

Abstract
Adenine-DNA glycosylase MutY of Escherichia coli catalyzes the cleavage of adenine when mismatched with 7,8-dihydro-8-oxoguanine (GO), an oxidatively damaged base. The biological outcome is the prevention of C/G→A/T transversions. The molecular mechanism of base excision repair (BER) of A/GO in mammals is not well understood. In this study we report stimulation of mammalian adenine-DNA glycosylase activity by apurinic/apyrimidinic (AP) endonuclease using murine homolog of MutY (Myh) and human AP endonuclease (Ape1), which shares 94% amino acid identity with its murine homolog Apex. After removal of adenine by the Myh glycosylase activity, intact AP DNA remains due to lack of an efficient Myh AP lyase activity. The study of wild-type Ape1 and its catalytic mutant H309N demonstrates that Ape1 catalytic activity is required for formation of cleaved AP DNA. It also appears that Ape1 stimulates Myh glycosylase activity by increasing formation of the Myh–DNA complex. This stimulation is independent of the catalytic activity of Ape1. Consequently, Ape1 preserves the Myh preference for A/GO over A/G and improves overall glycosylase efficiency. Our study suggests that protein–protein interactions may occur in vivo to achieve efficient BER of A/GO.
INTRODUCTION
7,8-Dihydro-8-oxoguanine (GO) is a common oxidative DNA lesion generated by direct modification via reactive oxygen species or by incorporation of d(GO)TP during DNA synthesis. GO lesions are mutagenic and can mispair with adenine during DNA replication (1–3). If the resulting A/GO is not repaired before the next round of DNA replication, a C/G→A/T transversion occurs and the opportunity for repair is lost. Defects in the A/GO repair system of Escherichia coli lead to a mutator phenotype (4,5).
In E.coli A/GO is repaired via base excision repair (BER), which is initiated by the DNA repair enzyme adenine-DNA glycosylase (MutY). MutY catalyzes hydrolysis of the N-glycosylic bond of the mispaired adenine base, generating an apurinic/apyrimidinic (AP) site opposite the GO base (AP/GO) (6). Subsequent steps to repair the AP site generated by MutY are initiated by another BER enzyme, AP endonuclease (7–9). AP endonuclease cleaves the sugar–phosphate backbone at the AP site yielding a 5′-deoxyribose 5-phosphate and a 3′-hydroxyl nucleotide residue. This abnormal 5′-abasic residue is later removed and the gap is repaired by other components, recreating the original state of the DNA and completing BER for A/GO. In E.coli the two major AP endonucleases, endonuclease IV (10,11) and exonuclease III (12,13), belong to two different conserved families.
In vitro biochemical studies have demonstrated that the purified MutY protein can cleave undamaged adenine from both A/GO and A/G mispairs (6,14,15). However, each shows distinctly different glycosylase kinetics (16–18). With A/GO substrate MutY causes an initial burst of product formation followed by very slow turnover. In contrast, with A/G substrate MutY lacks the initial burst of product formation and displays a more rapid turnover. In the case of A/GO the slow turnover that MutY exhibits is due to enhanced binding to GO-containing DNA, which dramatically slows down release of the product AP/GO. Physiologically, this tight binding may prevent events that would otherwise lead to the loss of genetic information on both DNA strands. The distinct glycosylase kinetics and binding properties of E.coli MutY for A/GO- versus A/G-containing substrates provide some biochemical evidence to suggest that A/GO is its biologically relevant substrate. This is also consistent with genetic studies (4). The 39 kDa adenine-DNA glycosylase MutY contains two functional domains, an N-terminal catalytic domain p26 and C-terminal domain p13, which can be separated by controlled trypsin proteolysis (19). Studies of p26 domain crystal structures demonstrated that the mechanism for adenine removal is via nucleotide flipping (20). The C-terminal domain, p13, is responsible for the distinct glycosylase kinetics and binding properties for A/GO- versus A/G-containing substrates (17,18).
MutY also possesses a residual uncoupled and controversial AP lyase activity (21–24), which results in strand breaks 3′ to the AP site, leaving a 3′-terminal unsaturated sugar derivative. Recent studies by Williams and David (23), using a quantitative glycosylase/lyase assay, have shown that the AP lyase activity of MutY is ∼10-fold less than its adenine glycosylase activity (23). These levels are similar to the known monofunctional enzyme uracil-DNA glycosylase (23), giving further support for the monofunctional nature of MutY. However, it was also shown that, unlike the monofunctional uracil-DNA glycosylase, MutY is capable of forming a Schiff base intermediate, which is characteristic of DNA glycosylases catalyzing a concomitant β-lyase reaction (22–25).
MutY homologs have been identified in Schizosaccharomyces pombe (SpMYH) and human (hMYH) (26–31), suggesting that BER is also involved in the repair of A/GO lesions in higher eukaryotic organisms. In humans an AP endonuclease-mediated repair process presumably completes BER of A/GO lesions. AP endonuclease, a homolog of E.coli exonuclease III, has been identified in bovine, murine and human cells (7). The crystal structure of the major human AP endonuclease, Ape1 (also called HAP1, APEX and REF1), reveals the mechanism for recognition and cleavage of AP DNA by the 35 kDa monomeric protein (32,33). Two alternative repair pathways have been demonstrated in an in vitro reconstituted system for the repair of AP lesions using human repair enzymes (34). The first pathway is DNA polymerase β-dependent and involves Ape1, DNA polymerase β and either DNA ligase I (35) or DNA ligase III (36). The second pathway is proliferating cell nuclear antigen (PCNA)-dependent (37,38) and involves Ape1, replication factor C, PCNA, DNA polymerase δ or ɛ, flap endonuclease and DNA ligase I. It is not clear which AP repair pathway is involved in BER of A/GO. Recently coupling between Ape1 and DNA-N-glycosylases in repair systems has been studied in vitro using human thymine-DNA glycosylase and human uracil-DNA glycosylase (39,40). In the case of both enzymes it was shown that Ape1 stimulates the glycosylase activity and proposed that it cleaves the AP site after displacement of the glycosylase. So far the effect of AP endonuclease on the adenine-DNA glycosylase has not been studied.
Our efforts to elucidate the role of adenine-DNA glycosylase in carcinogenesis have been two-fold: creating an animal knockout model and characterization of the molecular mechanism of the A/GO repair process. As a first step towards in vitro reconstitution of the mammalian A/GO repair process we looked for a functional interaction between murine adenine-DNA glycosylase (Myh) and AP endonuclease, which is likely the next step in BER of an A/GO lesion. We utilized Ape1 in this study due to its close homology to the murine homolog Apex (94% amino acid identity; 41) and previous characterization of both wild-type and mutant proteins (42). We demonstrate that Ape1 not only cleaves the AP DNA generated by Myh, but also stimulates Myh glycosylase activity by enhancing formation of Myh–DNA complexes. As a consequence of their individual enzyme attributes, Myh and Ape1 in concert preserve the substrate preference for A/GO and improve the efficiency of the glycosylase activity. Our data suggest that an interaction between the glycosylase and AP endonuclease may occur in vivo to facilitate mammalian BER of A/GO.
MATERIALS AND METHODS
Identification and cloning of Myh
The hMYH cDNA sequence (27) was used to search the NCBI non-redundant database. Two murine cDNA sequences (accession nos AA409964 and AA409965) with substantial sequence homology to the hMYH gene were identified. Primers made from these two sequences were used to amplify the Myh cDNA sequence by PCR from a murine liver cDNA library (Stratagene, La Jolla, CA). The PCR products were cloned using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA) and sequenced using a Sequitherm Cycle Sequencing Kit (Epicentre Technologies, Madison, WI). The 5′-end of the Myh sequence was amplified by rapid amplification of cDNA ends (5′-RACE) using Marathon Ready cDNA libraries from liver and testis (Clontech, Palo Alto, CA). The PCR products were then cloned using a TOPO TA cloning kit (Invitrogen) and sequenced using a Sequitherm Cycle Sequencing Kit (Epicentre Technologies). The above experiments were performed according to the manufacturers’ protocols.
Generation of rabbit polyclonal antibodies to Myh
A truncated Myh protein (Ser104–Pro316) was subcloned between BamHI and SalI sites of pQE30 (Qiagen, Chatsworth, CA) for the purpose of generating antibodies against Myh. This truncated protein was expressed as inclusion bodies following isopropyl β-d-thiogalactoside (IPTG) induction, purified (43) and then dissolved by boiling in phosphate-buffered saline (PBS) containing 0.1% SDS for 5 min. The dissolved truncated protein was used to generate rabbit polyclonal antibodies (Covance, Denver, PA).
To prepare the polyclonal antibody for western blot analysis, the antibody (1 µl) was first incubated on ice for 1 h with 50 mg E.coli CC104mutY acetone powder in 5 ml TTBS (20 mM Tris–HCl, pH 7.6, 137 mM NaCl and 0.1% Tween 20). This was followed by centrifugation at 4°C for 30 min to obtain a clear suspension. Usually a 1:10 000 dilution of the antibody was used in the western blots.
Expression in E.coli and purification of Myh and Ape1
The Myh cDNA, lacking the first 28 N-terminal amino acids, was subcloned into the pQE30 vector (Qiagen) at the BamHI site. Transformants of E.coli CC104mutY/pREP4 with pQE30Myh were grown at 37°C in 100 ml of LB medium with 200 µg/ml ampicillin and 25 µg/ml kanamycin. When growth reached an A600 of 0.5, the 100 ml cell culture was used to inoculate 2 l of LB medium with 200 µg/ml ampicillin, 25 µg/ml kanamycin and 10 µM IPTG. This culture was harvested after overnight growth. The bacterial lysate was prepared in a French press (16 000 p.s.i.) in buffer I (20 mM potassium phosphate, pH 7.6, 10% glycerol and 0.5 mM PMSF). After centrifugation the clear lysate was poured onto an SP Sepharose column. The column was washed with 0.1 M KCl in buffer I and was eluted with 0.5 M KCl in buffer I. The peak Myh-containing fractions, as determined by western blot, were combined. The KCl concentration was adjusted to 0.3 M by addition of buffer I. This was poured onto a Ni2+–NTA column. The column was washed with buffer I plus 0.3 M KCl and 10 mM imidazole and then eluted with buffer I plus 0.3 M KCl with 0.2 M imidazole. The fractions were analyzed by western blot and peak fractions containing Myh were combined and dialyzed overnight in buffer II (50 mM Tris–HCl, pH 7.6, 30 mM KCl, 1 mM EDTA, 1 mM DTT and 50% glycerol). A clear protein sample was obtained after centrifugation of the dialyzed sample and stored at –80°C in small aliquots. The protein concentration was determined to be 0.887 mg/ml by Bio-Rad Protein Assay (Bio-Rad, Hercules, CA).
The APE1 open reading frame (ORF) was PCR amplified with Pfu polymerase using plasmid pCW26 (44) as template. APE1 was subcloned into pET28b (Novagen, Madison, WI) behind the sequence for the hexa-histidine tag using BamHI and XhoI sites at the 5′- and 3′-ends of the ORF, respectively, to generate pET28b-APE1. The construct was verified by DNA sequencing. To purify histidine-tagged Ape1 protein, overnight cultures of E.coli BL21(DE3) harboring vector pET28b-APE1 were used to inoculate 2 l of LB supplemented with 30 µg/ml kanamycin. Protein expression was induced by addition of IPTG to 400 µM during mid log phase, followed by incubation for 3 h at 37°C. The cell pellet was washed once with PBS and then resuspended in 40 ml of buffer A (50 mM HEPES–KOH, pH 7.5, 0.5 M KCl, 10% glycerol, 0.1 mM EDTA, 0.1 mM DTT and 1 mM imidazole) which contained 1 mM PMSF, 2 µg/ml aprotinin, 2 µg/ml leupeptin and 1 µg/ml pepstatin A. Cells were lysed by two passages through a French press cell. After clarification by centrifugation at 30 000 g for 30 min the supernatant was loaded onto a 10 ml Ni+2–NTA column (Qiagen) and washed with 10 column volumes each of buffer A supplemented with 1 and 20 mM imidazole. Proteins were eluted with a linear gradient of 20–100 mM imidazole. Peak fractions were pooled, dialyzed against buffer B (50 mM HEPES–KOH, pH 7.5, 100 mM KCl, 10% glycerol, 1 mM EDTA and 0.1 mM DTT) and loaded onto a Mono-S HR 5/5 column (Pharmacia, Piscataway, NJ). After washing with 10 column volumes of buffer B the proteins were eluted with a linear gradient of 100–250 mM KCl. Peak fractions were pooled and dialyzed against buffer B for storage at –80°C. Ape1, at a final concentration of 0.5 mg/ml as determined by Bio-Rad Protein Assay, was purified to >94% homogeneity.
Escherichia coli MutY was purchased from Trevigen (Gaithersburg, MD). Ape1 mutant H309N was purified in its native form as described by Masuda et al. (42).
Glycosylase activity assay
Double-stranded DNA substrates (96mer) containing A/GO, A/G or T/GO mispairs at position 60 were prepared in an annealing buffer containing 70 mM Tris–HCl (pH 7.6) and 10 mM MgCl2, as described previously (45). The A-containing strand was 5′-32P-end-labeled.
The glycosylase activity assay was carried out essentially as described previously (6) except that EDTA was omitted from the reaction buffer. The standard reaction mixture contained 20 mM Tris–HCl (pH 7.6), 50 µg/ml bovine serum albumin (BSA), 1 µl of 32P-labeled 96mer double-stranded DNA substrate (final concentration 0.2 nM unless otherwise noted), and 2 µl of purified recombinant Myh in a total volume of 20 µl. In the reactions with no Myh, 2 µl of buffer was added. The glycosylase activity assay was carried out at 37°C over the indicated durations. Unless otherwise stated, the reaction products were then treated with 4 µl of 1 N NaOH and heated at 90°C for 4 min to completely convert the AP/GO (apurinic site opposite GO) product to cleaved product prior to electrophoresis. The reaction products were mixed with 8 µl of loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue and 0.05% xylene cyanol FF), heated at 94°C for 1 min and finally analyzed on a denaturing 15% polyacrylamide gel. Gels were dried and visualized by a PhosphorImager 445 SI (Sunnyville, CA), and quantified by Molecular Dynamics ImageQuaNT v. 4.2a. The percent cleaved product was determined by dividing the intensity of the cleaved product band by the total intensity. Total intensity is the sum of the intensities of the intact band and the cleaved product band minus the background intensity (obtained from blank areas of identical size).
The experiments with Ape1 protein, its catalytic mutant H309N and BSA (New England Biolabs, Beverly, MA) were done under the conditions described above but with an additional 40 mM NaCl. An aliquot of 2 µl of Ape1, H309N or BSA of the desired concentration was added to the reaction mixture to a final volume of 20 µl. In the reactions with no Ape1, H309N or BSA, 2 µl of buffer was added instead.
In time course experiments 20 µl samples were taken at the times indicated in the figure legends. The reaction was stopped either by addition of loading buffer and cooling to 4°C or by addition of 4 µl of 1 N NaOH and incubation at 90°C for 4 min followed by addition of loading buffer. Heating and gel running conditions were the same as for other glycosylase reactions.
Electrophoretic mobility shift assay (EMSA)
The Myh–DNA complex was studied using EMSA. The glycosylase reaction mixture was split into two aliquots at the beginning of the reaction. After the glycosylase reaction, one aliquot was used for EMSA and the other for glycosylase analysis (as described above). The EMSA samples were immediately run on an 8% native polyacrylamide gel at 100 V (14 V/cm) until the first dye front reached the bottom. The 8% gel was equilibrated at 37°C and run for 5 min prior to loading of the samples. The EMSA gel was dried, visualized and quantified with a PhosphorImager using the same procedure as described above.
To generate a product of free cleaved AP/GO (cAP/GO), 0.5 nM Myh was incubated with 0.2 nM A/GO substrate in the presence of 50 nM Ape1 at 37°C for 30 min. The free cAP/GO DNA could then be converted to a Myh–cAP/GO bound form by adding increasing amounts of Myh and incubating the reaction at 37°C for an additional 15 min. The reaction mixture was then analyzed on an 8% native polyacrylamide gel equilibrated at 37°C.
RESULTS
Cloning and purification of the murine MutY homolog Myh
The composite PCR sequence contains an ORF coding for a murine homolog of adenine-DNA glycosylase (type 2) (29). It has the alternative N-terminal sequence that begins with a second methionine residue (Fig. 1). We designated it Myh (type 2). The putative Myh shares 78% amino acid identity and 86% similarity with hMYH, as determined by BestFit (46). Both Myh and hMYH contain a putative PCNA-binding sequence (QxxLxxFF; 47) (Fig. 1).
Figure 1.

Alignment of Myh protein with its homologs, hMYH, SpMYH and E.coli MutY. Adenine-DNA glycosylase sequences (protein symbol and GenBank accession no. in parentheses): Myh of Mus musculus (mMYH, AY007717); hMYH of Homo sapiens (hMYH, U63329); SpMYH of S.pombe (SpMYH, AF053340); MutY of E.coli (EcMutY, P17802). The conserved amino acid residues among these four proteins are shaded gray. The strictly conserved amino acid residues among these four proteins are shaded black. The conserved motifs (20) are indicated. The four Cys residues for binding of the [4Fe-4S] cluster are marked with asterisks. The strictly conserved Asp residue, which is critical for catalytic activity, is marked with a dot. The putative PCNA binding sequence (QxxLxxFF; 46) is boxed. The amino acid residue Ser29 is marked with a triangle and marks the beginning of the recombinant Myh (see text).
We expressed the recombinant Myh starting at Ser29 (Fig. 1). This eliminates four rare Arg codons (AGA and AGG, coding for Arg9, Arg20, Arg21 and Arg22), which in turn facilitates better expression of the recombinant protein. Low expression levels of hMYH, which contains several rare arginine codons, were encountered previously (28). The hexa-histidine-tagged recombinant Myh, expressed in E.coli mutY, was purified by SP Sepharose and Ni2+–NTA chromatographic separation (Fig. 2). The recombinant Myh was purified to near homogeneity with an estimated molecular weight in close agreement with the predicted size of 56 kDa. The purified fraction also contained ∼10% proteolytic degraded fragments of ∼40 kDa. The yellow color in the Myh-containing fraction (data not shown) was consistent with the presence of a [4Fe-4S] cluster in the protein.
Figure 2.
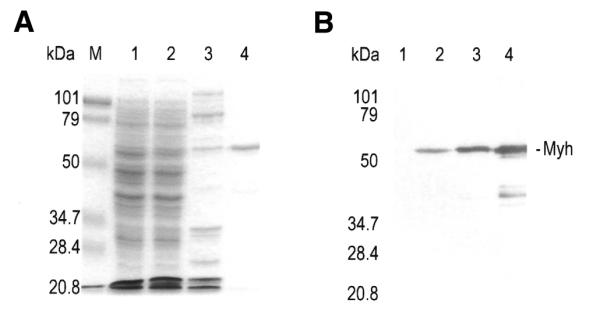
Expression and purification of Myh protein. The 10% SDS–polyacrylamide gel contains the following samples: lysate of CC104mutY/pREP4 cells containing pQE30 (lane 1) or pQE30/Myh (lane 2) after induction of Myh protein expression by IPTG; fraction eluted from an SP Sepharose column in the presence of 0.5 M KCl (lane 3); Myh protein eluted from a Ni2+–NTA column in the presence of 0.2 M imidazole (lane 4). The gel was visualized by Coomassie blue staining (A) and by western blot using polyclonal anti-Myh antibody (B). Myh protein is indicated on the right. Lane M contains molecular mass standards (Bio-Rad) as indicated on the left.
Ape1 cleavage of AP DNA generated by Myh
Before we investigated Ape1 cleavage of AP sites generated by Myh, we first needed to determine whether Myh possessed an AP lyase activity of its own. The procedure was similar to that reported for E.coli MutY (23). Figure 3 shows that in the presence of a saturating concentration of Myh, ∼80% of A/GO substrate was converted to glycosylase product within the first 5 min of the reaction. This was measured by the amount of cleaved product (cAP/GO) in alkaline-treated samples. In contrast, without alkaline treatment <10% of the substrate was converted to cAP/GO, with no significant increase over 60 min. These results suggest that Myh lacks efficient AP lyase activity, resulting in the majority of the product remaining in the intact (uncleaved) AP/GO form. Similar results were obtained with the A/G substrate (data not shown).
Figure 3.

Lack of efficient Myh AP lyase activity. The glycosylase reaction was carried out at 37°C with 0.2 nM A/GO substrate and 5 nM Myh. Samples were taken at different time points as indicated, with or without alkaline treatment, and were analyzed on a 15% denaturing polyacrylamide gel. (A) Phosphoimage of the reaction. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left. An asterisk indicates the 5′-end-labeled strand. (B) Plot of the fraction cleaved versus time.
We next addressed whether Ape1 can cleave Myh-generated AP sites. To determine if the catalytic activity of Ape1 is responsible, the catalytic mutant H309N was also used (42). The H309N protein has ∼10–4 of the wild-type catalytic activity, but is still capable of binding to AP DNA (data not shown; 42). The reactions in Figure 4 were performed with a saturating concentration of Myh with A/GO substrate and in the absence of alkaline treatment. They show that the combination Myh/Ape1 produced cAP/GO, whereas Myh/H309N was unable to produce the cleaved product. Therefore, Ape1 can cleave AP/GO generated by Myh and its catalytic function is likely the mechanism of cleavage rather than stimulation of potential Myh AP lyase activity.
Figure 4.

Cleavage of AP DNA by Ape1 and its catalytic mutant H309N. The glycosylase reactions were carried out at 37°C for 30 min with 0.2 nM A/GO substrate, 5 nM Myh and increasing amounts of Ape1 as indicated. Then each reaction mixture, without alkaline treatment, was analyzed on a 15% denaturing polyacrylamide gel. A plot of the fraction cleaved by wild-type Ape1 or catalytic mutant H309N versus concentration of Ape1 protein is shown.
Ape1 stimulation of Myh glycosylase activity
Since substrate preference has been seen with E.coli MutY and other MutY homologs, we wanted to see if it was also present in the Myh/Ape1 system. First, substrate preference with Myh alone was determined by analysis of glycosylase product formation with both A/GO and A/G substrates (Fig. 5A). Samples were treated with alkaline solution to completely cleave the AP products prior to electrophoresis. Approximately 3 nM Myh was required to achieve maximum cleavage of the A/GO substrate. We consistently observed higher adenine glycosylase activity on the A/GO substrate than on the A/G substrate over the Myh concentration range tested. We concluded that Myh alone showed a preference for A/GO over A/G.
Figure 5.


Substrate preference of Myh and Myh/Ape1. Substrate DNA (0.2 nM) was incubated with Myh alone or Myh/Ape1 at a constant molar ratio of 1:100. The glycosylase reaction was carried out at 37°C for 30 min. Then each reaction mixture, with alkaline treatment, was analyzed on a 15% denaturing polyacrylamide gel. An asterisk indicates the 5′-end-labeled strand. (A) Plot of the fraction of glycosylase product versus Myh. Each individual point is represented with error bars as an average of data from two to four experiments. (B) Plot of the fraction of glycosylase product versus Myh/Ape1 (only Myh concentrations are indicated; Ape1 concentrations can be calculated based on a constant 1:100 molar ratio).
The substrate preference of Myh/Ape1 for A/GO and A/G was then determined and compared with the substrate preference observed for Myh alone (Fig. 5B). The total amounts of glycosylase product after alkaline treatment were analyzed with increasing amounts of Myh alone or Myh/Ape1, where the latter was maintained at a constant molar ratio of 1:100. In the range 0–1 nM Myh we observed that Ape1 significantly stimulated glycosylase activity on the A/GO substrate. This stimulation was less significant with the A/G substrate. The preference for A/GO over A/G with Myh/Ape1 was ∼10:1 and was similar to the preference seen with Myh alone. Overall, Ape1 maintains the Myh substrate preference while stimulating glycosylase activity.
In order to determine whether the catalytic activity of Ape1 is required for glycosylase stimulation, the catalytic mutant H309N was tested (Fig. 6). Myh alone, Myh/50 nM Ape1 or Myh/50 nM H309N were incubated with A/GO substrate in the range 0–0.625 nM Myh. The total amount of glycosylase product was analyzed after alkaline treatment. Myh/50 nM BSA was also included as a non-specific protein control. Increased glycosylase product was seen for both Myh/Ape1 and Myh/H309N over Myh alone and both showed a greater increase than that seen with Myh/BSA. At 0.5 nM Myh an increase in cleavage of 40–45% was observed with Myh/Ape1 and Myh/H309N, compared with only 5% seen with Myh/BSA. The fact that the Ape1 catalytic mutant H309N retains the ability to stimulate Myh glycosylase activity on the A/GO substrate suggests that protein–protein interaction between Myh and Ape1 may be the mechanism of stimulation. This concept will be discussed later in this paper. The stimulation of Myh glycosylase activity by increasing concentrations of Ape1 is shown in Figure 7. The highest concentration of Ape1 resulted in a product level similar to that produced by Myh alone at a 10-fold higher concentration (Fig. 7, lanes 3 and 9).
Figure 6.

Glycosylase stimulation by Ape1 and H309N. The glycosylase reactions were carried out at 37°C for 30 min with 0.2 nM A/GO substrate and increasing concentrations of Myh in the presence of 50 nM wild-type Ape1 (wt Ape1), 50 nM catalytic mutant H309N (H309N mutant), 50 nM BSA or buffer. Then each reaction mixture, with alkaline treatment, was analyzed on a 15% denaturing polyacrylamide gel. A plot of the fraction of glycosylase product versus [Myh] is shown.
Figure 7.
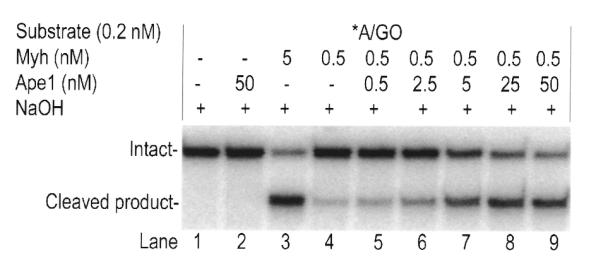
Effect of Ape1 concentration on glycosylase stimulation. The glycosylase reactions were carried out at 37°C for 30 min with 0.2 nM A/GO and 0.5 nM Myh in the presence of increasing amounts of Ape1. 5 nM Myh is also included. Then each reaction mixture, with alkaline treatment, was analyzed on a 15% denaturing polyacrylamide gel. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left. An asterisk indicates the 5′-end-labeled strand.
Kinetics of A/GO cleavage in the presence of Myh/Ape1
Accumulation of both glycosylase product and cleaved product was monitored simultaneously over the course of 60 min reactions containing Myh, Myh/Ape1 or Myh/H309N with A/GO substrate (Fig. 8). Figure 8A shows accumulation of glycosylase product (AP/GO). At 0.5 nM Myh generated only 16% AP/GO within the first 10 min and this level was maintained over the course of the 60 min reaction. However, 0.5 nM Myh/5 nM Ape1 and 0.5 nM Myh/5 nM H309N generated 55 and 42% AP/GO, respectively, within the first 10 min and also maintained this level over the course of the 60 min reaction. Figure 8B shows accumulation of the cleaved product (cAP/GO). cAP/GO, which is only seen with Myh/Ape1, gradually accumulated over the course of the 60 min reaction. The kinetics of Myh alone (Fig. 8A) are similar to those shown for E.coli MutY (16) and are due to tight binding of Myh to its glycosylase product AP/GO (17,18). The stimulation of glycosylase activity with Myh/Ape1 and Myh/H309N takes place at the beginning of the reaction, providing a significant amount of AP/GO product. Therefore, it seems that the rate limiting step in the formation of cAP/GO could be either release of the AP/GO product by Myh or the endonuclease activity of Ape1, but not the glycosylase activity of Myh.
Figure 8.


Accumulation of the glycosylase product AP/GO (A) and the cleaved product cAP/GO (B) over time. The glycosylase reactions were carried out at 37°C with 0.2 nM A/GO substrate and 0.5 nM Myh in the presence of 5 nM wild-type Ape1 (wt Ape1), 5 nM catalytic mutant (H309N mutant) or buffer. Samples were taken at different time points as indicated, with or without alkaline treatment, and were analyzed on a 15% denaturing polyacrylamide gel. (A) Plot of the fraction of glycosylase product AP/GO versus time. Individual points represented with error bars are the average from two experiments. (B) Plot of the fraction of cleaved product cAP/GO versus time.
Protein–DNA complexes in the presence of Myh/Ape1
We looked at protein–DNA interactions using EMSA to help explain the stimulation of glycosylase activity with the Myh/Ape1 system. The reactions were carried out at 37°C for 10 min with 0.5 nM Myh alone, 0.5 nM Myh/50 nM Ape1, 0.5 nM Myh/50 nM H309N or 0.5 nM Myh/50 nM BSA. Formation of protein–DNA complexes was analyzed (Fig. 9A). Simultaneously, the glycosylase reaction products were monitored (Fig. 9B). While no protein–DNA complexes were observed in the reactions containing only Ape1 or H309N (Fig. 9A, lanes 2–4), increased amounts of the Myh–DNA complex were observed in the Myh/Ape1 and Myh/H309N reactions when compared to Myh alone (Fig. 9A, lanes 5–7). These increases correlated with the increase in glycosylase products in the corresponding samples (Fig. 9B, lanes 5–7). Ape1 stimulation of Myh–DNA complex formation was further demonstrated using a non-cleavable DNA substrate, T/GO (18,29). Figure 10 shows that while Ape1 alone does not bind to T/GO, increased binding of Myh to T/GO was achieved in the presence of Ape1 (Fig. 10, lanes 7–9). A similar stimulatory effect was also observed in the presence of the catalytic mutant H309N (Fig. 10, lanes 12–14), but was not seen in the presence of BSA (Fig. 10, lanes 2–4). Cleavage of T/GO was monitored over the course of the experiment and no cleavage was detected (data not shown). A double-stranded DNA containing C/GO was also used as a non-specific DNA control and no Myh–DNA complex was observed under these experimental conditions (data not shown). These results show that Ape1, even in its non-catalytic state, can stimulate formation of Myh–DNA complexes and suggest that this may be the mechanism of glycosylase stimulation.
Figure 9.
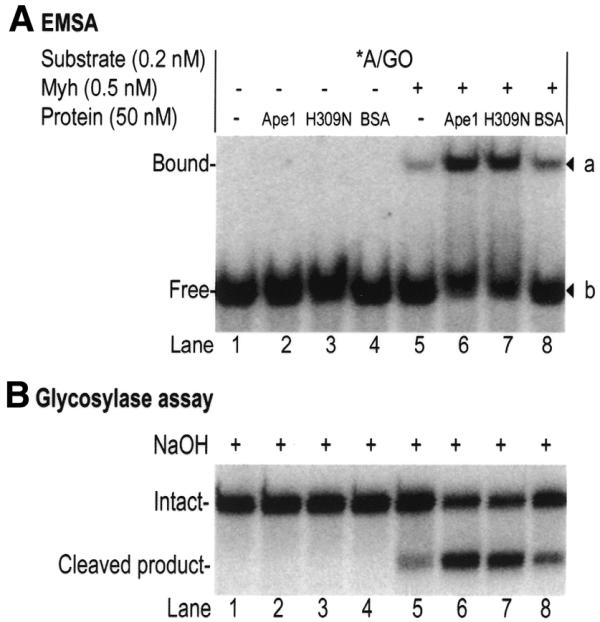
Stimulation of Myh–DNA complex formation by Ape1 and H309N. The glycosylase reactions were carried out at 37°C for 10 min with 0.2 nM A/GO substrate and 0.5 nM Myh in the presence of 50 nM wild-type Ape1 (wt Ape1), 50 nM catalytic mutant H309N (H309N mutant), 50 nM BSA or buffer. One aliquot of each reaction was directly analyzed by EMSA on an 8% native polyacrylamide gel. The other aliquot, with alkaline treatment, was analyzed on a 15% denaturing polyacrylamide gel. An asterisk indicates the 5′-end-labeled strand. (A) Phosphoimage of Myh–DNA complexes. The observed bands a and b are marked on the right. The bound and free DNA forms are indicated on the left. (B) Phosphoimage of accumulation of glycosylase product. The intact 96mer DNA (Intact) and cleaved 60mer product (Cleaved product) are indicated on the left.
Figure 10.

Stimulation of Myh binding to non-cleavable T/GO substrate by Ape1 and H309N. The binding reactions were carried out at 37°C for 10 min with 50 pM T/GO DNA in the presence of Myh, Myh/Ape1, Myh/H309N or Myh/BSA at the concentrations indicated. The binding products were analyzed by EMSA on an 8% native polyacrylamide gel. An asterisk indicates the 5′-end-labeled strand. The observed bands a and b are marked on the right. The bound and free DNA forms are indicated on the left.
In the above reactions we did not observe formation of an Ape1–DNA complex, which has a faster electrophoretic mobility than the Myh–DNA complex (data not shown). Nor did we observe a Myh–Ape1–DNA tertiary complex. However, the amounts of cleaved product cAP/GO generated within the first 10 min of the reaction were minimal (Fig. 8B) and perhaps too low for these other complexes to form. Therefore, we studied the protein–DNA interaction under conditions of excess cAP/GO. EMSA was then performed after a 30 min reaction using the same samples as in Figure 7 but without alkaline treatment (Fig. 11A). Accumulation of free cleaved product cAP/GO (band bb), which migrates slightly slower than the free intact DNA (band b) (40), was observed in the presence of increasing amounts of Ape1. Accumulation of Myh–DNA complex (band a) was also observed, which may be due to the ability of Ape1 to enhance Myh–DNA complex formation. Even under these conditions, where significant amounts of cAP/GO DNA are formed, we still did not observe formation of an Ape1–DNA or a tertiary complex. The inability to observe an Ape1–DNA complex allows us to speculate that it may be unstable under our conditions. This instability is consistent with a previous study showing that the t1/2 of Ape1–DNA is only ∼30 s in solution (48). Interestingly, the free cAP/GO DNA generated by 0.5 nM Myh/50 nM Ape1 can form a stable complex with Myh (Fig. 11B), which is demonstrated when excess Myh is present, causing an increase in band a and a corresponding decrease in band bb (Fig. 11B, lanes 2–6).
Figure 11.

Accumulation of free cAP/GO and subsequent binding to Myh. The glycosylase reactions were carried out at 37°C for 30 min with 0.2 nM A/GO substrate and (A) 0.5 nM Myh (sub-saturating concentration) in the presence of increasing amounts of Ape1 or (B) 0.5 nM Myh/50 nM Ape1 with addition of more Myh after the 30 min glycosylase reaction. Myh–DNA complexes were analyzed by EMSA on an 8% native polyacrylamide gel. The observed bands a, b and bb are marked on the right. The bound and free DNA forms are indicated on the left. An asterisk indicates the 5′-end-labeled strand.
The reactions depicted in Figure 12 occurred under a saturating Myh concentration which produces maximum amounts of AP (60–70%) or cAP (45–60%) products (data not shown). Figure 12 shows that Myh is unable to bind AP/G or cAP/G (Fig. 12, lanes 3 and 4) but binds to both AP/GO and cAP/GO DNA (Fig. 12, lanes 7 and 8). To further confirm that band a was composed of protein–DNA (AP/GO or cAP/GO) complexes, proteinase K was added. This resulted in complex disruption and an increase in band b (free AP/GO) or band bb (free cAP/GO), respectively (data not shown). In addition, Myh cannot bind to C/GO or C/G (data not shown). We conclude that Myh binding is specific for AP/GO and cAP/GO DNA.
Figure 12.
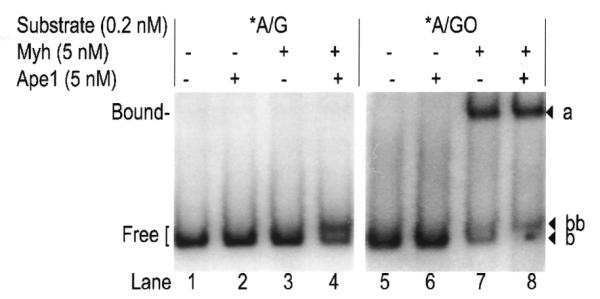
Myh–DNA complex formation. The glycosylase reaction was carried out at 37°C for 30 min with 0.2 nM substrate DNA and 5 nM Myh (saturating concentration) in the presence of 5 nM Ape1. Myh–DNA complexes were analyzed by EMSA on an 8% native polyacrylamide gel. The observed bands a, b and bb are marked on the right. The bound and free DNA forms are indicated on the left. An asterisk indicates the 5′-end-labeled strand.
Turnover of Myh in Myh/Ape1
A study of Myh turnover with or without Ape1 was performed using a substrate:Myh ratio of 10:1. A representative experiment is shown in Figure 13; 5 nM A/GO substrate was incubated with 0.5 nM Myh alone or 0.5 nM Myh/50 nM Ape1. Both AP/GO products (with alkaline treatment) and cAP/GO products (without alkaline treatment) were analyzed over a 5 h time course. Myh alone at 0.5 nM generated ∼0.12 nM glycosylase product within the first 10 min and this level was maintained over 5 h. However, in the presence of 50 nM Ape1, 0.5 nM Myh was capable of generating ∼0.31 nM glycosylase product over the course of 5 h. If we assume that the plateau reached in the absence of Ape1 is a functional measure of the active Myh protein present (0.12 nM), then all further activity seen is due to stimulation by Ape1. Most of this stimulation occurred in the interval 10–120 min.
Figure 13.

Turnover of Myh. The glycosylase reactions were carried out at 37°C with 5 nM A/GO substrate and 0.5 nM Myh in the presence of 5 nM Ape1 or buffer. Samples were taken at different time points, with or without alkaline treatment, and analyzed on a 15% denaturing polyacrylamide gel. A plot of the formation of AP/GO with alkaline treatment (+NaOH) or cAP/GO without alkaline treatment (–NaOH) versus time is shown.
DISCUSSION
A defect in MutY leads to a mutator phenotype in E.coli, qualifying MutY homologs as candidates for cancer involvement in higher eukaryotes. In addition to constructing an animal model defective in this enzyme activity to elucidate the roles of MutY in carcinogenesis, it is also important to understand the molecular mechanism of BER for A/GO. In this work we present evidence of a functional interaction between adenine-DNA glycosylase and AP endonuclease. The murine homolog Myh is 515 amino acids in length and corresponds to hMYH (type 2) (29), which is translated from the second methionine. While purification of recombinant hMYH from E.coli has been difficult (28–31), we were able to purify the hexa-histidine-tagged recombinant murine homolog Myh to near homogeneity relatively easily. The recombinant Myh complements the E.coli mutY phenotype (data not shown), showing that neither the hexa-histidine tag nor lack of the first 28 N-terminal amino acids prevents biological function.
We demonstrated that the recombinant Myh lacks efficient AP lyase activity as judged by a comparison of the amount of cleaved product (in the absence of alkaline treatment) to the amount of glycosylase product (in the presence of alkaline treatment). Our results with Myh are consistent with recent work on E.coli MutY, which demonstrated that the AP lyase activity is much weaker than, and uncoupled from, the adenine glycosylase activity of MutY (23). This lack of efficient AP lyase activity of Myh leads to the formation of intact (uncleaved) AP products, which then wait to be converted to the cleaved product by the next enzyme in BER.
The AP endonuclease Ape1 can cleave the AP product generated by Myh. The catalytic activity of Ape1 is essential for this cleavage, as a catalytically impaired mutant, H309N, while capable of binding to AP DNA, is unable to generate the cleaved product. In addition to endonuclease activity, Ape1 also stimulates Myh glycosylase activity on an A/GO substrate. The initial stimulation does not require catalytic function of Ape1, since it is retained in the mutant H309N. Using a non-cleavable DNA substrate we demonstrate that Ape1 decreases the Myh–substrate DNA dissociation constant, thereby promoting efficient formation of Myh–DNA complexes and, as a consequence, generating more glycosylase product. After the initial stimulation we observed a slow stimulatory effect over the course of several hours in the presence of a 100-fold excess of Ape1 over Myh. This slow stimulatory effect was not observed when the Ape1 concentration was reduced to 10-fold excess (data not shown). The mechanism of this slow stimulatory effect is not clear at this point. Ape1 may increase the rate of Myh product release or, alternatively, this effect could simply be explained by an attenuation of product inhibition by Ape1 endonuclease activity. The substrate preference of Myh and stimulatory effect of Ape1 lead to significantly more AP/GO product over AP/G product in the Myh/Ape1 system when compared to Myh alone, supporting the concept that A/GO is the biologically preferred substrate for Myh.
Myh protein not only forms a stable Myh–DNA complex with its own product, AP/GO DNA, but also with the Ape1 product, cAP/GO DNA. In the Myh/Ape1 in vitro system the state of the cAP/GO, bound or free, depends on the concentrations of both Myh and Ape1. In the presence of a sufficient amount of Ape1 to convert AP/GO to cAP/GO, low concentrations of Myh allow free cAP/GO to accumulate, while high concentrations of Myh lead to bound cAP/GO. Formation of stable Myh–cAP/GO complexes may be inhibitory to Myh turnover in our in vitro repair system. Physiologically, Myh binding to cAP/GO in vivo may protect DNA from loss of the unpaired base catalyzed by non-specific nucleases and subsequent generation of a double-strand DNA break. Alternatively, Myh binding to cAP/GO may suggest that Myh has a role to play in BER after Ape1 cleavage.
In our normal EMSA we did not observe the formation of an Ape1–DNA complex, which suggests that the Ape1–DNA complex is unstable. This is consistent with published results (48). We also did not observe a Myh–Ape1–DNA tertiary structure, even with prolonged gel runs (data not shown), suggesting that either the structure was not formed or that it is too unstable for detection. However, it is still possible that a Myh–Ape1–DNA complex formed but that its mobility is very similar to the Myh–DNA complex (band a) and therefore cannot be separated under our experimental conditions. Due to the inability to observe Ape1–DNA or Myh–Ape1–DNA complexes, it is not clear whether the displacement of Myh occurs prior to Ape1 cleaving the AP/GO substrate.
Overall, our study suggests that protein–protein interaction may play an important role in achieving efficient BER of A/GO in vivo. However, since we have only studied the interaction of the first two enzymes in the process, the characteristics demonstrated here may not hold true when the entire complement of enzymes is utilized. So far, two BER pathways for repair of AP lesions have been identified in an in vitro reconstituted system using human repair enzymes (34). It is not clear at this point whether Myh may be involved in one or both of these pathways in vivo. One piece of evidence that suggests that Myh may be involved in the PCNA-dependent pathway of BER is the presence of a putative PCNA-binding sequence, shown in Figure 1. Since the other enzymes in both BER pathways have been identified, the next logical step is to piece together all the BER components and determine their connection to Myh, so that a complete characterization of the molecular mechanism of the repair of A/GO can be achieved.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Claudia Baikalov and Isabella T. Phan for technical assistance on the project. We thank Michael S. DeMott for helpful discussions and comments on the manuscript. The phosphoimaging analysis was done at the UCLA-DOE Biochemistry Instrumentation facility. This work was supported by USHHS Institutional National Research Service Award T32 CA09056 (to H.Y.), grant 5 T32 ES07155 from the National Institute of Environmental Health Sciences, NIH (to D.W.) and by NIH grants GM32184 (to J.H.M.) and GM40000 (to B.D.).
DDBJ/EMBL/GenBank accession no. AY007717
References
- 1.Shibutani S., Takeshita,M. and Grollman,A.P. (1991) Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature, 349, 431–434. [DOI] [PubMed] [Google Scholar]
- 2.Moriya M., Ou,C., Bodepudi,V., Johnson,F., Takeshita,M. and Grollman,A.P. (1991) Site-specific mutagenesis using a gapped duplex vector: a study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat. Res., 254, 281–288. [DOI] [PubMed] [Google Scholar]
- 3.Pavlov Y.I., Minnick,D.T., Izuta,S. and Kunkel,T.A. (1994) DNA replication fidelity with 8-oxodeoxyguanosine triphosphate. Biochemistry, 33, 4695–4701. [DOI] [PubMed] [Google Scholar]
- 4.Nghiem Y., Cabrera,M., Cupples,C.G. and Miller,H.J. (1988) The mutY gene: a mutator locus in Escherichia coli that generates G.C→T.A transversions. Proc. Natl Acad. Sci. USA, 85, 2709–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 7.Friedberg E.C., Walker,G.C. and Siede,W. (1995) DNA Repair and Mutagenesis. ASM Press, Washington, DC, pp. 135–190.
- 8.Parikh S.S., Mol,C.D., Hosfield,D.J. and Tainer,J.A. (1999) Envisioning the molecular choreography of DNA base excision repair. Curr. Opin. Struct. Biol., 9, 37–47. [DOI] [PubMed] [Google Scholar]
- 9.Mol C.D., Parikh,S.S., Putnam,C.D., Lo,T.P. and Tainer,J.A. (1999) DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu. Rev. Biophys. Biomol. Struct., 28, 101–128. [DOI] [PubMed] [Google Scholar]
- 10.Saporito S.M. and Cunningham,R.P. (1988) Nucleotide sequence of the nfo gene of Escherichia coli K-12. J. Bacteriol ., 170, 5141–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hosfield D.J., Guan,Y., Haas,B.J., Cunningham,R.P. and Tainer, JA. (1999) Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell, 98, 397–408. [DOI] [PubMed] [Google Scholar]
- 12.Weiss B. (1976) Endonuclease II of Escherichia coli is exonuclease III. J. Biol. Chem., 251, 1896–1901. [PubMed] [Google Scholar]
- 13.Mol C.D., Kuo,C.F., Thayer,M.M., Cunningham,R.P. and Tainer,J.A. (1995) Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature, 374, 381–386. [DOI] [PubMed] [Google Scholar]
- 14.Lu A.L. and Chang,D.Y. (1988) A novel nucleotide excision repair for the conversion of an A/G mismatch to C/G base pair in E. coli. Cell, 54, 805–812. [DOI] [PubMed] [Google Scholar]
- 15.Au K.G., Clark,S., Miller,J.H. and Modrich,P. (1989) Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs. Proc. Natl Acad. Sci. USA, 86, 8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Porello S.L., Leyes,A.E. and David,S.S. (1998) Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry, 37, 14756–14765. [DOI] [PubMed] [Google Scholar]
- 17.Noll D.M., Gogos,A., Granek,J.A. and Clarke,N.D. (1999) The C-terminal domain of the adenine-DNA glycosylase MutY confers specificity for 8-oxoguanine.adenine mispairs and may have evolved from MutT, an 8-oxo-dGTPase. Biochemistry, 38, 6374–6379. [DOI] [PubMed] [Google Scholar]
- 18.Li X., Wright,P.M. and Lu,A.-L. (2000) The C-terminal domain of MutY glycosylase determines the 7,8-dihydro-8-oxo-guanine specificity and is crucial for mutation avoidance. J. Biol. Chem., 275, 8448–8455. [DOI] [PubMed] [Google Scholar]
- 19.Manuel R.C., Czerwinski,E.W. and Lloyd,R.S. (1996) Identification of the structural and functional domains of MutY, an Escherichia coli DNA mismatch repair enzyme. J. Biol. Chem., 271, 16218–16226. [DOI] [PubMed] [Google Scholar]
- 20.Guan Y., Manuel,R.C., Arvai,A.S., Parikh,S.S., Mol,C.D., Miller,J.H., Lloyd,S. and Tainer,J.A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct. Biol., 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- 21.Tsai-Wu J.J., Liu,H.F. and Lu,A.L. (1992) Escherichia coli MutY protein has both N-glycosylase and apurinic/apyrimidinic endonuclease activities on A.C and A.G mispairs. Proc. Natl Acad. Sci. USA, 89, 8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zharkov D.O. and Grollman,A.P. (1998) MutY DNA glycosylase: base release and intermediate complex formation. Biochemistry, 37, 12384–12394. [DOI] [PubMed] [Google Scholar]
- 23.Williams S.D. and David,S.S. (1998) Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA. Nucleic Acids Res., 26, 5123–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams S.D. and David,S.S. (1999) Formation of a Schiff base intermediate is not required for the adenine glycosylase activity of Escherichia coli MutY. Biochemistry, 38, 15417–15424. [DOI] [PubMed] [Google Scholar]
- 25.Dodson M.L., Michaels,M.L. and Lloyd,R.S. (1994) Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem., 269, 32709–32712. [PubMed] [Google Scholar]
- 26.Lu A.L. and Fawcett,W.P. (1998) Characterization of the recombinant MutY homolog, an adenine DNA glycosylase, from yeast Schizosaccharomyces pombe. J. Biol. Chem., 273, 25098–25105. [DOI] [PubMed] [Google Scholar]
- 27.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.-H., Wei,Y.-F. and Miller,J.H. (1996) Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Slupska M.M., Luther,W.M., Chiang,J.-H., Yang,H. and Miller,J.H. (1999) Functional expression of hMYH, a human homolog of the Escherichia coli mutY protein. J. Bacteriol ., 181, 6210–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takao M., Zhang,Q.M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohtsubo T., Nishioka,K., Imaiso,Y., Iwai,S., Shimokawa,H., Oda,H., Fujiwara,T. and Nakabeppu,Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res., 28, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsai-Wu J.-J., Su,H.-T., Wu,Y.-L., Hsu,S.-M. and Wu,C.H.H. (2000) Nuclear localization of the human mutY homologue hMYH. J. Cell Biochem., 77, 666–677. [PubMed] [Google Scholar]
- 32.Gorman M.A., Morera,S., Rothwell,D.G., de La Fortelle,E., Mol,C.D., Tainer,J.A., Hickson,I.D. and Freemont,P.S. (1997) The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J., 16, 6548–6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 DNA repair and coordination. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- 34.Wilson D.M. and Thompson,L.H. (1997) Life without DNA repair. Proc. Natl Acad. Sci. USA, 94, 12754–12757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prasad R., Singhal,R.K., Srivastava,D.K., Molina,J.T., Tomkinson,A.E. and Wilson,S.H. (1996) Specific interaction of DNA polymerase beta and DNA ligase I in a multiprotein base excision repair complex from bovine testis. J. Biol. Chem., 271, 16000–16007. [DOI] [PubMed] [Google Scholar]
- 36.Kubota Y., Nash,R.A., Klungland,A., Schär,P., Barnes,D.E. and Lindahl,T. (1996) Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 37.Pascucci B., Stucki,M., Jónsson,Z.O., Dogliotti,E. and Hübscher,U. (1999) Long patch base excision repair with purified human proteins. DNA ligase I as patch size mediator for DNA polymerases delta and epsilon. J. Biol. Chem., 274, 33696–33702. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto Y., Kim,K., Hurwitz,J., Gary,R., Levin,D.S., Tomkinson,A.E. and Park,M.S. (1999) Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins. J. Biol. Chem., 274, 33703–33708. [DOI] [PubMed] [Google Scholar]
- 39.Parikh S.S., Mol,C.D., Slupphaug,G., Bharati,S., Krokan,H.E. and Tainer,J.A. (1998) Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J., 17, 5214–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waters T.R., Gallinari,P., Jiricny,J. and Swann,P.F. (1999) Human thymine DNA glycosylase binds to apurinic sites in DNA but is displaced by human apurinic endonuclease 1. J. Biol. Chem., 274, 67–74. [DOI] [PubMed] [Google Scholar]
- 41.Seki S., Akiyama,K., Watanabe,S., Hatsushika,M., Ikeda,S. and Tsutsui,K. (1991) cDNA and deduced amino acid sequence of a mouse DNA repair enzyme (APEX nuclease) with significant homology to Escherichia coli exonuclease III. J. Biol. Chem., 266, 20797–20802. [PubMed] [Google Scholar]
- 42.Masuda Y., Bennett,R.A. and Demple,B. (1998) Rapid dissociation of human apurinic endonuclease (Ape1) from incised DNA induced by magnesium. J. Biol. Chem., 273, 30352–30359. [DOI] [PubMed] [Google Scholar]
- 43.Harlow E. and Lane,D. (1998) Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 90–91.
- 44.Demple B., Herman,T. and Chen,D.S. (1991) Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc. Natl Acad. Sci. USA, 88, 11450–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang H., Fitz-Gibbon,S., Marcotte,E.M., Tai,J.H., Hyman,E.C. and Miller,J.H. (2000) Characterization of a thermostable DNA glycosylase specific for U/G and T/G mismatches from the hyperthermophilic archaeon Pyrobaculum aerophilum. J. Bacteriol., 182, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Genetics Computer Group (1997) Wisconsin Package 9.1. GCG, Madison, WI.
- 47.Otterlei M., Warbrick,E., Nagelhus,T.A., Haug,T., Slupphaug,G., Akbari,M., Aas,P.A., Steinsbekk,K., Bakke,O. and Krokan,H.E. (1999) Post-replicative base excision repair in replication foci. EMBO J., 18, 3834–3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wilson D.M., Takeshita,M. and Demple,B. (1997) Abasic site binding by the human apurinic endonuclease, Ape, and determination of the DNA contact sites. Nucleic Acids Res., 25, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]