

Abstract
We aimed to determine whether an increased rate of long-chain fatty acid (LCFA) transport and/or a reduction in mitochondrial oxidation contributes to lipid deposition in hearts, as lipid accumulation within cardiac muscle has been associated with heart failure. In hearts of lean and obese Zucker rats we examined: (a) triacylglycerol (TAG) and mitochondrial content and distribution using transmission electron microscopy (TEM), (b) LCFA oxidation in cardiac myocytes, and in isolated subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria, and (c) rates of LCFA transport into cardiac vesicles. Compared to lean rats, in obese Zucker rats, lipid droplet size was similar but there were more (P < 0.05) droplets, and TAG esterification rates and contents were markedly increased. TEM analyses and biochemical determinations showed that SS and IMF mitochondria in obese animals did not appear to be different in their appearance, area, density and number, nor in citrate synthase, β-hydroxy-acyl-CoA dehydrogenase and carnitine palmitoyl-transferase-I enzymatic activities, electron transport chain proteins, nor in their rates of LCFA oxidation either in cardiac myocytes or in isolated SS and IMF mitochondria (P > 0.05). In contrast, sarcolemmal plasma membrane fatty acid binding protein (FABPpm) and fatty acid translocase (FAT/CD36) protein and palmitate transport rates into cardiac vesicles were increased (P < 0.05; +50%) in obese animals. Collectively these data indicate that mitochondrial dysfunction in LCFA oxidation is not responsible for lipid accumulation in obese Zucker rat hearts. Rather, increased sarcolemmal LCFA transport proteins and rates of LCFA transport result in a greater number of lipid droplets within cardiac muscle.
Non-technical summary
The storage of fat within the heart muscle has been associated with reductions in force production, which has implications for the ability of the heart to adequately pump blood. With the assistance of membrane proteins known as transport proteins, fats from the blood can be moved into heart muscle cells, where they can either be stored or used for generating energy (within a structure called mitochondria) to pump blood. We provide evidence that in obese animals more fat accumulates within the heart as a result of their increased transport across the membranes of heart cells, not due to reductions in mitochondrial number or function. The knowledge of why fat accumulates in the heart may provide insight into novel treatments/therapies, and the current study suggests therapies focused on limiting the entry of fats into the heart may restore the ability of the heart to pump blood.
Introduction
Obesity, insulin resistance and dyslipidaemia are strong risk factors for the development of cardiovascular disease and the pathogenesis of atherosclerosis. While dyslipidaemia and elevated plasma lipid profiles have well-defined roles in the generation of atherosclerotic plaque formation, recent evidence also suggests that lipid accumulation within cardiac muscle may participate in the progression of heart failure.
In healthy hearts, approximately 70–90% of the fatty acids taken up are immediately oxidized (Wisneski et al. 1987; Chandler et al. 2003), while only a small proportion is esterified to triacylglycerol (TAG). Nevertheless, an accumulation of TAG has been associated with cardiac contractile dysfunction (reviewed in Stanley et al. 2005). The exact mechanism(s) responsible for TAG accumulation remains unknown, but may involve (1) a dysfunction in mitochondrial fatty acid oxidation, and/or (2) an increase in sarcolemmal long-chain fatty acid (LCFA) transport. It has previously been shown in obese Zucker rats that rates of LCFA transport across cardiac sarcolemmal membranes (Coort et al. 2004) are increased, but whether there is also a dysfunction in mitochondrial LCFA oxidation is more controversial. Such a dysfunction in mitochondrial LCFA oxidation appears to be present in Akita mice, a model of type 1 diabetes, as fatty acid oxidation in isolated mitochondria is reduced, along with reductions in electron transport chain (ETC) proteins (Bugger et al. 2009). In contrast, in some models of insulin resistance cardiac LCFA oxidation is not impaired (Coort et al. 2004), and is occasionally increased (Belke et al. 2000; Neitzel et al. 2003; Mazumder et al. 2004; Wang et al. 2005). Whether these observations reflect concurrent changes in the capacity of mitochondrial LCFA oxidation and/or their number is unknown. Thus, whether cardiac TAG accumulation can be attributed to a dysfunction in mitochondrial LCFA oxidation remains uncertain.
Mitochondria exist in two sub-cellular compartments, creating another level of complexity in determining a role for mitochondrial dysfunction-induced lipid accumulation. Based on their spatial locations, these distinct mitochondrial populations have been termed subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria, and are located directly beneath the sarcolemmal membrane and between myofibrils, respectively. SS and IMF mitochondria have different characteristics in their size, enzymatic activities and rates of LCFA oxidation (Palmer et al. 1985; Hoppel et al. 2002; Kelley et al. 2002; Holloway et al. 2009). In addition, these mitochondrial sub-populations respond differently to various physiological perturbations, including ischaemia-reperfusion injury (Lesnefsky et al. 2001) and streptozotocin-induced diabetes (Dabkowski et al. 2009). However, it is not currently known whether or not dysfunction in LCFA oxidation in either of these mitochondrial sub-populations contribute to TAG accumulation.
The purpose of the current study was to determine the extent of a possible imbalance between LCFA transport and oxidation in the generation of intramuscular lipid depots. To this end, we have measured in hearts of lean and obese Zucker rats: (1) plasma membrane LCFA transport and transport proteins, (2) rates of LCFA oxidation in cardiac myocytes and isolated SS and IMF mitochondria, (3) markers of mitochondrial LCFA transport (carnitine palmitoyl-tranferase I activity), (4) matrix enzymes (citrate synthase and β-hydroxyacyl-CoA dehydrogenase activities), (5) ETC proteins, and (6) mitochondrial morphology and lipid content using transmission electron microscopy (TEM). We have not found any evidence that alterations in mitochondrial fatty acid oxidation contribute to TAG accumulation within obese Zucker rat hearts.
Methods
Animals
Female lean (n = 15, weighing ∼225 g) and obese Zucker (fa/fa; n = 15, weighing ∼350 g) rats were purchased from Charles River, and all efforts were made to minimize the number of rats used (Drummond, 2009). Animals were housed in a climate- and temperature-controlled room, on a 12:12 h light–dark cycle, with rat chow and water provided ad libitum for 2 weeks prior to the start of experiments. Animals were anaesthetized with an intraperitoneal injection of sodium pentobarbital (60 mg kg−1) and the heart was excised. The left ventricles were rapidly dissected: (1) a small (3 × 1 × 1 mm) section from the lumen of the ventricle was placed into fixing buffer (described below) for TEM analysis, (2) a 30–50 mg section was rapidly frozen for TAG analysis, (3) a 5–10 mg section was used to measure citrate synthase (CS) and β-hydroxyacyl-CoA dehydrogenase (β-HAD) activities, and (4) the remainder was used to isolate mitochondria for palmitate oxidation, carnitine palmitoyl-tranferase I activity (CPTI) activity, Western blotting, the generation of giant sarcolemmal vesicles and cardiac myocytes. This study was approved by the University of Guelph Animal Care Committee, and conforms to the guide for the care and use of laboratory animals published by the US National Institutes of Health.
Transmission electron microscope analysis of mitochondria
Samples were rapidly immersed in a fixing buffer (2.5% glutaraldehyde, 1.0% parafermaldehyde in PBS, BioRad, Mississauga, ON) and incubated at 4°C overnight. Tissue was then washed 3 times in 0.1 m Hepes and subsequently suspended in 1.0% osmium tetroxide for 4 h. Thereafter, tissue was washed 3 times in 100 mm Hepes and suspended in 2% uranyl acetate for 3 h, washed 3 times in 0.1% Hepes, and dehydrated by incubating in a graded ethanol series (i.e. 25, 50, 75, 95 and twice in 100% ethanol). Tissue was infiltrated with resin by suspending in 50/50 ethanol/resin (London Resin Company White) for 4 h on a rotating mixer, and subsequently suspended in pure resin for 4 h on a rotating mixer. Tissue was then placed in an embedding capsule containing pure resin, and incubated overnight at 60°C to polymerize. Sections (100 nm) were cut and laid onto 200 mesh formvar–carbon copper grids and then stained with 2% uranyl acetate and Reynold's lead citrate. A minimum of three sections were laid onto each grid (range 3–5), and labelled with serial numbering such that individuals quantifying images were blinded to the phenotype of the animals. Two investigators quantified the images, and the inter- and intra-investigator variability tests showed no signs of bias and low coefficients of variation (<10%). From each fibre 20 images were obtained in a randomized systematic order, including 12 at 64,000× magnification, 4 at 10,500× magnification and 4 at 5800× magnification. Samples were viewed on a Philips CM 10 TEM at 80 kV, and images obtained with an Olympus/SIS Morada CCD camera using the Olympus/SIS iTEM software. Images were analysed using the measurement tools provided by this software. Individual lipid droplet and mitochondrial sizes were determined repeatedly and averaged for a given animal. Mitochondrial sub-population densities were determined by measuring the total mitochondrial area within a defined 10 μm2 box (3.33 μm × 3.33 μm) that was identical for all measures and was randomly placed in a minimum of eight locations within an image, similar to methodologies we (Holloway et al. 2010) and others (Marchand et al. 2007) have previously published. Lipid droplet number was normalized to the area of the muscle visualized. Several cardiac muscle fibres (3–5) were imaged in this manner for each animal and averaged.
Intramuscular triacylglycerol content
Total TAG content was determined in heart homogenates from lean and obese Zucker rats. Lipids were extracted and separated by high-performance thin-layer chromatography (HPTLC), and quantified as previously described (Coort et al. 2004).
Cardiac myocytes
Cardiac myocytes from lean and obese Zucker rats were isolated using a Langendorff perfusion system and a modified Krebs–Henseleit bicarbonate medium (Fischer et al. 1991; Luiken et al. 1997), and used to determine rates of [1-14C]palmitate oxidation and TAG esterification as previously described (Glatz et al. 1984).
Preparation of giant vesicles and palmitate transport
Giant vesicles were generated from hearts, and subsequently used for the measurement of palmitate uptake and Western blotting as previously described (Bonen et al. 1998; Luiken et al. 2001, 2002; Koonen et al. 2002).
Mitochondrial enzymatic activities
Samples (∼10 mg) were homogenized in 100 vol/wt of a 100 mm potassium phosphate buffer (Srere, 1969) and used for the measurements of CS and β-HAD. Total CS activity was assayed spectrophotometrically at 412 nm (37°C) (Srere, 1969), and β-HAD activity was measured at 340 nm (37°C) (Bergmeyer, 1974). CS and β-HAD activities were also determined in isolated mitochondria after lysing the mitochondria with 0.04% Triton X-100 and repeated freeze–thawing. Homogenate values were expressed per gram wet weight, while mitochondrial values were normalized to mitochondrial protein.
Isolation of mitochondria
Differential centrifugation was used to obtain both SS and IMF mitochondrial fractions, as previously published by our group (Holloway et al. 2009).
Carnitine palmitoyl-transferase I activity
The forward radioisotope assay was used for the determination of CPTI activity as described by McGarry et al. (1983) with minor modifications as we have previously reported (Holloway et al. 2009).
Western blotting
All samples were analysed for total protein (BCA protein assay), and 5 μg of isolated mitochondria and 10 μg sarcolemmal vesicles were loaded. Samples were separated by electrophoresis on SDS–polyacrylamide gels and transferred to polyvinylidene difluoride membranes. The polyclonal antibody to detect plasma membrane fatty acid binding protein (FABPpm) was produced in J. Calles-Escandon's laboratory, and the MO-25 antibody used to detect fatty acid translocase (FAT/CD36) was produced in N. N. Tandon's laboratory. A commercially available antibody was used to detect cytochrome c oxidase subunit IV (COXIV; Invitrogen, Burlington, ON, Canada), and an antibody cocktail was used to detect complexes I–V of the electron transport chain (Mitosciences, Eugene, OR, USA). All samples for a given protein were transferred and developed on the same membrane to limit variation. Blots were quantified using chemiluminescence and the ChemiGenius 2 Bioimaging system (SynGene, Cambridge, UK).
Mitochondrial palmitate oxidation
Labelled 14CO2 production from palmitate oxidation and acid-soluble trapped 14C were measured in a sealed system, as previously described by our laboratory (Holloway et al. 2009).
Statistics
All data are presented as the mean ± standard error of the mean (s.e.m.). Unpaired t tests and two-way analysis of variance were used where appropriate, and when significance was obtained a Fisher's LSD post hoc analysis was completed. Statistical significance was accepted at P < 0.05.
Results
Intramyocellular triacylglycerols
To examine TAG in the hearts of lean and obese Zucker rats we have examined the subcellular TAG deposition in TEM images in combination with biochemical determinations of cardiac TAG content and rates of esterification. These approaches have revealed that the size of each lipid droplet is not different between lean and obese animals (Fig. 1A–C); however, the number of lipid droplets (lean, 0.019 ± 0.004 lipid droplets μm−2vs. obese, 0.033 ± 0.003 lipid droplets μm−2), TAG content (Fig. 1D) and esterification rates (Fig. 1E) are all increased (P < 0.05) in hearts of obese animals.
Figure 1. TEM images (A–C) and biochemical determination of triacylglycerol (TAG) content (D) and TAG esterification rates (E) in heart muscle from lean and obese Zucker rats.
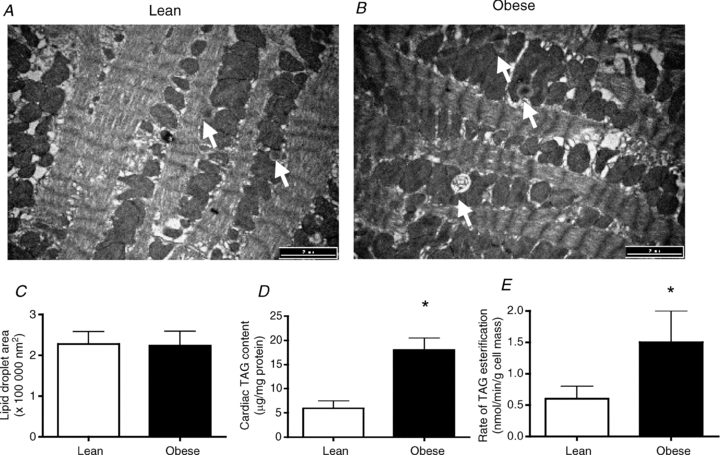
Data are expressed as the mean ± s.e.m. Images were taken at 10,500× magnification, and the black scale bar is 2 μm. n = 5–6 animals for each experiment. White arrows indicate lipid droplets. *Significantly different (P < 0.05) from lean animals.
Plasma membrane fatty acid transport proteins
The content of FABPpm (+50%; Fig. 2A) and FAT/CD36 (+60%; Fig. 2B), and rates of palmitate transport (∼2-fold; Fig. 2C) were higher (P < 0.05) in giant sarcolemmal vesicles generated from obese Zucker rats. However, palmitate oxidation in isolated cardiac myocytes generated from lean and obese Zucker rats was not different (Fig. 2D).
Figure 2. Plasma membrane fatty acid binding protein (FABPpm; A), fatty acid translocase (FAT/CD36; B), and rates of palmitate transport (C) in giant sarcolemmal vesicles, as well as rates of palmitate oxidation in cardiac myocytes (D), from lean and obese Zucker rats.
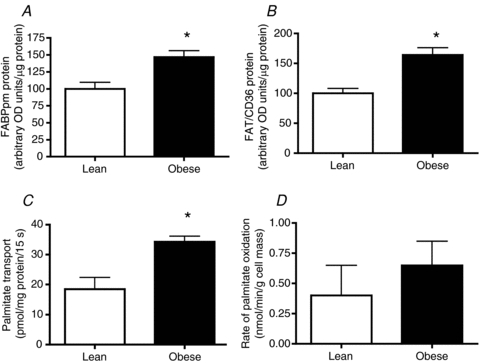
Data are expressed as the mean ± s.e.m.n = 4–5 animals for each experiment. *Significantly different (P < 0.05) from lean animals.
Mitochondrial morphology and content
Qualitative assessment of mitochondria in lean and obese animals did not reveal any obvious differences with respect to cristae density (Fig. 3A) and general appearance (Figs 1A and B, and 3A and B). In addition, the area of individual mitochondria (Fig. 3C) as well as the overall mitochondrial density (Fig. 3B and D) were not altered (P > 0.05) with obesity. The similarity in the activities of mitochondrial marker enzymes β-HAD and CS (Fig. 4A and B, respectively) indicates that total content is not altered in obese Zucker rats. Our analyses could not quantify the number of SS and IMF mitochondria within a given area.
Figure 3. Subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondrial area (A and C) and density (B and D) in heart muscle from lean and obese Zucker rats.
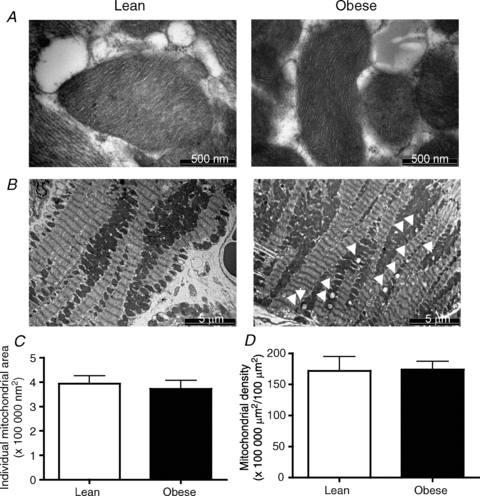
Data are expressed as the mean ± s.e.m. Images were taken either at 64,000× (A) or 5,800× (B) magnification, and the black scale bar is 500 nm (A) or 5 μm (B), respectively. n = 6 animals for each experiment. White arrowheads indicate lipid droplets.
Figure 4. β-hydroxyacyl-CoA dehydrogenase (β-HAD), citrate synthase (CS) and carnitine palmitoyl-transferase I (CPTI) enzymatic activities in whole muscle (A and B) and isolated subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria (C–E) in lean and obese Zucker rats.
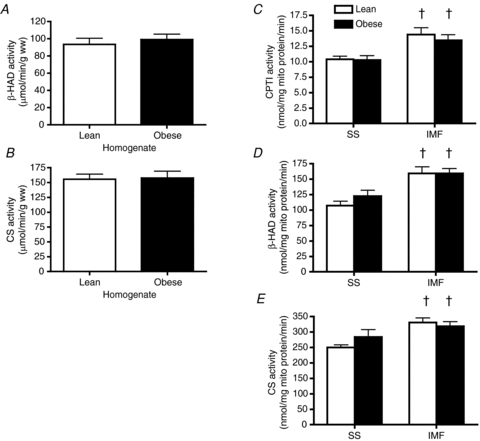
Data are expressed as the mean ± s.e.m.n = 6 animals for each experiment. †Significantly different (P < 0.05) from SS mitochondria.
Mitochondrial enzymatic activities in SS and IMF mitochondria
In isolated mitochondria, CPTI (Fig. 4C), β-HAD (Fig. 4D) and CS (Fig. 4E) enzymatic activities were higher (P < 0.05) in IMF compared to SS mitochondria. However, these enzymatic activities were not different in lean and obese rats.
Contents of electron transport chain proteins in SS and IMF mitochondria
In isolated mitochondria the content of nuclear transcribed ETC proteins, complexes I (Fig. 5A) II (Fig. 5B), III (Fig. 5C), IV subunit 4 (Fig. 5E), and V (Fig. 5F), and mitochondrial transcribed proteins, complex IV subunit 1 (Fig. 5D), were not affected by obesity in either SS or IMF mitochondria. While not affected by obesity, complex III was lower (P < 0.05) in IMF mitochondria, whereas complexes I, II, IV (subunits I and IV) and V were not different in mitochondrial subpopulations.
Figure 5. Content of electron transport chain proteins in subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria in lean (L) and obese (O) Zucker rats.
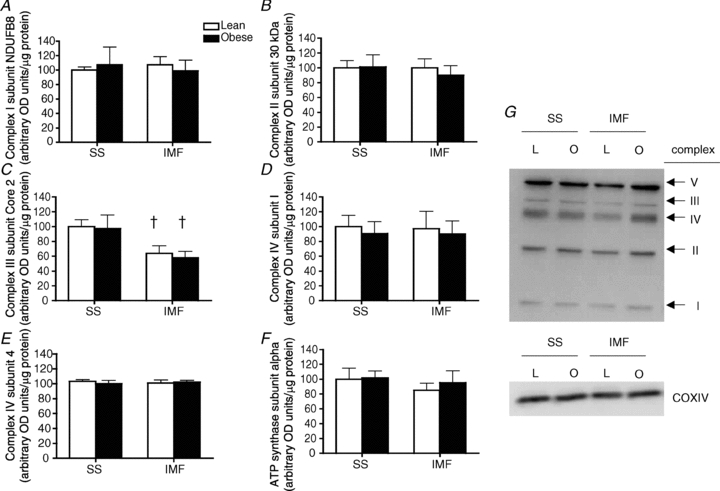
Complex 1 subunit NDUFB8 (A); complex II subunit 30 kDa (B); complex III subunit Core 2 (C); complex IV subunit 1 (D); complex IV subunit 4 (E); ATP synthase subunit α (F); and representative blots (G). Data are expressed as the mean ± s.e.m.n = 6 animals for each experiment. †Significantly different (P < 0.05) from SS mitochondria
Mitochondrial fatty acid oxidation rates
Rates of palmitate oxidation were higher in IMF compared to SS mitochondria in both lean and obese rats; however, obesity did not alter rates of palmitate oxidation (Fig. 6A). Ratios of acid-soluble intermediates to CO2 production have previously been used to infer incomplete oxidation of fatty acids and mitochondrial dysfunction (i.e. an imbalance between TCA and ETC fluxes) in skeletal (Koves et al. 2008) and cardiac (Ussher et al. 2009) muscle of insulin-resistant animals. However, in both SS and IMF mitochondria CO2 production (Fig. 6B), acid-soluble intermediates (Fig. 6C) and subsequently the ratio of acid-soluble intermediates/CO2 (Fig. 6D) were not altered with obesity.
Figure 6. Subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondrial palmitate oxidation (A), contribution of CO2 (B), acid soluble intermediates (ASM; C) and the ratio of ASM/CO2 (D) in lean and obese Zucker rats.

Data are expressed as the mean ± s.e.m.n = 6 animals for each experiment. †Significantly different (P < 0.05) from SS mitochondria.
Discussion
We demonstrate in cardiac muscle from obese Zucker rats that: (1) TAG concentration is increased as a result of an increased number of lipid droplets, not from an increase in the size of the lipid droplet. This may result from (2) an increase in LCFA transport across the plasma membrane, as a result of (3) increased plasma membrane LCFA transport proteins. However, SS and IMF mitochondria were unaltered with respect to; (4) morphological appearance, (5) content, (6) rates of LCFA oxidation, (7) CPTI, β-HAD and CS activities, and (8) ETC protein contents. Therefore, it appears that lipids accumulate from an increased rate of LCFA transport into hearts of obese Zucker rats, not from alterations in SS or IMF mitochondrial LCFA oxidation.
Intramyocellular lipids are more prevalent in number, not size, in obese Zucker rats
As expected, we show that TAG content is increased substantially (∼3-fold) in obese Zucker rat cardiac muscle. This finding is supported by previous literature in rodent models of insulin resistance (Sharma et al. 2004), in addition to recent findings in humans that show cardiac muscle TAG concentrations are increased with insulin resistance (Marfella et al. 2009). This TAG increase is a result of an increase in the number of lipid droplets, not their increased size. In addition, it appears that the majority of the lipid droplets are associated with IMF mitochondria, similar to skeletal muscle (Hoppeler, 1986; Tarnopolsky et al. 2007). While this relationship may suggest indirectly that lipid oxidation in IMF mitochondria is reduced, such that lipids accumulate in IMF mitochondrial-associated lipid droplets, direct measurements in isolated mitochondria did not support this notion.
Mitochondrial content and LCFA oxidation are not altered in obese Zucker rats
Rates of mitochondrial LCFA oxidation can be influenced by both the content of mitochondria, and the intrinsic function of mitochondria (i.e. palmitate oxidation in isolated mitochondria). We have used TEM images of mitochondria and biochemical enzymatic activities of mitochondrial proteins (CS and β-HAD) to determine cardiac mitochondrial content. Both approaches suggest that mitochondrial content is unaltered in obese Zucker rat cardiac muscle. Furthermore, TEM images of individual mitochondria indicated these to be normal, as we did not observe swelling or alterations in mitochondrial size or cristae density in either SS or IMF mitochondria, suggesting unaltered function. This was confirmed with biochemical measurements, including unaltered CPTI, CS and β-HAD enzymatic activities, ETC proteins (complexes I–V) and palmitate oxidation rates in isolated SS and IMF mitochondria. Altogether these data suggest that mitochondrial content and the intrinsic ability of mitochondria to oxidize fatty acids are not altered in obese Zucker rats, suggesting that total cardiac LCFA oxidation rates would not be altered. This was confirmed using cardiac myocytes (Fig. 2D). Unaltered CS activity and ETC protein contents, along with an unaltered acid-soluble intermediate/CO2 ratio in SS and IMF mitochondria, a possible surrogate measure for an imbalance between the TCA cycle and the ETC (Koves et al. 2008), would suggest that flux through the TCA cycle and the ETC is not disproportionate in obese Zucker rats.
In the current study, rates of palmitate oxidation and enzymatic rates of CPTI, CS and β-HAD were all greater in IMF mitochondria. These data are supported by studies that have reported similarly greater respiration rates in IMF mitochondria in the presence of a variety of substrates in both skeletal (Cogswell et al. 1993; Ferreira et al. 2010) and cardiac muscle (Chen et al. 2008). In contrast, in the current study all components of the ETC examined were either not different (complexes I, II, IV and V) or reduced (complex III) in IMF mitochondria. However, each component of the ETC is comprised of multiple proteins (e.g. complex I has 46 proteins), and given the dissociation between IMF mitochondrial palmitate oxidation and the abundance of specific ETC proteins, it is likely that the subunits examined in the current study are not limiting for electron flux.
The current data in insulin-resistant obese Zucker rats support recent work in type I diabetic Akita mice, in which state III mitochondrial respiration in the presence of palmitoyl-carnitine is unaltered (Bugger et al. 2008). In addition, mitochondria isolated from Akita mice are unaltered with respect to ATP synthesis rates, coupling, proton leak and number (Bugger et al. 2008). Surprisingly, this same group has recently reported that state III respiration rates in the presence of either glutamate or succinate are reduced in Akita mice (Bugger et al. 2009). This may suggest that while maximal flux through the ETC is diminished in this animal model, fatty acid oxidation is limited by processes proximal to the ETC, including β-oxidation and/or electron flux through electron transfer flavoprotein (ETF) and ETF-ubiquinone oxidoreductase. This may suggest a central role of the ETC proteins in mediating mitochondrial dysfunctions in oxidative respiration. Importantly, we did not observe alterations in ETC proteins or rates of fatty acid oxidation in the current study. However, more severe exposure to a lipotoxic environment may, through reactive oxygen species (ROS) -mediated events, induce a dysfunction in mitochondrial fatty acid oxidation. In this respect it may be feasible to speculate that the IMF mitochondria, given the close proximity to the lipid droplets, may be more susceptible to intrinsic alterations in LCFA oxidation. In support of this, a recent study has reported that in type I diabetic hearts, while SS mitochondria remain largely unaltered, in IMF mitochondria superoxide production and oxidative damage are increased, while ETC proteins are reduced (Dabkowski et al. 2009). Regardless of the mitochondrial role in ROS-mediated events, the current study does not support the notion that mitochondrial dysfunction in fatty acid oxidation induces lipid accumulation in cardiac muscle. A similar situation appears to exist in skeletal muscle, as several groups have also determined that mitochondrial dysfunction is not responsible for lipid accumulation in this tissue (Turner et al. 2007; Bonnard et al. 2008; Hancock et al. 2008; Kraegen et al. 2008).
Plasma membrane LCFA transport is increased in obese Zucker rats
The transport of LCFAs is primarily a protein-mediated event, involving several transport proteins including FAT/CD36 and FABPpm (reviewed in Luiken et al. 1999). These two transport proteins may work in a concerted effort to transport LCFAs into cardiac muscle (Chabowski et al. 2007), and both transport proteins have been shown to translocate from an intracellular depot to the plasma membrane in response to various stimuli (Luiken et al. 2001, 2003; Koonen et al. 2004; Jain et al. 2009). Here we show that the plasma membrane LCFA transport (∼3-fold) and contents of both FABPpm (∼50%) and FAT/CD36 (∼60%) are increased in obese Zucker rat cardiac muscle. These findings substantiate our previous observations in both cardiac (Luiken et al. 2001) and skeletal muscle (Luiken et al. 2001; Holloway et al. 2009) of obese Zucker rats. In all of these previous studies we have not observed a change in the total protein content of either transport protein, which suggests that these transport proteins have been redistributed to the plasma membrane. While the mechanism(s) responsible for redistributing these transport proteins to the plasma membrane remain(s) to be elucidated, the result is an increase in plasma membrane LCFA transport, which would be exacerbated in vivo given the higher circulating plasma free fatty acid concentrations. Given that mitochondrial content and oxidation rates of LCFA remained unaltered in obese Zucker rats, we suggest that TAGs do not accumulate from alterations in mitochondria, but as a result of increased membrane transport. This is consistent with our recent work showing inhibition of mitochondrial LCFA oxidation in healthy hearts (via etomoxir CPTI inhibition) does not induce TAG accumulation (Luiken et al. 2009), and is supported by the observation that the insulin-sensitizing effect of piaglitazone is not mediated through an increase in LCFA oxidation (van der Meer et al. 2009). Given that cardiac TAG accumulation has been associated with contractile dysfunction (Sharma et al. 2004), therapeutic strategies that decrease TAG content may be beneficial. In this respect, internalization of plasma membrane transport proteins may be particularly advantageous.
Summary
We have shown that mitochondrial content, morphology, LCFA oxidation rates (cardiac myocytes and isolated SS and IMF mitochondria), CPTI, β-HAD and CS activities, ETC protein contents are not altered in cardiac muscle of obese Zucker rats, while plasma membrane LCFA transport is substantially increased. The imbalance and subsequent accumulation of lipid droplets in the IMF region therefore appears to result solely from the pronounced increase in plasma membrane LCFA transport as a direct result of increased plasma membrane contents of both FAT/CD36 and FABPpm, and not from any alteration in mitochondria.
Acknowledgments
This work was funded by the Heart and Stroke Foundation of Ontario (A.B.) and the Natural Sciences and Engineering Research Council of Canada (G.P.H. and A.B.), The Netherlands Heart Foundation grant 2002.T049, the Netherlands Organization for Health Research and Development (NWO-ZonMw grant 40-00812-98-03075), and the European Commission (Integrated Project LSHM-CT-2004-005272, Exgenesis). J.J.F.P.L. was the recipient of a VIDI-Innovational Research Grant from the Netherlands Organization of Scientific Research (NWO-ZonMw Grant 016.036.305). J.F.C.G. is the Netherlands Heart Foundation Professor of Cardiac Metabolism. A.B. is the Canada Research Chair in Metabolism and Health.
Glossary
Abbreviations
- β-HAD
β-hydroxyacyl-CoA dehydrogenase
- BCA
bicinchoninic acid
- CPTI
carnitine palmitoyl-transferase I
- CS
citrate synthase
- COXIV
cytochrome c oxidase subunit IV
- ETC
electron transport chain
- ETF
electron transfer flavoprotein
- FAT/CD36
fatty acid translocase
- IMF
intermyofibrillar
- LCFA
long-chain fatty acid
- FABPpm
plasma membrane fatty acid binding protein
- ROS
reactive oxygen species
- SS
subsarcolemmal
- TEM
transmission electron microscopy
- TAG
triacylglycerol
- TCA
tricarboxylic acid cycle
Author contributions
G.P.H. contributed to study design and data interpretation, performed experiments and wrote the manuscript. L.A.S., R.J.H., J.F.C.G. and J.J.F.P.L. performed experiments and edited the manuscript. A.B. contributed to study design, data interpretation and editing of the manuscript. All authors approved the final version of this manuscript.
References
- Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- Bergmeyer HU. Methods of Enzymatic Analysis. Vol. 4. New York: Verlag Chemie Weinheim; 1974. [Google Scholar]
- Bonen A, Luiken JJ, Liu S, Dyck DJ, Kiens B, Kristiansen S, Turcotte LP, Van Der Vusse GJ, Glatz JF. Palmitate transport and fatty acid transporters in red and white muscles. Am J Physiol Endocrinol Metab. 1998;275:E471–E478. doi: 10.1152/ajpendo.1998.275.3.E471. [DOI] [PubMed] [Google Scholar]
- Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57:2924–2932. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugger H, Chen D, Riehle C, Soto J, Theobald HA, Hu XX, Ganesan B, Weimer BC, Abel ED. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58:1986–1997. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabowski A, Gorski J, Luiken JJ, Glatz JF, Bonen A. Evidence for concerted action of FAT/CD36 and FABPpm to increase fatty acid transport across the plasma membrane. Prostaglandins Leukot Essent Fatty Acids. 2007;77:345–353. doi: 10.1016/j.plefa.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Chandler MP, Chavez PN, McElfresh TA, Huang H, Harmon CS, Stanley WC. Partial inhibition of fatty acid oxidation increases regional contractile power and efficiency during demand-induced ischemia. Cardiovasc Res. 2003;59:143–151. doi: 10.1016/s0008-6363(03)00327-4. [DOI] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol. 2008;294:C460–C466. doi: 10.1152/ajpcell.00211.2007. [DOI] [PubMed] [Google Scholar]
- Cogswell AM, Stevens RJ, Hood DA. Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am J Physiol Cell Physiol. 1993;264:C383–C389. doi: 10.1152/ajpcell.1993.264.2.C383. [DOI] [PubMed] [Google Scholar]
- Coort SL, Hasselbaink DM, Koonen DP, Willems J, Coumans WA, Chabowski A, Van Der Vusse GJ, Bonen A, Glatz JF, Luiken JJ. Enhanced sarcolemmal FAT/CD36 content and triacylglycerol storage in cardiac myocytes from obese zucker rats. Diabetes. 2004;53:1655–1663. doi: 10.2337/diabetes.53.7.1655. [DOI] [PubMed] [Google Scholar]
- Dabkowski ER, Williamson CL, Bukowski VC, Chapman RS, Leonard SS, Peer CJ, Callery PS, Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol. 2009;296:H359–H369. doi: 10.1152/ajpheart.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA, Amado F. Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics. 2010;10:3142–3154. doi: 10.1002/pmic.201000173. [DOI] [PubMed] [Google Scholar]
- Fischer Y, Rose H, Kammermeier H. Highly insulin-responsive isolated rat heart muscle cells yielded by a modified isolation method. Life Sci. 1991;49:1679–1688. doi: 10.1016/0024-3205(91)90310-8. [DOI] [PubMed] [Google Scholar]
- Glatz JF, Jacobs AE, Veerkamp JH. Fatty acid oxidation in human and rat heart. Comparison of cell-free and cellular systems. Biochim Biophys Acta. 1984;794:454–465. doi: 10.1016/0005-2760(84)90012-2. [DOI] [PubMed] [Google Scholar]
- Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci U S A. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway GP, Benton C, Mullen KL, Yoshida Y, Snook LA, Han XX, Glatz JF, Luiken JJ, Lally J, Dyck DJ, Bonen A. In obese rat muscle transport of palmitate is increased and is channeled to triacylglycerol storage despite an increase in mitochondrial palmitate oxidation. Am J Physiol Endocrinol Metab. 2009;296:E738–E747. doi: 10.1152/ajpendo.90896.2008. [DOI] [PubMed] [Google Scholar]
- Holloway GP, Gurd BJ, Snook LA, Lally J, Bonen A. Compensatory increases in nuclear PGC1a protein are primarily associated with subsarcolemmal mitochondrial adaptations in ZDF rats. Diabetes. 2010;59:819–828. doi: 10.2337/db09-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppel CL, Moghaddas S, Lesnefsky EJ. Interfibrillar cardiac mitochondrial comples III defects in the aging rat heart. Biogerontology. 2002;3:41–44. doi: 10.1023/a:1015251212039. [DOI] [PubMed] [Google Scholar]
- Hoppeler H. Exercise-induced ultrastructural changes in skeletal muscle. Int J Sports Med. 1986;7:187–204. doi: 10.1055/s-2008-1025758. [DOI] [PubMed] [Google Scholar]
- Jain SS, Chabowski A, Snook LA, Schwenk RW, Glatz JF, Luiken JJ, Bonen A. Additive effects of insulin and muscle contraction on fatty acid transport and fatty acid transporters, FAT/CD36, FABPpm, FATP1, 4 and 6. FEBS Lett. 2009;583:2294–2300. doi: 10.1016/j.febslet.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51:2944–2950. doi: 10.2337/diabetes.51.10.2944. [DOI] [PubMed] [Google Scholar]
- Koonen DP, Benton CR, Arumugam Y, Tandon NN, Calles-Escandon J, Glatz JF, Luiken JJ, Bonen A. Different mechanisms can alter fatty acid transport when muscle contractile activity is chronically altered. Am J Physiol Endocrinol Metab. 2004;286:E1042–E1049. doi: 10.1152/ajpendo.00531.2003. [DOI] [PubMed] [Google Scholar]
- Koonen DP, Coumans WA, Arumugam Y, Bonen A, Glatz JF, Luiken JJ. Giant membrane vesicles as a model to study cellular substrate uptake dissected from metabolism. Mol Cell Biochem. 2002;239:121–130. [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, Bain J, Stevens R, Dyck JR, Newgard CB, Lopaschuk GD, Muoio DM. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Kraegen EW, Cooney GJ, Turner N. Muscle insulin resistance: a case of fat overconsumption, not mitochondrial dysfunction. Proc Natl Acad Sci U S A. 2008;105:7627–7628. doi: 10.1073/pnas.0803901105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnefsky EJ, Slabe TJ, Stoll MS, Minkler PE, Hoppel CL. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H2770–H2778. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Arumugam Y, Dyck DJ, Bell RC, Pelsers MM, Turcotte LP, Tandon NN, Glatz JF, Bonen A. Increased rates of fatty acid uptake and plasmalemmal fatty acid transporters in obese Zucker rats. J Biol Chem. 2001;276:40567–40573. doi: 10.1074/jbc.M100052200. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Coort SL, Willems J, Coumans WA, Bonen A, Van Der Vusse GJ, Glatz JF. Contraction-induced fatty acid translocase/CD36 translocation in rat cardiac myocytes is mediated through AMP-activated protein kinase signaling. Diabetes. 2003;52:1627–1634. doi: 10.2337/diabetes.52.7.1627. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Koonen DP, Willems J, Zorzano A, Becker C, Fischer Y, Tandon NN, Van Der Vusse GJ, Bonen A, Glatz JF. Insulin stimulates long-chain fatty acid utilization by rat cardiac myocytes through cellular redistribution of FAT/CD36. Diabetes. 2002;51:3113–3119. doi: 10.2337/diabetes.51.10.3113. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Niessen HE, Coort SL, Hoebers N, Coumans WA, Schwenk RW, Bonen A, Glatz JF. Etomoxir-induced partial carnitine palmitoyltransferase-I (CPT-I) inhibition in vivo does not alter cardiac long-chain fatty acid uptake and oxidation rates. Biochem J. 2009;419:447–455. doi: 10.1042/BJ20082159. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, Schaap FG, van Nieuwenhoven FA, Van Der Vusse GJ, Bonen A, Glatz JF. Cellular fatty acid transport in heart and skeletal muscle as facilitated by proteins. Lipids. 1999;34:S169–S175. doi: 10.1007/BF02562278. [DOI] [PubMed] [Google Scholar]
- Luiken JJ, van Nieuwenhoven FA, America G, Van Der Vusse GJ, Glatz JF. Uptake and metabolism of palmitate by isolated cardiac myocytes from adult rats: involvement of sarcolemmal proteins. J Lipid Res. 1997;38:745–758. [PubMed] [Google Scholar]
- McGarry JD, Mills SE, Long CS, Foster DW. Observations on the affinity for carnitine, and malonyl-CoA sensitivity, of carnitine palmitoyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem J. 1983;214:21–28. doi: 10.1042/bj2140021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand I, Tarnopolsky M, Adamo KB, Bourgeois JM, Chorneyko K, Graham TE. Quantitative assessment of human muscle glycogen granules size and number in subcellular locations during recovery from prolonged exercise. J Physiol. 2007;580:617–628. doi: 10.1113/jphysiol.2006.122457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, Cacciapuoti F, Rossi F, D’Amico M, Paolisso G. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J Lipid Res. 2009;50:2314–2323. doi: 10.1194/jlr.P900032-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- Neitzel AS, Carley AN, Severson DL. Chylomicron and palmitate metabolism by perfused hearts from diabetic mice. Am J Physiol Endocrinol Metab. 2003;284:E357–E365. doi: 10.1152/ajpendo.00380.2002. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical differences between subsarcolemmal and interfibrillar mitochondria from rat cardiac muscle: effects of procedural manipulations. Arch Biochem Biophys. 1985;236:691–702. doi: 10.1016/0003-9861(85)90675-7. [DOI] [PubMed] [Google Scholar]
- Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- Srere PA. Methods in Enzymology. New York: Academid; 1969. Citrate synthase. [Google Scholar]
- Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak-Tarnopolsky SN, Devries MC, Hamadeh MJ. Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1271–R1278. doi: 10.1152/ajpregu.00472.2006. [DOI] [PubMed] [Google Scholar]
- Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- Ussher JR, Koves TR, Jaswal JS, Zhang L, Ilkayeva O, Dyck JR, Muoio DM, Lopaschuk GD. Insulin-stimulated cardiac glucose oxidation is increased in high-fat diet-induced obese mice lacking malonyl CoA decarboxylase. Diabetes. 2009;58:1766–1775. doi: 10.2337/db09-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Meer RW, Rijzewijk LJ, de Jong HW, Lamb HJ, Lubberink M, Romijn JA, Bax JJ, de Roos A, Kamp O, Paulus WJ, Heine RJ, Lammertsma AA, Smit JW, Diamant M. Pioglitazone improves cardiac function and alters myocardial substrate metabolism without affecting cardiac triglyceride accumulation and high-energy phosphate metabolism in patients with well-controlled type 2 diabetes mellitus. Circulation. 2009;119:2069–2077. doi: 10.1161/CIRCULATIONAHA.108.803916. [DOI] [PubMed] [Google Scholar]
- Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2005;288:H2102–H2110. doi: 10.1152/ajpheart.00935.2004. [DOI] [PubMed] [Google Scholar]
- Wisneski JA, Gertz EW, Neese RA, Mayr M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J Clin Invest. 1987;79:359–366. doi: 10.1172/JCI112820. [DOI] [PMC free article] [PubMed] [Google Scholar]