhagen WIM, Wuyts FL, Huygen PLM, D’Haese P, Robertson NG, Morton CC, McGuirt WT, Smith RJH, Declau F, Heyning PH, Camp GV. High prevalence of symptoms of Meniere’s disease in three families with a mutation in the COCH gene. Hum Mol Genet. 1999;8:1425–9. doi: 10.1093/hmg/8.8.1425. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Bergstrom RA, Schimenti JC, Jones TA. Vestibular responses to linear acceleration are absent in otoconia-deficient C57BL/6JEi-het mice. Hear Res. 1999a;135:56–60. doi: 10.1016/s0378-5955(99)00090-8. [DOI] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Johnson KR, Yu H, Jones TA. Gravity receptor function in mice with graded otoconial deficiencies. Hear Res. 2004;191:34–40. doi: 10.1016/j.heares.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Jones SM, Subramanian G, Avniel W, Guo Y, Burkard RF, Jones TA. Stimulus and recording variables and their effects on mammalian vestibular evoked potentials. J Neurosci Methods. 2002;118:23–31. doi: 10.1016/s0165-0270(02)00125-5. [DOI] [PubMed] [Google Scholar]
- Jones TA, Jones SM. Short latency compound action potentials from mammalian gravity receptor organs. Hear Res. 1999b;136:75–85. doi: 10.1016/s0378-5955(99)00110-0. [DOI] [PubMed] [Google Scholar]
- Kamarinos M, McGill J, Lynch M, Dahl H. Identification of a novel COCH mutation, I109N, highlights the similar clinical features observed in DFNA9 families. Hum Mutat. 2001;17:351. doi: 10.1002/humu.37. [DOI] [PubMed] [Google Scholar]
- Kemperman MH, De Leenheer EMR, Huygen PLM, van Duijnhoven G, Morton CC, Robertson NG, Cremers FPM, Kremer H, Cremers CWRJ. Audiometric, vestibular, and genetic aspects of a DFNA9 family with a G88E COCH mutation. Otol Neurotol. 2005;26:926–33. doi: 10.1097/01.mao.0000185062.12458.87. [DOI] [PubMed] [Google Scholar]
- Kharkovets T, Dedek K, Maier H, Schweizer M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T, Jentsch TJ. Mice with altered KCNQ4 K+ channels implicate sensory outer hair cells in human progressive deafness. Embo J. 2006;25:642–52. doi: 10.1038/sj.emboj.7600951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–4. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- Maison SF, Rosahl TW, Homanics GE, Liberman MC. Functional role of GABAergic innervation of the cochlea: phenotypic analysis of mice lacking GABA(A) receptor subunits alpha 1, alpha 2, alpha 5, alpha 6, beta 2, beta 3, or delta. J Neurosci. 2006;26:10315–26. doi: 10.1523/JNEUROSCI.2395-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maison SF, Liu XP, Vetter DE, Eatock RA, Nathanson NM, Wess J, Liberman MC. Muscarinic signaling in the cochlea: presynaptic and postsynaptic effects on efferent feedback and afferent excitability. J Neurosci. 2010;30:6751–62. doi: 10.1523/JNEUROSCI.5080-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima T, Rodriguez CI, Robertson NG, Morton CC, Stewart CL, Griffith AJ. Targeted disruption of mouse Coch provides functional evidence that DFNA9 hearing loss is not a COCH haploinsufficiency disorder. Hum Genet. 2005;118:29–34. doi: 10.1007/s00439-005-0001-4. [DOI] [PubMed] [Google Scholar]
- Muller M, von Hunerbein K, Hoidis S, Smolders JW. A physiological place-frequency map of the cochlea in the CBA/J mouse. Hear Res. 2005;202:63–73. doi: 10.1016/j.heares.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Nagy I, Horvath M, Trexler M, Repassy G, Patthy L. A novel COCH mutation, V104del, impairs folding of the LCCL domain of cochlin and causes progressive hearing loss. J Med Genet. 2004;41:e9. doi: 10.1136/jmg.2003.012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noben-Trauth K, Johnson KR. Inheritance patterns of progressive hearing loss in laboratory strains of mice. Brain Res. 2009;1277:42–51. doi: 10.1016/j.brainres.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB, Jr., Miyamoto RT, Linthicum FH, Jr., Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nature Genet. 1998;20:299–303. doi: 10.1038/3118. [DOI] [PubMed] [Google Scholar]
- Robertson NG, Jones SM, Sivakumaran TA, Giersch AB, Jurado SA, Call LM, Miller CE, Maison SF, Liberman MC, Morton CC. A targeted Coch missense mutation: a knock-in mouse model for DFNA9 late-onset hearing loss and vestibular dysfunction. Hum Mol Genet. 2008;17:3426–34. doi: 10.1093/hmg/ddn236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Street VA, Kallman JC, Robertson NG, Kuo SF, Morton CC, Phillips JO. A novel DFNA9 mutation in the vWFA2 domain of COCH alters a conserved cysteine residue and intrachain disulfide bond formation resulting in progressive hearing loss and site-specific vestibular and central oculomotor dysfunction. Am J Med Genet A. 2005;139:86–95. doi: 10.1002/ajmg.a.30980. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Asamura K, Kikuchi Y, Takumi Y, Abe S, Imamura Y, Hayashi T, Aszodi A, Fassler R, Usami S. Type IX collagen knock-out mouse shows progressive hearing loss. Neurosci Res. 2005;51:293–8. doi: 10.1016/j.neures.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Tebo AE, Szankasi P, Hillman TA, Litwin CM, Hill HR. Antibody reactivity to heat shock protein 70 and inner ear-specific proteins in patients with idiopathic sensorineural hearing loss. Clin Exp Immunol. 2006;146:427–32. doi: 10.1111/j.1365-2249.2006.03227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami S, Takahashi K, Yuge I, Ohtsuka A, Namba A, Abe S, Fransen E, Patthy L, Otting G, Van Camp G. Mutations in the COCH gene are a frequent cause of autosomal dominant progressive cochleo-vestibular dysfunction, but not of Meniere’s disease. Eur J Hum Genet. 2003;11:744–8. doi: 10.1038/sj.ejhg.5201043. [DOI] [PubMed] [Google Scholar]
- Yao J, Py BF, Zhu H, Bao J, Yuan J. Role of protein misfolding in DFNA9 hearing loss. J Biol Chem. 2010;285:14909–19. doi: 10.1074/jbc.M110.106724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan HJ, Han DY, Sun Q, Yan D, Sun HJ, Tao R, Cheng J, Qin W, Angeli S, Ouyang XM, Yang SZ, Feng L, Cao JY, Feng GY, Wang YF, Dai P, Zhai SQ, Yang WY, He L, Liu XZ. Novel mutations in the vWFA2 domain of COCH in two Chinese DFNA9 families. Clin Genet. 2008;73:391–4. doi: 10.1111/j.1399-0004.2008.00972.x. [DOI] [PubMed] [Google Scholar]
- Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130:94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QY, Yan D, Ouyang XM, Du LL, Yu H, Chang B, Johnson KR, Liu XZ. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum Mol Genet. 2005;14:103–11. doi: 10.1093/hmg/ddi010. [DOI] [PMC free article] [PubMed] [Google Scholar]