

Abstract
The lung is unique being exposed directly to the atmospheric environment containing xenobiotics, pathogens, and other agents which are continuously inhaled on a daily basis. Additionally, the lung is exposed to higher ambient oxygen levels which can promote the formation of a complex number of reactive oxygen and nitrogen species. Due to this constant barrage of potential damaging agents, the lung has developed a high degree of plasticity in dealing with ever changing conditions. In the present commentary, we will focus on glutathione (GSH) as a key antioxidant in the lung airways and discuss mechanisms by which the lung uses GSH to adapt to its rapidly changing environment. We will then examine the evidence on how defective and inadequate adaptive responses can lead to lung injury, inflammation and disease. Lastly, we will examine some of the recent attempts to alter lung GSH levels with therapies in a number of human lung diseases and discuss some of the limitations of such approaches.
Keywords: Cystic fibrosis, chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, γ-glutamylcysteine ligase, infection and cigarette smoke
The lung’s first line of defense, the airway fluid
The lung epithelial lining fluid (ELF) is a continuous thin fluid containing a heterogeneous mixture of macromolecules that includes proteins, mucous, and surfactants as well as a number of low molecular weight antioxidants [1]. One of the functions of the many ELF components are to act as a barrier against inhaled agents and detoxify potentially damaging reactive species that are abundant in the atmospheric environment [2]. The lung also has the additional challenge of being able to quickly change the components of the ELF to adapt to a constantly shifting composition of inhaled agents in the atmospheric environment. How the lung does this and the sensors it uses to do this are largely unexplored and unknown. An equally understudied area is how these adaptive responses are altered with disease and aging and the long-term consequence of maladaptive responses to environmental challenges.
Protective Macromolecules and Enzymes
To date numerous proteins and other factors have been identified in both normal and disease states as potential biomarkers based on the type and amount of specific proteins found in the airways. A common approach to sample the ELF is to perform a bronchoalveolar lavage (BAL) that instills a small amount of saline into the lung airways which mixes with the ELF and is then aspirated out. Recent work has utilized a proteomic approach to identify protein constituents of the BAL fluid (BALF) [3]. A detailed description of all of the BAL proteins identified would be a full review in itself and is thus beyond the scope of the current commentary. Therefore we will limit this commentary to a few pertinent antioxidant proteins found in the BALF. Some of the more commonly measured enzymes range from catalase, superoxide dismutase (SOD), thioredoxin, glutathione-S-transferase, glutathione peroxidase, peroxiredoxins and glutathione reductase [4]. Some of these enzymes, such as extracellular SOD (SOD3), are known to be secreted by cells and require no additional cofactors to be active [5]. There is evidence that shows the recovered enzymes in the BALF are indeed intact and functional when provided proper substrates and cofactors, however the source(s) of some enzymes is controversial [1]. One theory is that the enzymes found in the BAL are leaked from the lung cells during either damage or normal turnover of the cells. A second explanation is that these enzymes are actively secreted by the lung cells themselves or originate from the plasma. Another confounding factor in the investigation of the role of these antioxidant enzymes is the availability of potential cofactors. For instance, glutathione reductase, although found to be functional ex vivo when recovered from the BAL, requires NADPH as a cofactor and yet NADPH is not found at high concentrations in the ELF nor is it known to be released by cells. A similar case exists for thioredoxin reductase which also requires NADPH for activity. In either case, the in vivo functionality of these extracellular enzymes is not fully understood and requires further investigation.
Low molecular weight antioxidants
There are a large number of different small molecule antioxidants in the ELF. The antioxidants in the ELF have not been characterized to the extent that they have been in the plasma due to the variability of the dilution of the ELF during BAL and the difficulty in obtaining both BAL and plasma from human subjects [6]. Despite the lack of a clear antioxidant profile there are a few molecules that are typically measured. GSH is one of the most characterized and abundant antioxidants found in the ELF while there are also substantial levels of cysteine, ascorbic acid, α-tocopherol, urate and thiocyanate that can act as antioxidants as well [6, 7]. Although urate can reach high levels in the ELF, its antioxidant benefit is not fully understood. Urate has been shown to be beneficial in models of ozone toxicity, but there is little other evidence of its protective function in the ELF [8]. Conversely, both ascorbic acid and α-tocopherol are found at much lower levels in the ELF and their levels are similar as those reported in the plasma [9, 10]. Thiocyanate is also a potentially important molecule found in the ELF at substantial levels that has interesting antioxidant and host defense properties [11]. However, very little is currently known about its source, synthesis and distribution in the lung.
GSH is unique in that it is one of the few antioxidants in the ELF that is expressed at higher levels than the plasma. Under normal conditions GSH in the ELF can range between 100–300 μM and increase to near millimolar levels under conditions of stress. A number of stimuli can elevate GSH levels including bacterial infection, disease, or smoking. Under these circumstances the elevated GSH may in fact be an adaptive response to these stimuli to avoid further damage to the lung. Conversely ELF GSH levels are decreased in many progressive lung diseases (Table 1) including idiopathic pulmonary fibrosis (IPF), acute respiratory distress syndrome (ARDS), cystic fibrosis (CF), lung transplantation, HIV infection, and late stage chronic obstructive pulmonary disease (COPD) [12, 13]. In many of these instances the lung’s ability to maintain a normal GSH basal level is compromised in some way and may potentially contribute to the progression of these particular conditions. However, due to our limited understanding of the mechanisms and pathways the lung uses to maintain airway GSH, it is unclear whether these events are linked directly with these various pathophysiologic conditions.
Table 1.
Reported ELF GSH levels in various disease states and the percent change from normal levels of the same study.*
|
ELF GSH levels (μM) |
||||
|---|---|---|---|---|
| Normal | Patients | Change | Reference | |
| Cigarette smokers | 429 ±34 | 775 ±119 | +55% | [7] |
| Chronic Obstructive pulmonary disease (COPD) | 114 ±44.5 | 72 ±24⧧ | −40% | [65] |
| Idopathic pulmonary diease (IPF) | 429 ±34 | 97 ±18 | −80% | [43] |
| Acute respiratory distress syndrome (ARDS) | 91.8 ±14.5 | 21.7 ±7.8 | −80% | [66] |
| Lung transplant | 302 ±40.8 | 94.0 ±9.7 | −70% | [67] |
| Cystic Fibrosis | 278 ±21 | 92 ±14 | −70% | [68] |
| HIV | 689 ±100.4 | 397 ±52.7 | −50% | [69] |
ELF estimate based on similar dilution factor as normal values
Data reported as mean ± SE in μM concentration of ELF GSH
Sources of ELF GSH
de Novo Synthesis and Systemic Supply
GSH is synthesized de novo in every organ to different degrees but can also be broken down and taken up by specific cell types. GSH is a tripeptide comprised of γ-glutamate, cysteine, and glycine. The initial energy dependent and rate limiting step catalyzes the peptide bond formation between γ-glutamate and cysteine through the enzyme γ-glutamylcysteine ligase (GCL) forming a dipeptide. In the second energy dependent step the glycine is added to the dipeptide by glutathione synthetase (GS). In varying tissue GSH can be exported into the extracellular space. In the liver, extracellular transport of GSH results in elevation of its levels in plasma and bile through multidrug resistance proteins (MRP) [14].
Plasma GSH levels have been widely considered to act as a more stable and less reactive source of systemic cysteine [15] which can be utilized via the plasma membrane associated enzyme γ-glutamyl transpeptidase (GGT) breakdown process. GGT is expressed to different degrees depending on the tissue, with the kidney expressing one of the highest levels [16]. The high expression of GGT in the kidney is thought to keep the plasma steady-state levels of GSH low.
In addition to recycling of GSH, there is evidence to suggest that intact GSH can be taken up via a Na+-dependant transport mechanism. This system has been demonstrated in renal proximal tubule cells, small intestinal enterocytes, and type II pneumocytes [17–19]. This uptake of intact GSH in certain cell types is intriguing and provides for an alternative rationale for the utilization of plasma GSH. GSH uptake independent of synthesis also suggests that some tissues may be dependent on other organs to supplement GSH directly, particularly in times of oxidative stress. This may be particularly important for the lung during acute exposures to oxidants in which case GSH could be rapidly taken up and then transported into the ELF. This would also indicate that secondary chronic diseases particularly of the liver, may contribute to the development or progression of lung disease if the lung depends on a systemic supply of GSH to establish and maintain an adaptive response.
Apical Transport
The lung maintains high levels of extracellular GSH in the apically located ELF in contrast to other tissues. The main apical transporter of GSH in the lung is the cystic fibrosis transmembrane conductance regulator (CFTR, ABCC7) protein, a member of the MRP family of transporters [20, 21]. While there is much debate on the exact mechanism of GSH transport through CFTR, it is clear in human CFTR deficient epithelial cells and in CFTR KO mice that the basal apical GSH levels are decreased by as much as 50% [21]. The CFTR KO mice also have defective GSH adaptive responses to stressors. During a lung infection with Pseudomonas aeruginosa (PA), the CFTR KO mice do not show the same increase in ELF GSH as seen in wild type mice [20]. In fact, the CFTR KO mice can only elevate their ELF GSH levels to that of the basal levels found in wild type mice during a PA infection. A similar phenomenon is observed with cigarette smoke exposure where CFTR deficient cells have blunted apical GSH levels compared to wild type cells [22]. CFTR appears to be involved in maintaining basal ELF GSH levels and is involved at least in part establishing a GSH adaptive response. While CFTR is the only known apical transporter of GSH in the lung, deletion of CFTR does not completely inhibit ELF GSH.
While the study of transporters in relation to lung diseases is still in its infancy, a recent study has shown that the breast cancer related protein (BCRP, ABCG2) transports GSH, a previously unknown function [23]. The implication is that there may be many more identified transporters that have dual functions, which may have direct implications on the ELF GSH and in turn several lung diseases. Clearly more work is needed in identifying transporters that may be involved in modulating the ELF GSH levels in the lung.
Role of GSH in protecting the lung
The antioxidant system may be one of the lung’s mechanisms to fine tune the magnitude of the inflammatory response to environmental triggers and also eventually resolve inflammatory responses. Many inflammatory responses are associated with increased release of reactive oxygen and nitrogen species and what better way to sense and control these mediators than the upregulation and release of antioxidants. GSH is a well studied low molecular weight thiol antioxidant that has important functions in maintaining a reducing environment in the lung despite its constant exposure to high ambient oxygen levels. Although GSH is classically linked to preventing oxidative stress, there is growing evidence that it may also be involved in modulating proinflammatory cytokine release as well [24]. Existing data suggests that GSH plays such a role in lung inflammatory responses by modulating a number of transcription factors that regulate inflammatory transcriptomes [25]. It is worth noting that extracellular and in some cases intracellular GSH levels are often abnormal in many chronic lung diseases and disorders (Figure 1).
Figure 1.
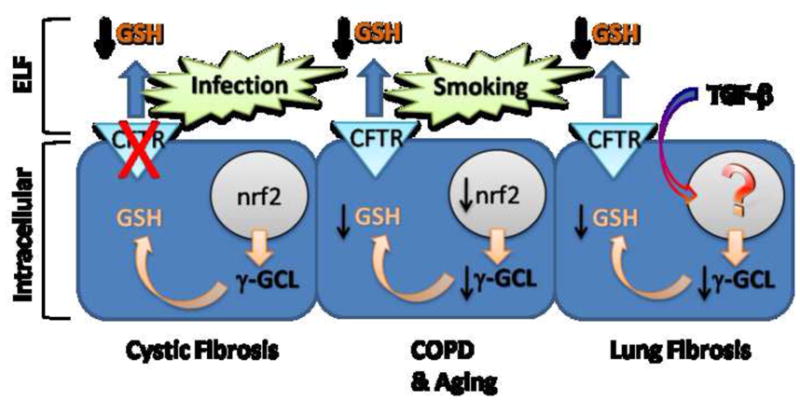
Several examples of maladaptive GSH responses in lung disease states. In Cystic Fibrosis lung disease, infections drive GSH adaptive responses, yet due to the CFTR mutation there is an inability to transport GSH into the ELF. In COPD and aging, chronic cigarette smoke drives GSH adaptive responses, however aging adversely affects Nrf2 activation which leads to deficient enzyme production and decreased intracellular GSH and well as decreased ELF GSH. In lung fibrosis, exaggerated TGF-β production in the airways produces affects on poorly identified transcriptional events leading to decreased γGCL levels and GSH synthesis.
Lung inflammation
Although GSH is not typically linked to immune responses, there is mounting evidence suggesting that GSH levels can modulate the amount and extent of proinflammatory cytokine release. Proinflammatory cytokines and chemokines like tumor necrosis factor alpha (TNFα) and interleukin-8 (IL-8) are commonly measured markers of inflammation in the airways. Alveolar type II cells release TNFα and IL-8 upon challenge with bacterial endotoxin and manipulation of intracellular GSH levels by either depletion using buthionine sulfoximine (BSO) or supplementation using glutamine results in increasing or decreasing the levels of TNFα and IL-8 released, respectively [24].
A casual relationship between lung GSH status and cytokine release is revealed among many common clinical lung diseases associated with chronic inflammation. A common lung disease associated with chronic inflammation is chronic obstructive pulmonary disease (COPD). In the airways of patients with late stage COPD and during exacerbations, the BAL IL-8 levels are significantly elevated while BAL GSH levels are significantly decreased [26]. Another example occurs with aging. Although aging is not a disease in the typical definition, there are correlated changes in both airway inflammation and ELF GSH with age [27]. During normal aging, the ELF GSH declines by as much as 50% compared to young controls, while at the same time there are increases in basal levels of TNFα in the airways. In addition, when aged mice are stressed by cigarette smoke, the resulting inflammatory response is exacerbated in the aged mice with lower ELF GSH as compared to the young mice. Aging is also a well characterized risk factor in the development of COPD [28]. These studies provide a rationale for targeting the mechanisms the lung uses to regulate intra and extracellular GSH as potential therapies to modulate and possibly shut off excessive inflammation processes that drive many lung diseases. While there are clear correlations between GSH and cytokine release, the exact mechanism by which this occurs, and any cross talk between GSH pathways and cytokine pathways, or whether other antioxidants can also have effects on inflammation directly has not fully been elucidated.
Lung Oxidation
Contrary to the role that GSH plays in inflammatory pathways, the role that GSH has in oxidation pathways has been investigated for decades. GSH can detoxify a wide range of pro-oxidants either through an enzymatic or direct reaction. GSH is a sulphydryl containing nucleophile that can react with a broad range of ROS, often resulting in less reactive species and oxidized glutathione (GSSG). The fact that GSH has a broad ability to react with oxidants is an important aspect in the ELF. An imbalance between proteases and protease inhibitors has been hypothesized to be important mechanisms in the development of cigarette smoke induced emphysema [29]. Under normal circumstances smokers have elevated ELF GSH levels, but as they age or develop disease ELF GSH levels decline. The decline in ELF GSH can lead to antiproteases including, alpha-1-antitrypsin, alpha-2-macroglobulin, and secretory leukoprotease inhibitor (SLPI), becoming oxidized resulting in decreased activity and a proteolytic imbalance and potential lung destruction and loss of lung function [30, 31]. For these types of studies the underlying factor is the ability to establish and maintain high levels of GSH in the ELF to detoxify the inhaled oxidants in cigarette smoke, thus protecting the resident antiproteases from oxidant deactivation. This field of research further highlights the important role of ELF GSH, and the importance of establishing and maintaining an adequate glutathione adaptive response to oxidant challenges.
Lung conditions associated with ELF GSH deficiency
Chronic obstructive pulmonary disease (COPD)
COPD is a disease characterized by the limitation in air flow in the lungs that is not fully reversible with bronchodilators. Both chronic bronchitis and emphysema are forms of COPD [32]. The main cause of COPD is chronic cigarette smoking, where nearly 90% of COPD cases can be attributed to smoking. Yet, only 20% of chronic cigarette smokers actually develop COPD [33, 34]. Cigarette smoke causes increases in ELF GSH, yet as COPD develops and progresses the ELF GSH tends to decline below normal levels [26]. This decline in ELF GSH makes individuals with moderate to severe COPD have increased inflammation and oxidative stress in their lungs. An interesting possibility is whether the chronic cigarette smokers that go on to develop lung disease are those who fail to develop an adequate antioxidant response or have marginal antioxidant responses which fall below a threshold during aging (Figure 1).
Aging
Although aging is not thought of as a disease, the normal process of aging does negatively impact the lung. Aging is associated with a gradual loss of lung function [35]. As individuals age they become more susceptible to a number of lung diseases, and it has been shown that in some individuals an emphysema-like phenotype can arise in the lung [36]. In models of aging there is a 50% decline in GSH levels in the liver, plasma, and lung tissues of aged rodents, as well as a 50% decline in ELF GSH [27, 37]. This decline stems from a decreased synthesis capacity as well as decreased Nrf2 nuclear translocation, a major contributor to the antioxidant adaptive and basal responses [38]. This global decline in antioxidant capacity leads to increased BALF proinflammatory cytokines, increased susceptibility to oxidants, and increased risk of developing a number of lung diseases.
Cystic Fibrosis (CF)
CF is a disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) protein. Individuals with this disease have little to no expression of functional CFTR. CFTR was originally identified as a chloride conductance channel which is also an apically expressed ATP binding cassette (ABC) transporter that is involved in the maintenance of ELF GSH. In CFTR KO mice, basal ELF GSH levels are roughly 50% of normal, and the capacity to increase ELF GSH also compromised [20]. The loss of CFTR has a stimulatory effect on the epithelial sodium channels (ENaC) in the airways which lead to increased sodium reabsorbtion effectively causing thick the hallmark thick mucus secretions which are an ideal environment for bacterial colonization [39]. The hallmark of CF lung disease is a persistent bacterial colonization of the lung with high numbers of neutrophils in the BAL [40]. Neutrophils are a major source of myeloperoxidase which produces a number of reactive species including the strong oxidizing agent hypochlorite [41].
The low basal ELF GSH in the CF lung along with the increase in neutrophil-mediated oxidative stress sets the stage for chronic inflammatory responses that contribute to the lung pathophysiology characteristic of this disease. It is interesting that normal mice respond to bacterial infections by elevating their ELF GSH levels and this ability is greatly impaired in CFTR KO mice. CF disease is a good example of how GSH adaptive responses are impaired due to a genetic defect in a GSH apical transporter that results in chronic inflammation and oxidative stress in the lung which is a major factor in the mortality and morbidity of this fatal genetic disease (Figure 1).
Idiopathic pulmonary fibrosis (IPF)
IPF is a disease characterized by excess remodeling, collagen deposition, and fibrosis within the parenchyma of the lung. The exact cause of IPF is not well understood but it is believed to be a combination of endogenous and exogenous stimuli that can adversely affect the alveoli [42]. In turn there may also be an abnormal repair process that leads to excessive scarring within the lung. While inflammation is typically low in individuals with IPF, excessive oxidative stress has been seen. In the BALF of patients with IPF there is a shift in the redox status of GSH with a higher degree of GSSG in the airways and lower basal levels of GSH [43]. In addition, BAL cells recovered from patients with IPF have shown increased levels of superoxide release and iNOS expression [44].
Recent data suggests that growth factors such as TGF-β can also modulate GSH biosynthesis in the lung. TGF-β is secreted as an inactive form that can be activated by proteases and reactive oxygen species. Dysregulation of TGF-β signaling has been heavily implicated in the fibrotic processes associated with IPF [45]. A number of studies have suggested that the excessive levels of active TGF-β create an imbalance by lowering both intracellular and extracellular levels of GSH in the lung [46]. TGF-β is thought to do this by negatively regulating γ-GCL expression resulting in low GSH levels and chronic lung oxidative stress which further drives TGF-β activation [47]. TGF-β can decrease intracellular GSH in fibroblasts which is associated with increases in collagen production, a hallmark of lung fibrosis [48]. This effect could be mitigated by restoration of intracellular GSH by applying exogenous GSH ester. IPF is a lung disease that has a strong association with an abnormal GSH response in the disease process (Figure 1).
Therapeutic approaches to ELF GSH deficiency
Oral GSH
GSH levels can readily be altered depending on a number of factors including diet and supplements. One therapeutic approach to increasing GSH levels has focused on orally administered GSH. A high dose of exogenous GSH has previously been shown to have some bioavailability when administered through an oral route and can increase plasma, lung tissue and ELF GSH levels [49, 50]. When mice are administered 300 mg/kg oral GSH, the ELF GSH peaks at roughly 800 μM within 60 min, a 4-fold increase over control levels [21]. Since there are a number of diseases that have characteristically low ELF GSH levels including advanced COPD, IPF, and ARDS; being able to modulate ELF GSH levels could be a powerful tool in improving quality of life for these individuals. However studies in humans administered lower concentrations of GSH did not find increases in GSH levels in the plasma (Table 2). Some issues with oral GSH administration are the short apparent half-life with increases in GSH occurring very rapidly, between 60–120 min, and return to basal levels within 4h. While increasing GSH levels through an oral route may help fight acute inflammation and oxidative stress, the transient nature of the increase does not lend itself well to the long term treatment of diseases. In addition, the availability of oral GSH is dependent on transporter expression to take up the GSH and transport it into the ELF, and individuals with transporter deficiencies like CF might not benefit from oral GSH delivery.
Table 2.
Summary of clinical trials of disease intervention using thiols.*
| Disease | Dose | Duration | Outcomes | Reference | |
|---|---|---|---|---|---|
| Oral GSH | |||||
| CF | 55–148 mg/kg/d | 5.5 mo | ↑FEV1, ↓BMI, ↓Infection | [70] | |
| N/A** | 46.1 mg/kg | 270 min | NC in systemic GSH | [71] | |
| Aerosol GSH | |||||
| CF | 600 mg, 2x/d | 3 d | ↓superoxide release, ↑BAL GSSG | [52] | |
| CF | 66 mg/kg 4x/d |
8 wk | ↑Mean peak flow, ↑perceived improvement | [51] | |
| IPF | 600 mg, 2x/d | 3 d | ↑BAL GSSG, ↓superoxide release | [44] | |
| CF | 300–450 mg, 3x/d | 14 d | ↑FEV1 | [72] | |
| Oral NAC | |||||
| COPD | 600 mg/d | 12 mo | ↓H2O2 in BAL, NC on lipid peroxides | [73] | |
| IPF | 600 mg 3x/d | 12 wk | ↑BAL GSH | [54] | |
| IPF | 600 mg 3x/d | 5 d | ↑BAL GSH, NC on bronchoscope parameters | [55] | |
| Aerosol NAC | |||||
| IPF | 352 mg/d | 12 mo | NC on pulmonary function or perceived quality of life | [57] |
Target disease indicates the disease in which the patients in the trials had been diagnosed
Abbreviations: N/A, no applicable disease; NC, no change; BAL, bronchoalveolar lavage; FEV1, Forced Expiratory volume in the first second; BMI, body mass index; GSSG, oxidized glutathione.
Aerosolized GSH
Since oral routes of GSH are dependent on transport and bioavailability issues, direct administration of aerosolized GSH has been examined in lung diseases such as COPD, IPF and CF [51] (Table 2). While this route seems to be the most direct, especially for treatment of lung diseases, there are still large doses administered, typically in the range of 600–1200 mg per day. While some clinical trials have shown positive effects of aerosolized GSH as measured by decreased superoxide release from alveolar macrophages, there have been very few positive results on lung function [44]. One study that examined CF patients administered aerosolized GSH have reported positive trends for GSH benefitting measures of FEV but significance was not reached due to low numbers of participants [51]. Another study of inhaled GSH in CF subjects did see any improvements in markers of inflammatory cytokines in the BALF but there was no measure of lung function [52]. While there are studies examining other cohorts of individuals with CF and IPF, there is no clear data on the benefits of aerosolized GSH. In several studies on individuals with CF or IPF, aerosolized GSH was shown to increase total ELF glutathione, but many of these increases were due to large increases in GSSG rather than GSH [44]. Increasing levels of GSSG in the ELF could actually counteract any therapeutic beneficial effect by decreasing the redox status of the ELF. In addition, large increases in GSSG in the airways could result in higher levels of oxidative stress leading to increased airway reactivity.
Oral N-acetylcysteine
N-acetylcysteine (NAC) can act as a thiol donor and as a precursor of cysteine for the synthesis of GSH. NAC has been shown to have some antioxidant effects of its own. As a precursor, NAC doesn’t have to be broken down or transported whole like GSH when given orally. The efficacy of oral NAC has been examined in individuals with COPD, IPF, and fibrosing alveolitis with varying degrees of success [53–55] (Table 2). In both COPD and IPF there were significant increases in ELF GSH, but absorption became a problem in the COPD patients. In patients with fibrosing alveolitis, the authors a trend toward improved lung function, measured as increased vital capacity and total lung capacity after 12 weeks of NAC treatment [54]. While there is some data available on the benefits of NAC on lung disease, some of it is conflicting and seems to be disease dependant. In addition, the same issues that plague oral GSH as a therapy hamper NAC, mainly the high doses (600–1800 mg/day), poor bioavailability, and the transient nature of the increases of either cysteine or GSH.
Aerosolized NAC
NAC has also been directly administration to the lung through aerosolized delivery. In mice subjected to belomycin induced lung fibrosis, aerosolized NAC showed marked improvements by decreasing the levels of airway chemokines as well as diminished lung fibrosis [56]. In clinical data, the effects have not been as promising (Table 2). In a pilot study of individuals with IPF, no significant differences in pulmonary function or quality of life were observed with inhaled NAC therapy [57].
Nrf2 inducers
A number of phase II antioxidant enzymes are controlled by the Nrf2 transcription factor, thus Nrf2 has become an attractive target to raise GSH levels and decrease inflammation. Sulforaphane, an isothiocyanate compound that is derived from cruciferous vegetables, has been shown to be an inducer of antioxidant response elements (ARE) that are typically upstream of many antioxidant genes including GCL, heme oxygenase-1, and NADPH quinine oxoreductase-1 [58]. Sulforaphane has been shown to be effective at inhibiting inflammatory cytokine release in airway cells [59]. While sulforaphane has shown positive effects in vitro studies, and many suggest that it may be beneficial in diseases like COPD, much of the focus on Nrf2 activators has shifted to synthetic triterpenoids.
The triterpenoid class of Nrf2 activators has been shown to be much more potent at activating Nrf2 and ARE than naturally derived compounds [60]. Various similar triterpenoids have been shown to be effective at decreasing inflammation in models of CF, emphysema, and hyperoxia induced lung injury [61–63]. They have also been shown to be effective at blocking TGF-β signaling, which would suggest that it may be effective for IPF [64]. Overall, the targeting of Nrf2 is an attractive prospect that would raise GSH levels and other antioxidant enzymes in a longer lasting fashion compared to thiol administration. While there is much research going into these types of compounds, most of the research is directed towards anticancer efficacy. Efficacy in clinical trials directed towards lung diseases like COPD, CF, or IPF have not yet been reported.
Conclusion
In summary, the ability to maintain proper levels of ELF antioxidants, primarily GSH, is a critical ability that is deficient or defective in many lung diseases. There is a dysregulation of ELF GSH in a number of diseases that can have profound effects on the ability to raise ELF GSH when the lung is stressed. There is promising, yet conflicting data on the benefits of either NAC or GSH as a viable therapy at raising ELF GSH and might be more beneficial for acute inflammation or oxidative stress due to the transient increases in GSH that are observed. While these therapies may not be as beneficial for chronic illnesses, this type of work is still important and highlights the potential benefits of drugs that can induce a longer lasting elevation in lung GSH.
Abbreviations
- BSO
L-buthionine sulfoximine
- γ-GCL
γ-glutamylcysteine ligase
- GSH
glutathione
- MRP
multi-drug resistant protein
- ELF
epithelial lining fluid
- BALF
bronchoalveolar lavage fluid
- SOD
superoxide dismutase
- IPF
idiopathic pulmonary fibrosis
- ARDS
acute respiratory distress syndrome
- CF
cystic fibrosis
- COPD
chronic obstructive pulmonary disease
- GCL
γ-glutamylcysteine ligase
- GGT
γ-glutamyl transpeptidase
- ABC
ATP binding cassette proteins
- MRP
multidrug resistance protein
- CFTR
cystic fibrosis transmembrane conductance regulator
- ENaC
Epithelial sodium channel
- PA
Pseudomonas aeruginosa
- BCRP
breast cancer related protein
- GSSG
glutathione disulfide
- NAC
N-acetylcysteine
- ARE
antioxidant response element
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cantin AM, Fells GA, Hubbard RC, Crystal RG. Antioxidant macromolecules in the epithelial lining fluid of the normal human lower respiratory tract. J Clin Invest. 1990;86:962–71. doi: 10.1172/JCI114798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelly FJ, Cotgrove M, Mudway IS. Respiratory tract lining fluid antioxidants: the first line of defence against gaseous pollutants. Cent Eur J Public Health. 1996;4 (Suppl):11–4. [PubMed] [Google Scholar]
- 3.Magi B, Bargagli E, Bini L, Rottoli P. Proteome analysis of bronchoalveolar lavage in lung diseases. Proteomics. 2006;6:6354–69. doi: 10.1002/pmic.200600303. [DOI] [PubMed] [Google Scholar]
- 4.Noel-Georis I, Bernard A, Falmagne P, Wattiez R. Database of bronchoalveolar lavage fluid proteins. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;771:221–36. doi: 10.1016/s1570-0232(02)00114-9. [DOI] [PubMed] [Google Scholar]
- 5.Kinnula VL, Crapo JD. Superoxide dismutases in the lung and human lung diseases. Am J Respir Crit Care Med. 2003;167:1600–19. doi: 10.1164/rccm.200212-1479SO. [DOI] [PubMed] [Google Scholar]
- 6.Cross CE, van der Vliet A, O’Neill CA, Louie S, Halliwell B. Oxidants, antioxidants, and respiratory tract lining fluids. Environ Health Perspect. 1994;102 (Suppl 10):185–91. doi: 10.1289/ehp.94102s10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. 1987;63:152–7. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- 8.Behndig AF, Blomberg A, Helleday R, Duggan ST, Kelly FJ, Mudway IS. Antioxidant responses to acute ozone challenge in the healthy human airway. Inhal Toxicol. 2009;21:933–42. doi: 10.1080/08958370802603789. [DOI] [PubMed] [Google Scholar]
- 9.Schock BC, Koostra J, Kwack S, Hackman RM, Van Der Vliet A, Cross CE. Ascorbic acid in nasal and tracheobronchial airway lining fluids. Free Radic Biol Med. 2004;37:1393–401. doi: 10.1016/j.freeradbiomed.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 10.Slade R, Crissman K, Norwood J, Hatch G. Comparison of antioxidant substances in bronchoalveolar lavage cells and fluid from humans, guinea pigs, and rats. Exp Lung Res. 1993;19:469–84. doi: 10.3109/01902149309064358. [DOI] [PubMed] [Google Scholar]
- 11.Fischer H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid Redox Signal. 2009;11:2453–65. doi: 10.1089/ars.2009.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantin AM, Begin R. Glutathione and inflammatory disorders of the lung. Lung. 1991;169:123–38. doi: 10.1007/BF02714149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris PE, Bernard GR. Significance of glutathione in lung disease and implications for therapy. Am J Med Sci. 1994;307:119–27. doi: 10.1097/00000441-199402000-00010. [DOI] [PubMed] [Google Scholar]
- 14.Leitner HM, Kachadourian R, Day BJ. Harnessing drug resistance: using ABC transporter proteins to target cancer cells. Biochem Pharmacol. 2007;74:1677–85. doi: 10.1016/j.bcp.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Compartmentation of glutathione: implications for the study of toxicity and disease. Toxicol Appl Pharmacol. 1996;140:1–12. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 16.Chikhi N, Holic N, Guellaen G, Laperche Y. Gamma-glutamyl transpeptidase gene organization and expression: a comparative analysis in rat, mouse, pig and human species. Comp Biochem Physiol B Biochem Mol Biol. 1999;122:367–80. doi: 10.1016/s0305-0491(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 17.Bai C, Brown LA, Jones DP. Glutathione transport by type II cells in perfused rat lung. Am J Physiol. 1994;267:L447–55. doi: 10.1152/ajplung.1994.267.4.L447. [DOI] [PubMed] [Google Scholar]
- 18.Lash LH, Hagen TM, Jones DP. Exogenous glutathione protects intestinal epithelial cells from oxidative injury. Proc Natl Acad Sci U S A. 1986;83:4641–5. doi: 10.1073/pnas.83.13.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lash LH, Jones DP. Transport of glutathione by renal basal-lateral membrane vesicles. Biochem Biophys Res Commun. 1983;112:55–60. doi: 10.1016/0006-291x(83)91796-5. [DOI] [PubMed] [Google Scholar]
- 20.Day BJ, van Heeckeren AM, Min E, Velsor LW. Role for cystic fibrosis transmembrane conductance regulator protein in a glutathione response to bronchopulmonary pseudomonas infection. Infect Immun. 2004;72:2045–51. doi: 10.1128/IAI.72.4.2045-2051.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kariya C, Leitner H, Min E, van Heeckeren C, van Heeckeren A, Day BJ. A role for CFTR in the elevation of glutathione levels in the lung by oral glutathione administration. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1590–7. doi: 10.1152/ajplung.00365.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kariya C, Chu HW, Huang J, Leitner H, Martin RJ, Day BJ. Mycoplasma pneumoniae infection and environmental tobacco smoke inhibit lung glutathione adaptive responses and increase oxidative stress. Infect Immun. 2008;76:4455–62. doi: 10.1128/IAI.00136-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brechbuhl HM, Gould N, Kachadourian R, Riekhof WR, Voelker DR, Day BJ. Glutathione transport is a unique function of the ATP-binding cassette protein ABCG2. J Biol Chem. 2010;285:16582–7. doi: 10.1074/jbc.M109.090506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haddad JJ. Glutathione depletion is associated with augmenting a proinflammatory signal: evidence for an antioxidant/pro-oxidant mechanism regulating cytokines in the alveolar epithelium. Cytokines Cell Mol Ther. 2000;6:177–87. doi: 10.1080/mccm.6.4.177.187. [DOI] [PubMed] [Google Scholar]
- 25.Haddad JJ, Harb HL. L-gamma-Glutamyl-L-cysteinyl-glycine (glutathione; GSH) and GSH-related enzymes in the regulation of pro- and anti-inflammatory cytokines: a signaling transcriptional scenario for redox(y) immunologic sensor(s)? Mol Immunol. 2005;42:987–1014. doi: 10.1016/j.molimm.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 26.Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, et al. Oxidative stress and airway inflammation in severe exacerbations of COPD. Thorax. 2005;60:293–300. doi: 10.1136/thx.2004.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gould NS, Min E, Gauthier S, Chu HW, Martin R, Day BJ. Aging adversely affects the cigarette smoke induced glutathione adaptive response in the lung. Am J Respir Crit Care Med. 2010 doi: 10.1164/rccm.201003-0442OC. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito K, Barnes PJ. COPD as a disease of accelerated lung aging. Chest. 2009;135:173–80. doi: 10.1378/chest.08-1419. [DOI] [PubMed] [Google Scholar]
- 29.Abboud RT, Vimalanathan S. Pathogenesis of COPD. Part I. The role of protease-antiprotease imbalance in emphysema. Int J Tuberc Lung Dis. 2008;12:361–7. [PubMed] [Google Scholar]
- 30.Cavarra E, Lucattelli M, Gambelli F, Bartalesi B, Fineschi S, Szarka A, et al. Human SLPI inactivation after cigarette smoke exposure in a new in vivo model of pulmonary oxidative stress. Am J Physiol Lung Cell Mol Physiol. 2001;281:L412–7. doi: 10.1152/ajplung.2001.281.2.L412. [DOI] [PubMed] [Google Scholar]
- 31.Gadek JE, Fells GA, Zimmerman RL, Rennard SI, Crystal RG. Antielastases of the human alveolar structures. Implications for the protease-antiprotease theory of emphysema. J Clin Invest. 1981;68:889–98. doi: 10.1172/JCI110344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cosio BG, Agusti A. Update in chronic obstructive pulmonary disease 2009. Am J Respir Crit Care Med. 181:655–60. doi: 10.1164/rccm.201001-0111UP. [DOI] [PubMed] [Google Scholar]
- 33.Peto R, Lopez AD, Boreham J, Thun M, Heath C., Jr Mortality from tobacco in developed countries: indirect estimation from national vital statistics. Lancet. 1992;339:1268–78. doi: 10.1016/0140-6736(92)91600-d. [DOI] [PubMed] [Google Scholar]
- 34.Snider GL. Chronic obstructive pulmonary disease: risk factors, pathophysiology and pathogenesis. Annu Rev Med. 1989;40:411–29. doi: 10.1146/annurev.me.40.020189.002211. [DOI] [PubMed] [Google Scholar]
- 35.Janssens JP, Pache JC, Nicod LP. Physiological changes in respiratory function associated with ageing. Eur Respir J. 1999;13:197–205. doi: 10.1034/j.1399-3003.1999.13a36.x. [DOI] [PubMed] [Google Scholar]
- 36.MacNee W. Accelerated lung aging: a novel pathogenic mechanism of chronic obstructive pulmonary disease (COPD) Biochem Soc Trans. 2009;37:819–23. doi: 10.1042/BST0370819. [DOI] [PubMed] [Google Scholar]
- 37.Teramoto S, Fukuchi Y, Uejima Y, Teramoto K, Ito H, Orimo H. Age-related changes in the antioxidant screen of the distal lung in mice. Lung. 1994;172:223–30. doi: 10.1007/BF00164439. [DOI] [PubMed] [Google Scholar]
- 38.Suh JH, Shenvi SV, Dixon BM, Liu H, Jaiswal AK, Liu RM, et al. Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid. Proc Natl Acad Sci U S A. 2004;101:3381–6. doi: 10.1073/pnas.0400282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berdiev BK, Qadri YJ, Benos DJ. Assessment of the CFTR and ENaC association. Mol Biosyst. 2009;5:123–7. doi: 10.1039/b810471a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heijerman H. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros. 2005;4 (Suppl 2):3–5. doi: 10.1016/j.jcf.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 41.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 42.American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 43.Cantin AM, Hubbard RC, Crystal RG. Glutathione deficiency in the epithelial lining fluid of the lower respiratory tract in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1989;139:370–2. doi: 10.1164/ajrccm/139.2.370. [DOI] [PubMed] [Google Scholar]
- 44.Borok Z, Buhl R, Grimes GJ, Bokser AD, Hubbard RC, Holroyd KJ, et al. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet. 1991;338:215–6. doi: 10.1016/0140-6736(91)90350-x. [DOI] [PubMed] [Google Scholar]
- 45.Liu RM, Gaston Pravia KA. Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic Biol Med. 2010;48:1–15. doi: 10.1016/j.freeradbiomed.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jardine H, MacNee W, Donaldson K, Rahman I. Molecular mechanism of transforming growth factor (TGF)-beta1-induced glutathione depletion in alveolar epithelial cells. Involvement of AP-1/ARE and Fra-1. J Biol Chem. 2002;277:21158–66. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- 47.Arsalane K, Dubois CM, Muanza T, Begin R, Boudreau F, Asselin C, et al. Transforming growth factor-beta1 is a potent inhibitor of glutathione synthesis in the lung epithelial cell line A549: transcriptional effect on the GSH rate-limiting enzyme gamma-glutamylcysteine synthetase. Am J Respir Cell Mol Biol. 1997;17:599–607. doi: 10.1165/ajrcmb.17.5.2833. [DOI] [PubMed] [Google Scholar]
- 48.Liu RM, Liu Y, Forman HJ, Olman M, Tarpey MM. Glutathione regulates transforming growth factor-beta-stimulated collagen production in fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2004;286:L121–8. doi: 10.1152/ajplung.00231.2003. [DOI] [PubMed] [Google Scholar]
- 49.Aw TY, Wierzbicka G, Jones DP. Oral glutathione increases tissue glutathione in vivo. Chem Biol Interact. 1991;80:89–97. doi: 10.1016/0009-2797(91)90033-4. [DOI] [PubMed] [Google Scholar]
- 50.Hagen TM, Wierzbicka GT, Sillau AH, Bowman BB, Jones DP. Bioavailability of dietary glutathione: effect on plasma concentration. Am J Physiol. 1990;259:G524–9. doi: 10.1152/ajpgi.1990.259.4.G524. [DOI] [PubMed] [Google Scholar]
- 51.Bishop C, Hudson VM, Hilton SC, Wilde C. A pilot study of the effect of inhaled buffered reduced glutathione on the clinical status of patients with cystic fibrosis. Chest. 2005;127:308–17. doi: 10.1378/chest.127.1.308. [DOI] [PubMed] [Google Scholar]
- 52.Roum JH, Borok Z, McElvaney NG, Grimes GJ, Bokser AD, Buhl R, et al. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J Appl Physiol. 1999;87:438–43. doi: 10.1152/jappl.1999.87.1.438. [DOI] [PubMed] [Google Scholar]
- 53.Pela R, Calcagni AM, Subiaco S, Isidori P, Tubaldi A, Sanguinetti CM. N-acetylcysteine reduces the exacerbation rate in patients with moderate to severe COPD. Respiration. 1999;66:495–500. doi: 10.1159/000029447. [DOI] [PubMed] [Google Scholar]
- 54.Behr J, Maier K, Degenkolb B, Krombach F, Vogelmeier C. Antioxidative and clinical effects of high-dose N-acetylcysteine in fibrosing alveolitis. Adjunctive therapy to maintenance immunosuppression. Am J Respir Crit Care Med. 1997;156:1897–901. doi: 10.1164/ajrccm.156.6.9706065. [DOI] [PubMed] [Google Scholar]
- 55.Meyer A, Buhl R, Magnussen H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur Respir J. 1994;7:431–6. doi: 10.1183/09031936.94.07030431. [DOI] [PubMed] [Google Scholar]
- 56.Hagiwara SI, Ishii Y, Kitamura S. Aerosolized administration of N-acetylcysteine attenuates lung fibrosis induced by bleomycin in mice. Am J Respir Crit Care Med. 2000;162:225–31. doi: 10.1164/ajrccm.162.1.9903129. [DOI] [PubMed] [Google Scholar]
- 57.Tomioka H, Kuwata Y, Imanaka K, Hashimoto K, Ohnishi H, Tada K, et al. A pilot study of aerosolized N-acetylcysteine for idiopathic pulmonary fibrosis. Respirology. 2005;10:449–55. doi: 10.1111/j.1440-1843.2005.00725.x. [DOI] [PubMed] [Google Scholar]
- 58.Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 1999;37:973–9. doi: 10.1016/s0278-6915(99)00082-4. [DOI] [PubMed] [Google Scholar]
- 59.Ritz SA, Wan J, Diaz-Sanchez D. Sulforaphane-stimulated phase II enzyme induction inhibits cytokine production by airway epithelial cells stimulated with diesel extract. Am J Physiol Lung Cell Mol Physiol. 2007;292:L33–9. doi: 10.1152/ajplung.00170.2006. [DOI] [PubMed] [Google Scholar]
- 60.Liby K, Hock T, Yore MM, Suh N, Place AE, Risingsong R, et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65:4789–98. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 61.Nichols DP, Ziady AG, Shank SL, Eastman JF, Davis PB. The triterpenoid CDDO limits inflammation in preclinical models of cystic fibrosis lung disease. Am J Physiol Lung Cell Mol Physiol. 2009;297:L828–36. doi: 10.1152/ajplung.00171.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reddy NM, Suryanaraya V, Yates MS, Kleeberger SR, Hassoun PM, Yamamoto M, et al. The triterpenoid CDDO-imidazolide confers potent protection against hyperoxic acute lung injury in mice. Am J Respir Crit Care Med. 2009;180:867–74. doi: 10.1164/rccm.200905-0670OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sussan TE, Rangasamy T, Blake DJ, Malhotra D, El-Haddad H, Bedja D, et al. Targeting Nrf2 with the triterpenoid CDDO-imidazolide attenuates cigarette smoke-induced emphysema and cardiac dysfunction in mice. Proc Natl Acad Sci U S A. 2009;106:250–5. doi: 10.1073/pnas.0804333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.To C, Kulkarni S, Pawson T, Honda T, Gribble GW, Sporn MB, et al. The synthetic triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid-imidazolide alters transforming growth factor beta-dependent signaling and cell migration by affecting the cytoskeleton and the polarity complex. J Biol Chem. 2008;283:11700–13. doi: 10.1074/jbc.M704064200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bridgeman MM, Marsden M, Selby C, Morrison D, MacNee W. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid, and lung tissue. Thorax. 1994;49:670–5. doi: 10.1136/thx.49.7.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest. 1991;100:1397–403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- 67.Baz MA, Tapson VF, Roggli VL, Van Trigt P, Piantadosi CA. Glutathione depletion in epithelial lining fluid of lung allograft patients. Am J Respir Crit Care Med. 1996;153:742–6. doi: 10.1164/ajrccm.153.2.8564127. [DOI] [PubMed] [Google Scholar]
- 68.Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol. 1993;75:2419–24. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- 69.Pacht ER, Diaz P, Clanton T, Hart J, Gadek JE. Alveolar fluid glutathione decreases in asymptomatic HIV-seropositive subjects over time. Chest. 1997;112:785–8. doi: 10.1378/chest.112.3.785. [DOI] [PubMed] [Google Scholar]
- 70.Visca A, Bishop CT, Hilton SC, Hudson VM. Improvement in clinical markers in CF patients using a reduced glutathione regimen: an uncontrolled, observational study. J Cyst Fibros. 2008;7:433–6. doi: 10.1016/j.jcf.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 71.Witschi A, Reddy S, Stofer B, Lauterburg BH. The systemic availability of oral glutathione. Eur J Clin Pharmacol. 1992;43:667–9. doi: 10.1007/BF02284971. [DOI] [PubMed] [Google Scholar]
- 72.Griese M, Ramakers J, Krasselt A, Starosta V, Van Koningsbruggen S, Fischer R, et al. Improvement of alveolar glutathione and lung function but not oxidative state in cystic fibrosis. Am J Respir Crit Care Med. 2004;169:822–8. doi: 10.1164/rccm.200308-1104OC. [DOI] [PubMed] [Google Scholar]
- 73.Kasielski M, Nowak D. Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir Med. 2001;95:448–56. doi: 10.1053/rmed.2001.1066. [DOI] [PubMed] [Google Scholar]