

Abstract
Frontotemporal dementia (FTD) syndromes comprise a heterogeneous group of neurodegenerative conditions characterized by atrophy in the frontal and temporal lobes. Three main clinical variants are recognized: Behavioral variant (bv-FTD), Semantic dementia (SD), and Progressive nonfluent aphasia (PNFA). However, logopenic/phonological (LPA) variant has been recently described, showing a distinctive pattern of brain atrophy and often associated to Alzheimer’s disease pathology. The diagnosis of FTD is challenging, since there is clinical, pathological, and genetic overlap between the variants and other neurodegenerative diseases, such as motoneuron disease (MND) and corticobasal degeneration (CBD). In addition, patients with gene mutations (tau and progranulin) display an inconsistent clinical phenotype and the correspondence between the clinical variant and its pathology is unpredictable. New cognitive tests based on social cognition and emotional recognition together with advances in molecular pathology and genetics have contributed to an improved understanding. There is now a real possibility of accurate biomarkers for early diagnosis. The present review concentrates on new insights and debates in FTD.
Keywords: Frontotemporal dementia, progressive nonfluent aphasia, semantic dementia, taupathies, TDP-43
Introduction
Frontotemporal dementia (FTD) encompasses a group of neurodegenerative diseases characterized by focal atrophy of frontal and anterior temporal lobes and non-Alzheimer pathology.[1,2] In people under 65 years of age, FTD is as common as Alzheimer’s disease (AD) and its prevalence has been estimated in 15 per 100,000 patients between 45 to 64 years of age.[3]
Patients with FTD display a heterogeneous clinical picture, which may include behavioral, cognitive, and motor manifestations.[4,5] However, based on the predominant initial symptoms, FTD can be readily separated into two groups: the behavioral variant (bv -FTD), which is characterized by loss of insight, personality changes, and disturbances in social cognition[1] and the language variant, also referred as primary progressive aphasia (PPA).[6] The latter can be further divided into a well-defined clinical-pathological entity, semantic dementia (SD),[7,8] and progressive nonfluent aphasia (PNFA).[1,8–11]
Despite this classification, there is a clinical, pathological, and genetic overlap. For instance, SD cases may develop features of bv-FTD,[12] and patients with the clinical variant often have common areas of brain atrophy[13] and family history of another variant.[14] Moreover, there is increasing evidence of overlap between FTD and other neurodegenerative disease, notably Motor Neuron disease (MND),[15] Progressive Supranuclear Palsy (PSP), and Corticobasal degeneration (CBD).[16,17] For example, cases initially diagnosed as PNFA may end up showing a clinical picture and pathology of CBD.[18] Indeed, some argue that those entities should all be included under the rubric of Pick’s complex.[19]
Differentiating one variant of FTD from another, as well as from other neurodegenerative and nondegenerative diseases (particularly psychiatric conditions) remains challenging.[20] Fortunately, recent advances in molecular pathology and genetics, improved imaging techniques, and better clinical descriptions have contributed enormously to our understanding of these conditions and are offering new insights, which we hope will be helpful for improved diagnosis and management of patients with these devastating disorders.
This review addresses the current concepts and advances in FTD.
Clinical Features
Behavioral variant (bv-FTD)
The clinical hallmark of bv-FTD is a disturbance in the personality and behavior, with changes of mood, motivation, and inhibition, leading to profound social disruption.[1,21,22] As the initial symptoms are neuropsychiatric, without impairment on cognitive screening tests, or overt changes on structural imaging,[23,24] these patients may be inappropriately diagnosed as suffering from a psychiatric disease, usually, depression or personality disorder.[20,25]
These changes become gradually evident to relatives, colleagues, and friends, because of disruption in their work performance, social, and family relationships. The effect on care is vivid with a high level of burden and stress.[26]
Patients may perform normally on standard neuropsychological tests of memory, language, attention, and visual spatial ability, but more recent tests designed to assess emotion processing,[27] social cognition,[28] theory of mind,[29] and complex decision making[30] are more sensitive and may show deficits in early cases, even if standard cognitive battery are normal.[24]
The most common features[31] of bv-FTD are shown in Table 1.
Table 1.
Most common symptoms in bv-FTD[31]
| Impaired Insight |
| Apathy |
| Disinhibition |
| Distractibility |
| Abnormal eating behavior |
| Stereotypic and ritualistic behavior |
| Impaired empathy |
| Mental rigidity |
| Dysexecutive symptoms |
| Speech adynamism |
In order to simplify the clinical picture, this myriad of neuropsychiatric manifestations may be classified in three main groups
Positive symptoms: These include disinhibition with lack of concern about social norms or embarrassment, impulsivity, outburst of violence, stereotypic and ritualistic behavior, abnormal appetite for sweets or gluttony, and impaired emotional judgment. When they are present, they strongly suggest the diagnosis of FTD.[22,24,32]
Negative symptoms: These include apathy and inertia, emotional blunting, impaired insight, lack of interest in usual or leisure activities, decline in the amount of speech (adynamism or laconic speech), and reduce self-care for complex instrumental activities.[33] These symptoms are less specific to FTD and also occur in depression.
Cognitive symptoms: These often appear later and include mental rigidity; loss of flexibility and abstraction; impairment in the pragmatic level of the discourse, with disorganization and distractibility; and poor planning and organization. At this stage, most patients fail in executive tasks and may show frontal release sign, such as grasping.[34,35]
This cluster of symptoms (positive, negative, and dysexecutive) have putative anatomical correlate to the orbitofrontal, medial, and dorsolateral frontal cortices, respectively.[33,36,37] Moreover, the progression of the atrophy, and consequently the clinical manifestations, may follow a predictable fashion, beginning in orbitofrontal and medial aspects of frontal cortex, and then involving the dorsolateral cortex and temporal anterior structures and basal ganglia.[38,39]
It has become increasingly apparent that some patients presenting with symptoms suggestive of bv-FTD fall to progress even over many years.[40] These “phenocopy” cases may actually constitute an unusual presentation of other conditions, such as late onset of bipolar disorder, personality disorder, or Asperger spectrum disorder. A number of features set apart these nonprogressive patients; normal performance on test of executive function[41] and of emotion processing;[27] better activities of daily living; and absence of brain atrophy.[40,42] The underlying pathophysiology in this group is unclear, but we assume that it reflects function disruption of orbitomesial frontal regions[43] in the absence of neurodegeneration and importantly a lack of atrophy on MRI[40] and hypometabolism on FDC-PET.[42] Table 2 displays characteristics that may distinguish one from another.
Table 2.
Comparison between Bv-FTD and phenocopy
| Feature | Bv-FTD | Phenocopy |
|---|---|---|
| Progression | +++ | − |
| Atrophy on MRI | ++ | − |
| FDG-PET Changes | +++ | − |
| Executive tasks impairment | ++ | +/− |
| Sleep disturbance | +++ | − |
Semantic dementia
Patients typically present with “loss of memory for words” and show impairment on tests of word comprehension, although the underlying deficit is the amodal store of semantic memory or knowledge about words, objects, people, and sounds.[7] [see Figure 1]. Patients show a gradual reduction of vocabulary and use high frequency terms (thing, boy), although speech is fluent and well articulated, without phonological or syntactic errors.[8,44,45]
Figure 1.
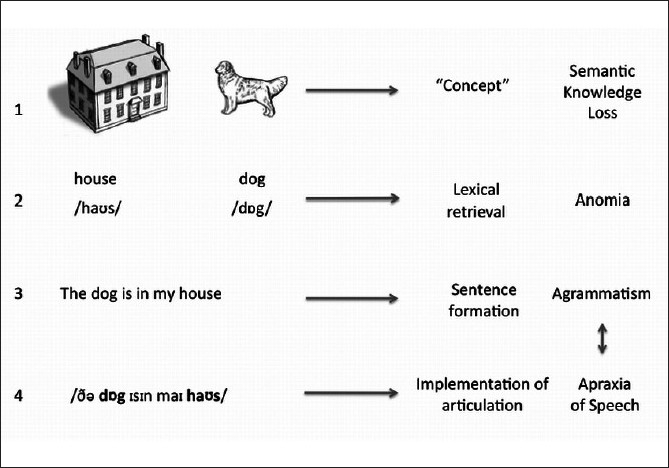
Stages of language production and PPA: In SD (1), there is a loss of semantic knowledge, so that the patient is not able to retrieve or comprehend words. In LPA (2), there are problems with word retrieval, but object recognition and word comprehension are intact. Whereas, in PNFA there is either disturbance in the word arrangement (agrammatism) (3), impairment in motor implementation of the speech (Apraxia of Speech) (4), or both (3 and 4).
A consistent feature is the impairment of naming objects or anomia. The performance is influenced by the level of familiarity and specificity of items asked. In other words, if the item is extensively encountered by the patient, it is likely to be forgotten later.[45] Likewise, the patient will tend to name objects that are prototypic of their category.[46] For instance, patients are able to name cat, dog, and horse, but not tiger or zebra, and use superordinate or general labels, calling the latter also a cat and horse, and may be just animal.[47]
Impairment of single word comprehension can be assessed by asking the patient to match the word with the corresponding object or to define the meaning of words.[45] There is a striking dissociation between repetition (preserved) and meaning (loss), best demonstrated by asking patients to repeat words such as “hippopotamus” or “catastrophe” and then to say what they mean.[48] These patients also show surface dyslexia where all words are read according to general rules of pronunciation, regardless of word meaning.[8]
In spite of the fact that word-based tests show clear deficits, actually there is a deterioration of central, amodal knowledge about objects or people which is apparent when nonverbal tests, such as “Pyramid and Palms test ” are employed, which requires subjects to match the two pictures that go together.[49]
Some SD patients present prominent deficits in identification of famous people. This deficit represents a general impairment on the “knowledge of people” than merely prosopagnosia (i.e., loss of ability to recognize faces), since patients are unable to produce any information from their name or voice.[50] Such patients typically show predominant right temporal atrophy[51] together with behavioral symptoms and poor insight.[52,53]
Progressive Nonfluent Aphasia
Unlike SD, the presenting features of PNFA are more varied and may reflect breakdown at various stages of speech production, from alterations in lexical retrieval, misarrangements of the words according to grammatical rules, or impaired motor programming of the intended utterance.[11]
Generally speaking, there are problems with the syntactic or motor aspects of speech, causing speech to be halting, slow, and distorted.[54]
Severe agrammatism causes oversimplification of the language production, lack of function words (e.g., prepositions, auxiliary verbs, or articles), or words inflections (i.e., endings of verb or noun according to conjugation or number, respectively).[10] But in the early stages, grammatical errors are subtle and may be difficult to distinguish from common errors or detect in a short interview. Syntactic problems are usually best assessed by testing sentence comprehension.[55]
Breakdown in the motor programming is referred to as apraxia of speech, which causes distortion of output with pauses, speech errors, and loss of melody. Patients may have difficulty in repeating or pronouncing polysyllabic words and strings of syllables (such as Pa-Ta-Ka), producing distortions and aprosodic intonation.[56]
Orobuccal apraxia may develop and some patients evolve into a picture of CBD or PSP.[57]
A third language variant: Logopenic/phonological
Gorno-Tempini et al. described cases of language variant that fulfilled neither SD nor PFNA criteria.[58] These cases showed reduced speech output with frequent pauses and impaired naming; preservation of grammar, motor speech, and semantic knowledge. A remarkable feature was profound impairment on repetition of sentences or string of words and difficulties in understanding complex instructions, despite of sparing single word repetition and comprehension. This has been attributed to a reduction of working memory resources, due to impairment of phonological loop.[59]
Interesting enough, this group showed a distinctive pattern of brain atrophy that involved the left temporoparietal junction. There is growing evidence that the underlying pathology is Alzheimer’s disease, suggesting that this variant is in fact, an atypical presentation of AD.[58,60,61]
Table 3contrasts the clinical features of the three language variants and Figure 1 shows the stages of language production involved.
Table 3.
Clinical features of language variants
| Feature | SD | PNFA | LPA |
|---|---|---|---|
| Agrammatism | − | +++/− * | − |
| Motor speech disorder | − | +++/− * | − |
| Anomia | +++ | + | +++ |
| Single word comprehension | +++ | − | − |
| Comprehension complex or sequential instructions | − | ++ | +++ |
| Single word-repetition | − | ++ | − |
| Sentence repetition | − | ++ | +++ |
| Surface dyslexia | +++ | − | − |
Either agrammatism or motor speech disorder must be included.
Imaging
The advent of high resolution MRI and of methods of automated qualification such as Voxel-based morphometry (VBM)[62] and cortical thickness measures[63] has enhanced our knowledge of the anatomical changes in the variants of FTD.
Patients with bv-FTD show atrophy of the orbitobasal and medialfrontal lobes, together with anterior temporal and insular involvement.[39,64] SD is associated with atrophy of the anterior temporal lobe involving particularly polar, anterior parahippocampal, and fusiform regions including the perirhinal cortex. The atrophy is bilateral, but typically asymmetric and often more severe on the left.[58,65] In PNFA, the changes are subtler and involve the left inferior frontal lobe and anterior insula cortex.[58,66,67] In logopenic/phonological variant the atrophy involves the left hemisphere, particularly the posterior temporal lobe (superior and middle temporal gyri) and inferior parietal lobe and lesser involvement of the precuneus.[58,59,68]
These changes can also be detected using simpler MRI-based visual rating scales, which simply use standard coronal cuts. These scales aid diagnosis and monitoring of progression.[69]
Pathology
Definitive diagnosis of FTD requires neuropathological examination. Unlike other dementia syndromes, notably AD, FTD encompasses considerable pathological heterogeneity.[2,18,70,71] The classification is based on the identification of intracellular protein inclusions by means of inmunohistochemistry.[71] Accordingly, three broad subdivisions have been recognized:
FTD with tau-positive inclusions: This includes classic Pick’s disease, Progressive Supranuclear Palsy, Corticobasal degeneration, argyrophilic grain disease, and patients with mutation of the Microtubule-associated protein tau (MAPT) gene on chromosome 17 (FTDP-17).[71]
FTD with tau-negative, ubiquitin-positive inclusions: This is the commonest pathological finding in FTD[70,72,73] and includes these with progranulin gene mutations.[74] The ubiquitinated protein has been identified as the transactive response DNA-binding protein with Mr 43 (TDP-43) which is also found in MND,[75,76] strengthening the association between FTD and MND. Interestingly, cases with ubiquitinated lesions without TDP-43[77] have been recently identified, which appear to have abnormal deposits of another protein called fused in sarcoma protein (FUS).[78]
Dementia lacking distinctive histology. Includes cases that do not show any particular or distinctive inclusion or histology, besides neuronal loss, superficial spongiosis, and gliosis. With the advent of newer inmunohistochemical techniques such cases are now increasingly rare.[71]
The correspondence between clinical phenotype and underlying pathological subtype has been a topic of considerable interest in recent years.[18,70,79] As shown in Figure 2, SD has been consistently associated with Ubiquitin (TDP-43) positive pathology.[9,72,80,81] In contrast, PNFA cases show variable results in different clinicopathology series, due probably to different diagnostic criteria employed as well as inclusion of logopenic cases in older series.[9,18,61,70,73,82] Despite that, PNFA more often associated to tau pathology, particularly if there is motor speech disorder.[83] In bv-FTD approximately a half has tau-positive and the other TDP-43-positive pathology.
Figure 2.
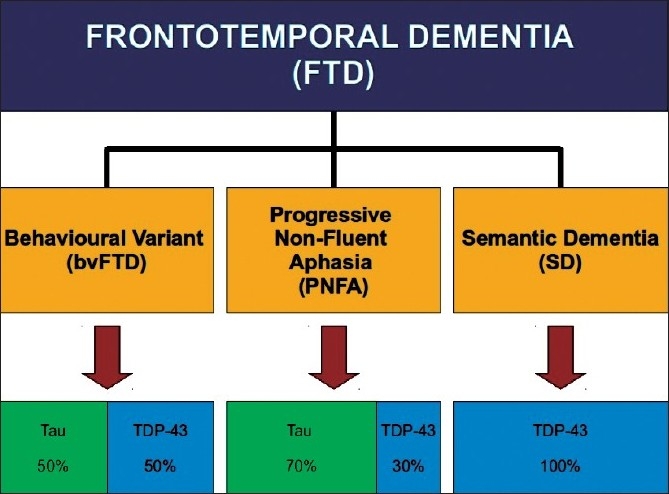
FTD syndromes and pathology
Genetic
Around 40% of patients report a family history of dementia, although in many instances this is almost certainly unrelated, but 10–20% have a clear pattern of autosomal dominant inheritance, with at least two relatives having young onset dementia or MND.[14,84,85] The heritability, however, varies according to the variant FTD: SD showing the least, whereas bv-FTD and FTD with MND the most inheritable.[14,81]
The commonest identified mutations are MAPT and progranuline (PGRN), both in chromosome 17q21.[86,87] Although the prevalence of mutations varies among studies, the two mutations have a similar frequency, being found in around 5–10% of patients.[88] Other mutations involve the Valosin-containing protein (VCP) and CHMP2B genes, but are very rare.[89]
The mutations of MAPT lead to abnormal intracellular accumulation of hyperphosphorylated tau.[90] While mutation of PGRN gene results in reduced expression of progranulin and is associated with Ubiquitin-TDP-43 pathology.[91] MAPT mutations typically give rise to the clinical phenotype called Frontotemporal dementia and parkinsonism linked to Chromosome 17 (FTDP-17). As its name suggests, the clinical picture embraces isolated behavioral and personality changes, initial extrapyramidal signs, suggesting of PSP or CBD, or a combination of behavioral and extrapyramidal syndromes.[92] In contrast, PGRN mutations appear to produce striking clinical heterogeneity which includes CBD, PNFA, or bv-FTD. Moreover, some cases depict an anmestic syndrome compatible with initial AD and a logopenic aphasia.[91]
Future directions
In the last twenty years, a great deal of progress on molecular genetic and imaging has led to new insights about FTD syndromes. New imaging methods, for instance VBM, has given a detailed account of pattern of brain atrophy, allowing an unbiased comparison of patients groups, while the development of radiotracers, such as PiB has enabled to identify the accumulation of extracellular beta-amyloid, and therefore, rule out cases of AD. Ligands specific to tau and TDP-43 are eagerly awaited.
Advances in neuropsychological assessment have also led a better understanding of the language and social cognitive difficulties seen in FTD.
Many issues remain unresolved. The relationship between genetic, pathologic, and clinical phenotype is of key importance as is the ability to identify pathological subtypes in vivo by the use of biomarkers. Eventually, it is hoped that biomarkers will be identified so that a specific therapy can be tailored according to the underlying pathology. The degree of overlap between MND and FTD syndromes is also a topic of keen current research interest.
Footnotes
Source of Support: Nil
Conflict of Interest: Nil
References
- 1.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, et al. Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–54. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 2.McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ, et al. Clinical and pathological diagnosis of frontotemporal dementia: Report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–9. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- 3.Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–21. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- 4.Kertesz A. Frontotemporal dementia: One disease, or many?: Probably one, possibly two. Alzheimer Dis Assoc Disord. 2005;19:S19–24. doi: 10.1097/01.wad.0000183080.69196.64. [DOI] [PubMed] [Google Scholar]
- 5.Josephs KA. Frontotemporal dementia and related disorders: Deciphering the enigma. Ann Neurol. 2008;64:4–14. doi: 10.1002/ana.21426. [DOI] [PubMed] [Google Scholar]
- 6.Mesulam MM. Primary progressive aphasia. Ann Neurol. 2001;49:425–32. [PubMed] [Google Scholar]
- 7.Hodges JR, Patterson K. Semantic dementia: A unique clinicopathological syndrome. Lancet Neurol. 2007;6:1004–14. doi: 10.1016/S1474-4422(07)70266-1. [DOI] [PubMed] [Google Scholar]
- 8.Hodges JR, Patterson K, Oxbury S, Funnell E. Semantic dementia. Progressive fluent aphasia with temporal lobe atrophy. Brain. 1992;115:1783–806. doi: 10.1093/brain/115.6.1783. [DOI] [PubMed] [Google Scholar]
- 9.Knibb JA, Xuereb JH, Patterson K, Hodges JR. Clinical and pathological characterization of progressive aphasia. Ann Neurol. 2006;59:156–65. doi: 10.1002/ana.20700. [DOI] [PubMed] [Google Scholar]
- 10.Hodges JR, Patterson K. Nonfluent progressive aphasia and semantic dementia: A comparative neuropsychological study. J Int Neuropsychol Soc. 1996;2:511–24. doi: 10.1017/s1355617700001685. [DOI] [PubMed] [Google Scholar]
- 11.Ash S, Moore P, Vesely L, Gunawardena D, McMillan C, Aderson C, Avants B, Grossman M. Non-fluent speech in frontotemporal lobar degeneration. J Neurolinguistics. 2009;22:370–83. doi: 10.1016/j.jneuroling.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosen HJ, Allison SC, Ogar JM, Amici S, Rose K, Dronkers N, et al. Behavioral features in semantic dementia vs other forms of progressive aphasias. Neurology. 2006;67:1752–6. doi: 10.1212/01.wnl.0000247630.29222.34. [DOI] [PubMed] [Google Scholar]
- 13.Schroeter ML, Raczka K, Neumann J, Yves von Cramon D. Towards a nosology for frontotemporal lobar degenerations: A meta-analysis involving 267 subjects. Neuroimage. 2007;36:497–510. doi: 10.1016/j.neuroimage.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 14.Goldman JS, Farmer JM, Wood EM, Johnson JK, Boxer A, Neuhaus J, et al. Comparison of family histories in FTLD subtypes and related tauopathies. Neurology. 2005;65:1817–9. doi: 10.1212/01.wnl.0000187068.92184.63. [DOI] [PubMed] [Google Scholar]
- 15.Bak TH, Hodges JR. Motor neurone disease, dementia and aphasia: Coincidence, co-occurrence or continuum? J Neurol. 2001;248:260–70. doi: 10.1007/s004150170199. [DOI] [PubMed] [Google Scholar]
- 16.Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol. 2003;54:S15–9. doi: 10.1002/ana.10570. [DOI] [PubMed] [Google Scholar]
- 17.Kertesz A, Blair M, McMonagle P, Munoz DG. The diagnosis and course of frontotemporal dementia. Alzheimer Dis Assoc Disord. 2007;21:155–63. doi: 10.1097/WAD.0b013e31806547eb. [DOI] [PubMed] [Google Scholar]
- 18.Kertesz A, McMonagle P, Blair M, Davidson W, Munoz DG. The evolution and pathology of frontotemporal dementia. Brain. 2005;128:1996–2005. doi: 10.1093/brain/awh598. [DOI] [PubMed] [Google Scholar]
- 19.Kertesz A. Pick complex: An integrative approach to fronto-temporal dementia: Primary progressive aphasia, corticobasal degeneration, and progressive supranuclear palsy. Neurologist. 2003;9:311–7. doi: 10.1097/01.nrl.0000094943.84390.cf. [DOI] [PubMed] [Google Scholar]
- 20.Rosness TA, Haugen PK, Passant U, Engedal K. Frontotemporal dementia: A clinically complex diagnosis. Int J Geriatr Psychiatry. 2008;23:837–42. doi: 10.1002/gps.1992. [DOI] [PubMed] [Google Scholar]
- 21.Boxer AL, Miller BL. Clinical features of frontotemporal dementia. Alzheimer Dis Assoc Disord. 2005;19:S3–6. doi: 10.1097/01.wad.0000183086.99691.91. [DOI] [PubMed] [Google Scholar]
- 22.Hodges JR. Frontotemporal dementia (Pick’s disease): Clinical features and assessment. Neurology. 2001;56:S6–10. doi: 10.1212/wnl.56.suppl_4.s6. [DOI] [PubMed] [Google Scholar]
- 23.Gregory CA, Serra-Mestres J, Hodges JR. Early diagnosis of the frontal variant of frontotemporal dementia: How sensitive are standard neuroimaging and neuropsychologic tests? Neuropsychiatry Neuropsychol Behav Neurol. 1999;12:128–35. [PubMed] [Google Scholar]
- 24.Wittenberg D, Possin KL, Rascovsky K, Rankin KP, Miller BL, Kramer JH. The early neuropsychological and behavioral characteristics of frontotemporal dementia. Neuropsychol Rev. 2008;18:91–102. doi: 10.1007/s11065-008-9056-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rankin KP, Santos-Modesitt W, Kramer JH, Pavlic D, Beckman V, Miller BL. Spontaneous social behaviors discriminate behavioral dementias from psychiatric disorders and other dementias. J Clin Psychiatry. 2008;69:60–73. doi: 10.4088/jcp.v69n0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mioshi E, Kipps CM, Dawson K, Mitchell J, Graham A, Hodges JR. Activities of daily living in frontotemporal dementia and Alzheimer disease. Neurology. 2007;68:2077–84. doi: 10.1212/01.wnl.0000264897.13722.53. [DOI] [PubMed] [Google Scholar]
- 27.Kipps CM, Nestor PJ, Acosta-Cabronero J, Arnold R, Hodges JR. Understanding social dysfunction in the behavioural variant of frontotemporal dementia: The role of emotion and sarcasm processing. Brain. 2009;132:592–603. doi: 10.1093/brain/awn314. [DOI] [PubMed] [Google Scholar]
- 28.Torralva T, Roca M, Gleichgerrcht E, Bekinschtein T, Manes F. A neuropsychological battery to detect specific executive and social cognitive impairments in early frontotemporal dementia. Brain. 2009;132:1299–309. doi: 10.1093/brain/awp041. [DOI] [PubMed] [Google Scholar]
- 29.Gregory C, Lough S, Stone V, Erzinclioglu S, Martin L, Baron-Cohen S, et al. Theory of mind in patients with frontal variant frontotemporal dementia and Alzheimer’s disease: Theoretical and practical implications. Brain. 2002;125:752–64. doi: 10.1093/brain/awf079. [DOI] [PubMed] [Google Scholar]
- 30.Torralva T, Kipps CM, Hodges JR, Clark L, Bekinschtein T, Roca M, et al. The relationship between affective decision-making and theory of mind in the frontal variant of fronto-temporal dementia. Neuropsychologia. 2007;45:342–9. doi: 10.1016/j.neuropsychologia.2006.05.031. [DOI] [PubMed] [Google Scholar]
- 31.Hodges JR. Frontotemporal dementia syndromes. Vol. 12. Cambridge:: Cambridge University Press; 2007. p. 334. [Google Scholar]
- 32.Rascovsky K, Hodges JR, Kipps CM, Johnson JK, Seeley WW, Mendez MF, et al. Diagnostic criteria for the behavioral variant of frontotemporal dementia (bvFTD): Current limitations and future directions. Alzheimer Dis Assoc Disord. 2007;21:S14–8. doi: 10.1097/WAD.0b013e31815c3445. [DOI] [PubMed] [Google Scholar]
- 33.Le Ber I, Guedj E, Gabelle A, Verpillat P, Volteau M, Thomas-Anterion C, et al. Demographic, neurological and behavioural characteristics and brain perfusion SPECT in frontal variant of frontotemporal dementia. Brain. 2006;129:3051–65. doi: 10.1093/brain/awl288. [DOI] [PubMed] [Google Scholar]
- 34.Libon DJ, Xie SX, Moore P, Farmer J, Antani S, McCawley G, et al. Patterns of neuropsychological impairment in frontotemporal dementia. Neurology. 2007;68:369–75. doi: 10.1212/01.wnl.0000252820.81313.9b. [DOI] [PubMed] [Google Scholar]
- 35.Hutchinson AD, Mathias JL. Neuropsychological deficits in frontotemporal dementia and Alzheimer’s disease: A meta-analytic review. J Neurol Neurosurg Psychiatry. 2007;78:917–28. doi: 10.1136/jnnp.2006.100669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cummings JL. Anatomic and behavioral aspects of frontal-subcortical circuits. Ann N Y Acad Sci. 1995;769:1–13. doi: 10.1111/j.1749-6632.1995.tb38127.x. [DOI] [PubMed] [Google Scholar]
- 37.Snowden JS, Neary D. Neuropsychiatric aspects of frontotemporal dementias. Curr Psychiatry Rep. 1999;1:93–8. doi: 10.1007/s11920-999-0015-z. [DOI] [PubMed] [Google Scholar]
- 38.Kril JJ, Halliday GM. Clinicopathological staging of frontotemporal dementia severity: Correlation with regional atrophy. Dement Geriatr Cogn Disord. 2004;17:311–5. doi: 10.1159/000077161. [DOI] [PubMed] [Google Scholar]
- 39.Perry RJ, Graham A, Williams G, Rosen H, Erzinçlioglu S, Weiner M, et al. Patterns of frontal lobe atrophy in frontotemporal dementia: a volumetric MRI study. Dement Geriatr Cogn Disord. 2006;22:278–87. doi: 10.1159/000095128. [DOI] [PubMed] [Google Scholar]
- 40.Davies RR, Kipps CM, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Progression in frontotemporal dementia: Identifying a benign behavioral variant by magnetic resonance imaging. Arch Neurol. 2006;63:1627–31. doi: 10.1001/archneur.63.11.1627. [DOI] [PubMed] [Google Scholar]
- 41.Hornberger M, Piguet O, Kipps C, Hodges JR. Executive function in progressive and nonprogressive behavioral variant frontotemporal dementia. Neurology. 2008;71:1481–8. doi: 10.1212/01.wnl.0000334299.72023.c8. [DOI] [PubMed] [Google Scholar]
- 42.Kipps CM, Hodges JR, Fryer TD, Nestor PJ. Combined magnetic resonance imaging and positron emission tomography brain imaging in behavioural variant frontotemporal degeneration: Refining the clinical phenotype. Brain. 2009;132:2566–78. doi: 10.1093/brain/awp077. [DOI] [PubMed] [Google Scholar]
- 43.Viskontas IV, Possin KL, Miller BL. Symptoms of frontotemporal dementia provide insights into orbitofrontal cortex function and social behavior. Ann N Y Acad Sci. 2007;1121:528–45. doi: 10.1196/annals.1401.025. [DOI] [PubMed] [Google Scholar]
- 44.Adlam AL, Patterson K, Rogers TT, Nestor PJ, Salmond CH, Acosta-Cabronero J, et al. Semantic dementia and fluent primary progressive aphasia: Two sides of the same coin? Brain. 2006;129:3066–80. doi: 10.1093/brain/awl285. [DOI] [PubMed] [Google Scholar]
- 45.Knibb JA, Hodges JR. Semantic dementia and primary progressive aphasia: A problem of categorization? Alzheimer Dis Assoc Disord. 2005;19:S7–14. doi: 10.1097/01.wad.0000183085.22562.13. [DOI] [PubMed] [Google Scholar]
- 46.Rogers TT, Lambon Ralph MA, Garrard P, Bozeat S, McClelland JL, Hodges JR, et al. Structure and deterioration of semantic memory: A neuropsychological and computational investigation. Psychol Rev. 2004;111:205–35. doi: 10.1037/0033-295X.111.1.205. [DOI] [PubMed] [Google Scholar]
- 47.Hodges JR, Graham N, Patterson K. Charting the progression in semantic dementia: Implications for the organisation of semantic memory. Memory. 1995;3:463–95. doi: 10.1080/09658219508253161. [DOI] [PubMed] [Google Scholar]
- 48.Hodges JR, Martinos M, Woollams AM, Patterson K, Adlam AL. Repeat and point: Differentiating semantic dementia from progressive non-fluent aphasia. Cortex. 2008;44:1265–70. doi: 10.1016/j.cortex.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 49.Bozeat S, Lambon Ralph MA, Patterson K, Garrard P, Hodges JR. Non-verbal semantic impairment in semantic dementia. Neuropsychologia. 2000;38:1207–15. doi: 10.1016/s0028-3932(00)00034-8. [DOI] [PubMed] [Google Scholar]
- 50.Snowden JS, Thompson JC, Neary D. Knowledge of famous faces and names in semantic dementia. Brain. 2004;127:860–72. doi: 10.1093/brain/awh099. [DOI] [PubMed] [Google Scholar]
- 51.Josephs KA, Whitwell JL, Vemuri P, Senjem ML, Boeve BF, Knopman DS, et al. The anatomic correlate of prosopagnosia in semantic dementia. Neurology. 2008;71:1628–33. doi: 10.1212/01.wnl.0000334756.18558.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan D, Anderson V, Pijnenburg Y, Whitwell J, Barnes J, Scahill R, et al. The clinical profile of right temporal lobe atrophy. Brain. 2009;132:1287–98. doi: 10.1093/brain/awp037. [DOI] [PubMed] [Google Scholar]
- 53.Thompson SA, Patterson K, Hodges JR. Left/right asymmetry of atrophy in semantic dementia: Behavioral-cognitive implications. Neurology. 2003;61:1196–203. doi: 10.1212/01.wnl.0000091868.28557.b8. [DOI] [PubMed] [Google Scholar]
- 54.Ogar JM, Dronkers NF, Brambati SM, Miller BL, Gorno-Tempini ML. Progressive nonfluent aphasia and its characteristic motor speech deficits. Alzheimer Dis Assoc Disord. 2007;21:S23–30. doi: 10.1097/WAD.0b013e31815d19fe. [DOI] [PubMed] [Google Scholar]
- 55.Grossman M, Rhee J, Moore P. Sentence processing in frontotemporal dementia. Cortex. 2005;41:764–77. doi: 10.1016/s0010-9452(08)70295-8. [DOI] [PubMed] [Google Scholar]
- 56.Ogar J, Slama H, Dronkers N, Amici S, Gorno-Tempini ML. Apraxia of speech: An overview. Neurocase. 2005;11:427–32. doi: 10.1080/13554790500263529. [DOI] [PubMed] [Google Scholar]
- 57.Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: A new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol. 2008;21:688–92. doi: 10.1097/WCO.0b013e3283168ddd. [DOI] [PubMed] [Google Scholar]
- 58.Gorno-Tempini ML, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335–46. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorno-Tempini ML, Brambati SM, Ginex V, Ogar J, Dronkers NF, Marcone A, et al. The logopenic/phonological variant of primary progressive aphasia. Neurology. 2008;71:1227–34. doi: 10.1212/01.wnl.0000320506.79811.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, et al. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mesulam M, Wicklund A, Johnson N, Rogalski E, Léger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Ann Neurol. 2008;63:709–19. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashburner J, Friston KJ. Voxel-based morphometry--the methods. Neuroimage. 2000;11:805–21. doi: 10.1006/nimg.2000.0582. [DOI] [PubMed] [Google Scholar]
- 63.Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000;97:11050–5. doi: 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosen HJ, Gorno-Tempini ML, Goldman WP, Perry RJ, Schuff N, Weiner M, et al. Patterns of brain atrophy in frontotemporal dementia and semantic dementia. Neurology. 2002;58:198–208. doi: 10.1212/wnl.58.2.198. [DOI] [PubMed] [Google Scholar]
- 65.Davies RR, Halliday GM, Xuereb JH, Kril JJ, Hodges JR. The neural basis of semantic memory: Evidence from semantic dementia. Neurobiol Aging. 2009;30:2043–52. doi: 10.1016/j.neurobiolaging.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 66.Rosen HJ, Kramer JH, Gorno-Tempini ML, Schuff N, Weiner M, Miller BL. Patterns of cerebral atrophy in primary progressive aphasia. Am J Geriatr Psychiatry. 2002;10:89–97. [PubMed] [Google Scholar]
- 67.Rohrer JD, Warren JD, Modat M, Ridgway GR, Douiri A, Rossor MN, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. 2009;72:1562–9. doi: 10.1212/WNL.0b013e3181a4124e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rohrer JD, Ridgway GR, Crutch SJ, Hailstone J, Goll JC, Clarkson MJ, et al. Progressive logopenic/phonological aphasia: Erosion of the language network. Neuroimage. 2010;49:984–93. doi: 10.1016/j.neuroimage.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kipps CM, Davies RR, Mitchell J, Kril JJ, Halliday GM, Hodges JR. Clinical significance of lobar atrophy in frontotemporal dementia: Application of an MRI visual rating scale. Dement Geriatr Cogn Disord. 2007;23:334–42. doi: 10.1159/000100973. [DOI] [PubMed] [Google Scholar]
- 70.Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, et al. Clinicopathological correlates in frontotemporal dementia. Ann Neurol. 2004;56:399–406. doi: 10.1002/ana.20203. [DOI] [PubMed] [Google Scholar]
- 71.Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: Clinical and pathological relationships. Acta Neuropathol. 2007;114:31–8. doi: 10.1007/s00401-007-0236-3. [DOI] [PubMed] [Google Scholar]
- 73.Shi J, Shaw CL, Du Plessis D, Richardson AM, Bailey KL, Julien C, et al. Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol. 2005;110:501–12. doi: 10.1007/s00401-005-1079-4. [DOI] [PubMed] [Google Scholar]
- 74.Bigio EH. Update on recent molecular and genetic advances in frontotemporal lobar degeneration. J Neuropathol Exp Neurol. 2008;67:635–48. doi: 10.1097/NEN.0b013e31817d751c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 76.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 77.Roeber S, Mackenzie IR, Kretzschmar HA, Neumann M. TDP-43-negative FTLD-U is a significant new clinico-pathological subtype of FTLD. Acta Neuropathol. 2008;116:147–57. doi: 10.1007/s00401-008-0395-x. [DOI] [PubMed] [Google Scholar]
- 78.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132:2922–31. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Weintraub S, Mesulam M. With or without FUS, it is the anatomy that dictates the dementia phenotype. Brain. 2009;132:2906–8. doi: 10.1093/brain/awp286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davies RR, Hodges JR, Kril JJ, Patterson K, Halliday GM, Xuereb JH. The pathological basis of semantic dementia. Brain. 2005;128:1984–95. doi: 10.1093/brain/awh582. [DOI] [PubMed] [Google Scholar]
- 81.Hodges JR, Mitchell J, Dawson K, Spillantini MG, Xuereb JH, McMonagle P, et al. Semantic dementia: Demography, familial factors and survival in a consecutive series of 100 cases. Brain. 2010;133:300–6. doi: 10.1093/brain/awp248. [DOI] [PubMed] [Google Scholar]
- 82.Mesulam M. Primary progressive aphasia pathology. Ann Neurol. 2008;63:124–5. doi: 10.1002/ana.20940. [DOI] [PubMed] [Google Scholar]
- 83.Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain. 2006;129:1385–98. doi: 10.1093/brain/awl078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, et al. Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71:1220–6. doi: 10.1212/01.wnl.0000319702.37497.72. [DOI] [PubMed] [Google Scholar]
- 85.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of frontotemporal dementia. Arch Neurol. 1999;56:817–22. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, et al. Association of missense and 5’-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–5. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 87.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 88.Pickering-Brown SM. Recent progress in frontotemporal lobar degeneration. Neuropathol Appl Neurobiol. 2010;36:4–16. doi: 10.1111/j.1365-2990.2009.01045.x. [DOI] [PubMed] [Google Scholar]
- 89.Sikkink S, Rollinson S, Pickering-Brown SM. The genetics of frontotemporal lobar degeneration. Curr Opin Neurol. 2007;20:693–8. doi: 10.1097/WCO.0b013e3282f1c961. [DOI] [PubMed] [Google Scholar]
- 90.Scarpini E, Galimberti D, Bresolin N. Genetics and neurobiology of frontotemporal lobar degeneration. Neurol Sci. 2006;27:S32–4. doi: 10.1007/s10072-006-0543-9. [DOI] [PubMed] [Google Scholar]
- 91.van Swieten JC, Heutink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008;7:965–74. doi: 10.1016/S1474-4422(08)70194-7. [DOI] [PubMed] [Google Scholar]
- 92.van Swieten J, Spillantini MG. Hereditary frontotemporal dementia caused by Tau gene mutations. Brain Pathol. 2007;17:63–73. doi: 10.1111/j.1750-3639.2007.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]