

Abstract
In the present study, we examined the effects of soluble epoxide hydrolase (sEH) inhibition on the development of angiotensin II-dependent hypertension and on renal function in transgenic rats with inducible expression of the mouse renin gene (strain name Cyp1a1-Ren-2). Hypertension was induced in these rats by indole-3-carbinol (I3C; 0.3% in the diet) for 12 days. The sEH inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (c-AUCB) was given in two doses (13 or 26 mg l−1) in drinking water. Blood pressure (BP), body weight (BW) and renal excretory parameters were monitored in conscious animals during the experiment. Renal haemodynamics was assessed at the end of treatment in anaesthetized rats. I3C administration resulted in severe hypertension with a rise in systolic BP from 118 ± 2 to 202 ± 3 mmHg, a loss of BW from 266 ± 5 to 228 ± 4 g and a rise in proteinuria from 14 ± 2 to 34 ± 3 mg day−1. Both doses of c-AUCB significantly attenuated the development of hypertension (systolic BP of 181 ± 4 and 176 ± 4 mmHg, respectively), the loss in BW (256 ± 4 and 259 ± 3 g, respectively) and the degree of proteinuria (27 ± 2 and 25 ± 3 mg day−1, respectively) to a similar extent. Moreover, c-AUCB prevented the reduction in renal plasma flow (5.4 ± 0.4 vs. 4.6 ± 0.3 ml min−1 g−1) and significantly increased sodium excretion (0.84 ± 0.16 vs. 0.38 ± 0.08 μmol min−1 g−1) during I3C administration. These data suggest that the oral administration of c-AUCB displays antihypertensive effects in Ren-2 transgenic rats with inducible malignant hypertension via an improvement of renal function.
Non-technical summary
Arachidonic acid metabolites called epoxyeicosatrienoic acids (EETs) influence vascular tone and renal tubular sodium and water transport and thus have been implicated in the control of blood pressure. Inhibition of the enzyme soluble epoxide hydrolase (sEH), which reduces EET degradation to the corresponding diols, leads to substantial attenuation of malignant hypertension in a transgenic rat strain harbouring the mouse renin gene particularly via an improvement of renal function. The observed antihypertensive and renoprotective effects of this novel pharmacological approach provide a potentially new direction in antihypertensive therapy.
Introduction
Cytochrome P450 (Cyp) metabolites of arachidonic and other fatty acids are increasingly recognized as important regulators of cardiovascular and renal function (Fleming, 2001; Roman, 2002; Imig, 2005; Capdevila et al. 2007). Cyp epoxygenases, specifically the 2C and 2J classes, metabolize arachidonic acid to epoxyeicosatrienoic acids (EETs) which influence vascular tone as well as renal tubular sodium and water transport. Thus they have been implicated in the control of blood pressure (Roman, 2002; Imig, 2006; Capdevila et al. 2007). Furthermore, by vasodilatation and the inhibition of distal sodium reabsorption, EETs may play an important antihypertensive role (Dos Santos et al. 2004; Wei et al. 2004; Sun et al. 2006; Imig, 2009). It has been suggested that the inability to increase the level of EETs contributes to the pathogenesis of hypertension (Imig et al. 2005; Chábová et al. 2007; Inceoglu et al. 2007). Since these compounds are metabolized by soluble epoxide hydrolase (sEH) to form the corresponding dihydroxyeicosatrienoic acids (DHETEs), which appear to have less biological effect, the bioavailability of EETs and other epoxidized fatty acids may be reduced by enhanced sEH activity (Yu et al. 2000; Zhao et al. 2004; Manhiani et al. 2009). Accordingly, it has been shown that pharmacological inhibition or gene disruption of sEH can increase the level of EETs, which may exhibit protective cardiovascular and antihypertensive properties in several models of hypertension (Dorrance et al. 2005; Imig et al. 2005; Jung et al. 2005; Huang et al. 2007; Loch et al. 2007). Since sEH inhibitors can be administered orally in amounts that clearly increase tissue bioavailability of EETs, this type of manipulation of their plasma and tissue levels may be a potentially useful new pharmacological approach (Hwang et al. 2007; Imig & Hammock, 2009).
It is well recognized that inappropriate activation of the renin–angiotensin system (RAS) is a major factor contributing to the pathophysiology of angiotensin II (ANG II)-dependent forms of hypertension (Hall et al. 1980; Navar et al. 1996; Cervenka et al. 1998). The inbred transgenic Cyp1a1-Ren-2 rat strain with inducible hypertension, which expresses the mouse renin gene under the control of the Cyp1a1 promoter, offers a new possibility to control the endogenous activation of the RAS, and thus the degree and duration of hypertension (Kantachuvesiri et al. 2001; Mitchell & Mullins, 2005; Erbanová et al. 2009). Although the Cyp1a1 promoter is not constitutively expressed, after exposure to various natural xenobiotics such as indole-3-carbinol (I3C), which can be easily administrated with the rat chow (Mitchell et al. 2006; Vanourková et al. 2006), the expression of the Cyp1a1 promoter is rapidly increased with a marked enhancement of the expression of the Ren-2 renin gene in the liver (Kantachuvesiri et al. 2001). This enhanced expression of the Ren-2 renin gene results in increased plasma renin activity with subsequently increased circulating ANG II levels leading to the development of ANG II-dependent form of hypertension (Vanourková et al. 2006). As described previously (Kantachuvesiri et al. 2001) the dietary intake of 0.3% I3C leads to the development of severe malignant hypertension in this transgenic model with associated renal injury (Graciano et al. 2007; Ortiz et al. 2007).
The role of eicosanoids as a compensatory system with protective effects against an enhanced RAS activity in models of malignant hypertension, however, remains poorly understood. Therefore, in this present study, we examined the hypothesis that administration of the novel sEH inhibitor cis-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyl-oxy]-benzoic acid (c-AUCB) (Hwang et al. 2007) would attenuate the development of hypertension and the impairment of renal function in transgenic rats with inducible expression of the mouse Ren-2 renin gene.
Methods
The studies were performed in accordance with guidelines and practices established by the Institute for Clinical and Experimental Medicine Animal Care and Use Committee, and the laws of the Czech Republic and European Union. All our experiments comply with the policies and regulations and meet the highest ethical standards of The Journal of Physiology (Drummond, 2009).
Experiments were performed in male Cyp1a1-Ren-2 transgenic rats aged 2 months. All animals used in the present study were bred at the Department of Experimental Medicine of the Institute for Clinical and Experimental Medicine from stock animals supplied from the Center for Cardiovascular Science, University of Edinburgh, UK. Standard rat chow (SEMED, Prague, Czech Republic) without (non-induced groups) or with 0.3% I3C (I3C-induced groups) has been used in the present study. It has been shown that I3C is a biological dietary supplement, which does not exhibit any harmful effects in transgene-negative rats, but strongly induces Cyp1a1 through activation of the aryl hydrocarbon receptor which is a basic helix–loop–helix transcription factor that binds to the Cyp1a1 promoter (Jellinck et al. 1993; Kantachuvesiri et al. 2001). Previous studies have demonstrated that the above dose of I3C in the rat chow resulted in a marked rise of the expression of the Ren-2 renin gene and the development of ANG II-dependent hypertension in transgene-positive animals, whereas the transgene-negative rats (Fisher F344 rats) remained normotensive (Kantachuvesiri et al. 2001; Mitchell et al. 2006).
Measurement of blood pressure and renal excretory function
Male Cyp1a1-Ren-2 transgenic rats, weighing 250 ± 20 g, were implanted with radiotransmitters TA11PA-C40 (Data Sciences, St. Paul, MN) as described previously (Imig et al. 2005; Ortiz et al. 2007; Husková et al. 2010) to continuously monitor blood pressure by a telemetric device throughout the experimental period. The animals were anaesthetized with a combination of tiletamine, zolazepam (Zoletil, Virbac SA, Carros Cedex, France; 8 mg kg−1), and xylasine (Rometar, Spofa, Czech Republic; 4 mg kg−1) intramuscularly. An abdominal midline incision was performed to expose the abdominal aorta, which was briefly occluded to allow insertion of the transmitter catheter. The catheter was secured in place with tissue glue. The transmitter body was sutured to the abdominal wall along the incision line as the incision was closed. The skin was finally sutured and the stitches were removed 7–10 days later after the incision was healed. After 10–12 days of recovery from surgery, data acquisition was initiated to determine basal blood pressure and data were collected daily as described previously (Husková et al. 2010).
The animals were randomly assigned to the following experimental groups: non-induced untreated control (n = 8), non-induced + c-AUCB (13 mg l−1; n = 6), non-induced + c-AUCB (26 mg l−1; n = 6), I3C-induced untreated (n = 8), I3C-induced + c-AUCB (13 mg l−1; n = 8), I3C-induced + c-AUCB (26 mg l−1; n = 8). Basal blood pressure was measured continuously for 7 days. Hypertension was induced in Cyp1a1-Ren-2 rats through dietary administration of I3C for 12 days. The sEH inhibitor c-AUCB was given in drinking water prepared freshly every third day. The crystaline c-AUCB (13 or 26 mg) was dissolved in ethanol (5 ml) and cyclodextrin (150 mg) after 5 min sonication and this solution was added to the 1 litre of drinking water. Hydrogencarbonate (3 ml l−1) was given to insure that the water did not become acidic since low pH can cause the compound to precipitate. Cyclodextrin and hydrogencarbonate were also given to untreated control rats in drinking water. These studies determined that these substances did not alter renal function in Cyp1a1-Ren-2 transgenic rats. The administration of c-AUCB was started 48 h before switching diets containing none or 0.3% I3C. The different doses of c-AUCB were used to determine possible dose-dependent effects of the treatment. Based on the previous study by Hwang et al. (2007), c-AUCB compound showed better water solubility and metabolic stability.
In the animals implanted with radiotransmitter, 24 h urine collections were performed in metabolic cages prior and during I3C administration (day 2, 4, 7 and 11) to assess daily sodium excretion and proteinuria, as described previously (Vanecková et al. 2007; Huskova et al. 2010). In another set of Cyp1a1-Ren-2 transgenic rats (n = 6 in each experimental group as above), urine collections were also performed to compare the renal excretory responses in intact animals with chronically catheterized animals after the surgical intervention. At the end of experiments, all rats were decapitated to collect blood and the kidneys were removed for further analysis.
Determination of ANG II, EETs, DHETEs and c-AUCB in plasma and renal tissue
Plasma and kidney ANG II levels were measured by radioimmunoassay using a commercially available kit (Euro-Diagnostica Co., Malmö, Sweden) as described previously (Kopkan et al. 2005; Vanourková et al. 2006, Husková et al. 2010). After decapitation, arterial blood was collected in chilled tubes containing a mixed inhibitor solution (5 mmol l−1 EDTA, 10 μmol l−1 pepstatin and 1.25 mmol l−1 1,10-phenanthroline) and the kidneys were immediately excised, drained, weighed and homogenized in chilled methanol. Blood samples and kidney homogenates were centrifuged at 4°C for 10 min at 3000 g, and plasma was separated and eluted in ethanol, centrifuged, evaporated using a Savant Speed Vac and stored at −80°C until assayed. Measurement of the total tissue content of ANG II is a generally accepted and extensively used approach to provide an index of overall tissue content that can be compared among experimental groups (Navar & Nishiyama, 2004). The levels of the arachidonic acid metabolites, EETs and DHETEs, were measured in the kidney cortex. Samples were extracted, separated by reverse-phase, high-performance liquid chromatography (HPLC), and analysed by negative-mode electrospray ionization and tandem mass spectroscopy as described previously (Imig et al. 2005; Huang et al. 2007). The concentration of c-AUCB in plasma was determined by mass spectrometry as described previously (Watanabe et al. 2003; Imig et al. 2005). Fluid intake of our rats approximated 15 ml of water per day, which corresponds roughly to a daily dose of 200 or 400 μg of c-AUCB. Plasma concentrations of c-AUCB were determined to validate an effective sEH inhibition which provided a maximal effect on the parameters monitored in the present study. It averaged 28 ± 3 ng/mL in non-induced rats and 39 ± 9 ng/mL in I3C-induced group after 14 days of treatment. These plasma concentrations of c-AUCB were above the range of the IC50 for the specific sEH inhibition and they were observed to inhibit sEH effectively in in vitro as well as in vivo studies (Hwang et al. 2007). A slightly higher c-AUCB level in hypertensive I3C-induced rats may be mainly attributed to the increased water intake.
Determination of CYP2C23 epoxygenase and sEH in kidney cortex and medulla
Immunoblot analysis was performed to determine protein expression of CYP2C23 epoxygenase and sEH in kidney cortex and medulla as previously described (Zhao et al. 2004; Ai et al. 2007; Huang et al. 2007). Detection was accomplished with an enhanced chemiluminescence Western blotting detection kit (ECL, GE Healthcare, Little Chalfont, UK), and blots were read using the luminescence image analyser LAS-3000, FUJI PHOTO FILM CO., Tokyo, Japan. Band intensity was measured densitometrically and the values were normalized for β-actin.
Determination of plasma renin activity and urinary nitrate/nitrite concentration
Plasma fractions were removed and assayed for plasma renin activity (PRA) by indirect radioimmunoassay (REN-CT2; CIS bio international, France). Results were expressed as nanograms per millilitre per hour of generated ANG I (Véniant et al. 1995; Campbell et al. 2009). Urine was collected into sterile tubes and nitrate/nitrite (NOx) concentration was measured colorimetrically (Assay Design, Ann Arbor, MI, USA) as reported previously (Kopkan & Majid, 2005).
Renal clearance studies to assess renal haemodynamics
In another set of Cyp1a1-Ren-2 transgenic rats without telemetry devices, the animals were randomly assigned to the following experimental groups: non-induced untreated control (n = 8), non-induced + c-AUCB (26 mg l−1; n = 9), I3C-induced untreated (n = 9), I3C-induced + c-AUCB (26 mg l−1; n = 9). These rats were subjected to the higher dose of sEH inhibitor for 13 days. Hypertension was induced also through dietary administration of 0.3% I3C for 11 days. At the end of the treatment, rats were anaesthetized with thiopental sodium (60 mg kg−1; intraperitoneally) and placed on a thermoregulated table to maintain body temperature at 37–37.5°C. A tracheostomy was performed to maintain a patent airway, and the exterior end of the tracheal cannula was placed inside a small plastic chamber, into which humidified 95% O2–5% CO2 mixture was continuously passed to improve the stability of arterial pressure in anaesthetized rats, as described previously (Kopkan et al. 2005; Mitchell et al. 2006). The right jugular vein was catheterized with PE-50 tubing for intravenous administration of solutions, additional anaesthetic as required and intravenous drugs. The right femoral artery was cannulated to allow continuous monitoring of arterial blood pressure via a pressure transducer and data-acquisition system (model MLT 1050; PowerLab/4SP; ADInstruments, Chalgrove, UK) and blood sampling. A suprapubic incision was made, and the bladder was exposed by blunt dissection through the abdominal wall. The bladder was catheterized for timed urine collections during the experimental protocol. During surgery, an isotonic saline solution containing bovine serum albumin (6%) (Sigma Chemical Co., Prague, Czech Republic) was infused at a rate of 20 μl min−1. The rats were allowed to recover for 1 h after completion of surgery when isotonic saline solution containing p-aminohippurate sodium (PAH; Merck, Sharp & Dohme, West Point, PA, USA) (1.5%), and polyfructosan (Inutest, Laevosan, Linz/Donau, Austria) (7.5%) was infused at the same infusion rate. The experimental protocol consisted of two 30 min urine collections to determine renal haemodynamic and excretory parameters in these rats as described previously (Kopkan et al. 2005).
Statistical analysis
Results are expressed as means ± s.e.m. One-way analysis of variance and two-way repeated-measures analysis of variance followed by the post hoc test were used when appropriate. Statistical significance was defined as a P value less than 0.05.
Results
Blood pressure and renal excretory function
Radiotelemetric blood pressure (BP) monitoring through the experiment confirmed that systolic blood pressure (SBP) in non-induced Cyp1a1-Ren-2 rats remained within the normotensive range (124 ± 6 to 126 ± 7 mmHg). Treatment with the sEH inhibitor c-AUCB at doses of 13 and 26 mg l−1, respectively, did not alter SBP in these non-induced normotensive animals (123 ± 4 to 121 ± 3 and 119 ± 5 to 118 ± 4 mmHg, respectively). In contrast, administration of the diet containing 0.3% I3C resulted in severe hypertension (Fig. 1A) which was associated with a significant loss of body weight (BW) in these rats (Fig. 1B). These animals also exhibited other signs of the malignant hypertension, such as polyuria, polydypsia, hunched posture and piloerection, as described previously (Kantachuvesiri et al. 2001; Mitchell et al. 2006). They did not reveal marked differences in food intake compared to non-induced animals (9.3 ± 1.1 vs. 11.7 ± 1.5 g day−1). In the groups of I3C-induced rats treated with 13 or 26 mg l−1 of c-AUCB, the development of hypertension was significantly attenuated to a similar extent (Fig. 1A) and the loss of BW was also markedly reduced (Fig. 1B). Other manifestations of malignant hypertension were also attenuated in these animals treated with c-AUCB. Diastolic blood pressure curves corresponded to SBP responses to I3C and c-AUCB administration (see online Supplemental Material). Heart rate remained unaltered by the sEH inhibitor in both non-induced and I3C-induced groups compared to untreated groups of rats (Supplemental Material).
Figure 1. Course of systolic blood pressure (A) and body weight (B) in I3C-induced Cyp1a1-Ren-2 rats, and effects of sEH inhibition by c-AUCB (13 and 26 mg l−1) in these rats (n = 8 in each group).
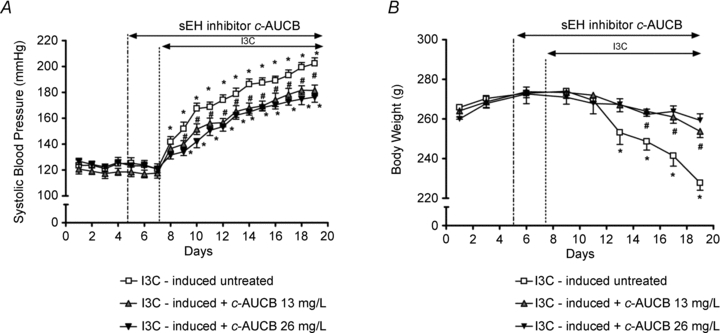
Data represent mean values ± s.e.m. *P < 0.05 vs. basal values; #P < 0.05 vs. I3C-induced untreated rats.
Sodium excretion (UNaV) and proteinuria were determined in 24 h urine samples collected in metabolic cages prior to and during treatment. Throughout the experiment daily UNaV in the non-induced untreated group (0.56 ± 0.04 to 0.64 ± 0.05 mmol day−1) was not significantly different when compared to groups treated with 13 and 26 mg l−1 of c-AUCB (0.53 ± 0.05 to 0.66 ± 0.06 and 0.58 ± 0.03 to 0.71 ± 0.07 mmol day−1, respectively). In the untreated I3C-induced group, UNaV was initially decreased by I3C administration on day 2 and was normalized thereafter (Fig. 2A). Interestingly, c-AUCB treatment not only attenuated the initial drop in UNaV induced by I3C but caused higher UNaV at day 4, 7 and 11 as compared to untreated rats (Fig. 2A). Both doses of c-AUCB altered UNaV to a similar extent. Although in non-induced control rats a slight proteinuria was present (15.2 ± 1.2 to 15.7 ± 0.9 mg day−1), treatment with c-AUCB at doses of both 13 and 26 mg l−1 did not cause any changes (14.5 ± 1.1 to 15.1 ± 1.7 and 14.2 ± 1.3 to 14.7 ± 1.1 mg day−1, respectively). However, c-AUCB significantly attenuated marked proteinuria induced by I3C feeding at day 7 and 11 (Fig. 2B) indicating renoprotective effects of sEH inhibition in hypertensive transgenic rats.
Figure 2. Course of daily sodium excretion (A) and proteinuria (B) in I3C-induced Cyp1a1-Ren-2 rats, and effects of sEH inhibition by c-AUCB (13 and 26 mg l−1) in these rats (n = 8 in each group).
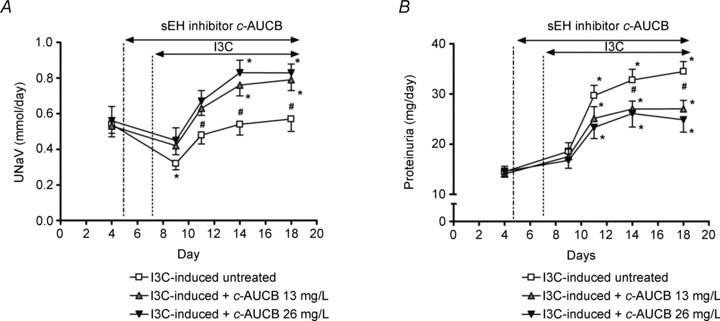
Data represent mean values ± s.e.m. *P < 0.05 vs. basal values; #P < 0.05 vs. I3C-induced untreated rats.
Daily diuresis (urine volume, UV) and water intake (WI) in untreated non-induced Cyp1a1-Ren-2 rats did not change significantly throughout the experiment. Treatment with c-AUCB did not affect UV and WI significantly in non-induced animals, although a slight increasing trend was observed (Table 1). Figure 3A represents changes in UV and Fig. 3B changes in WI in I3C-induced untreated and treated rats. Cyp1a1-Ren-2 rats administered I3C had a significant increase in UV and WI that was greater during c-AUCB treatment at day 3, 7 and 11.
Table 1.
Body weight (BW), sodium excretion (UNaV), urine volume (UV), nitrate/nitrite excretion (UNOxV) and plasma renin activity (PRA) in groups of intact Cyp1a1-Ren-2 rats (without telemetry devices) at the end of the experimental protocol
| Group | n | BW (g) | UNaV (mmol day−1) | UV (ml day−1) | UNOxV (μmol day−1) | PRA (ANG I ng ml−1 h−1) |
|---|---|---|---|---|---|---|
| Non-induced untreated | 6 | 284 ± 5 | 0.64 ± 0.05 | 7.8 ± 1.6 | 3.6 ± 0.2 | 26.8 ± 5.3 |
| Non-induced + c-AUCB 13 mg l−1 | 6 | 278 ± 4 | 0.69 ± 0.04 | 8.2 ± 1.4 | 3.2 ± 0.1 | 42.8 ± 8.1 |
| Non-induced + c-AUCB 26 mg l−1 | 6 | 281 ± 5 | 0.72 ± 0.06 | 8.5 ± 1.7 | 3.9 ± 0.2 | 44.1 ± 11.6 |
| I3C-induced untreated | 6 | 232 ± 3* | 0.58 ± 0.06 | 17.6 ± 2.4* | 2.1 ± 0.2* | 98.2 ± 14.3* |
| I3C-induced + c-AUCB 13 mg l−1 | 6 | 249 ± 4*# | 0.79 ± 0.04# | 24.4 ± 2.7*# | 2.4 ± 0.1* | 106.8 ± 16.3* |
| I3C-induced + c-AUCB 26 mg l−1 | 6 | 258 ± 5*# | 0.85 ± 0.05# | 26.7 ± 2.9*# | 2.6 ± 0.3* | 132.8 ± 18.7* |
Values are mean ± s.e.m.
P < 0.05 vs. non-induced groups;
P < 0.05 vs. I3C-induced untreated rats.
Figure 3. Course of daily urine volume (A) and water intake (B) in I3C-induced Cyp1a1-Ren-2 rats, and effects of sEH inhibition by c-AUCB (13 and 26 mg l−1) in these rats (n = 8 in each group).
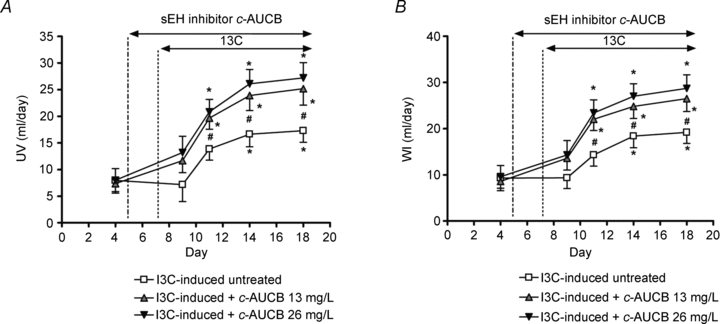
Data represent mean values ± s.e.m. *P < 0.05 vs. basal values; #P < 0.05 vs. I3C-induced untreated rats.
In another set of intact Cyp1a1-Ren-2 transgenic rats, basal values and temporal changes in BW, UNaV, UV and WI during I3C administration and c-AUCB treatment were similar to those observed in chronically catheterized animals with radiotransmitters. Thus the functional responses cannot be attributed to any confounding effects due to surgical intervention and chronic catheterization. Therefore, for the clarity of this study, Table 1 summarizes differences between groups at the end of the experimental period. In addition, significantly lower urinary NOx excretion (UNOxV) in I3C-induced rats was not affected by c-AUCB treatment and I3C-induced increases in PRA were not also influenced by c-AUCB treatment in Cyp1a1-Ren-2 transgenic rats (Table 1).
Concentrations of ANG II, EET, DHETE levels and protein expression of epoxygenases and sEH
ANG II levels were determined in plasma (Fig. 4A) and kidney cortex at the end of the experimental protocol (Fig. 4B). Administration of I3C significantly increased the levels of ANG II in plasma and kidney cortex compared to non-induced groups indicating that this model represents an ANG II-dependent form of hypertension. Treatment with the c-AUCB had no effects on plasma or kidney tissue ANG II levels in either non-induced or I3C-induced groups when compared to untreated groups of rats (Fig. 4).
Figure 4. Angiotension II (ANG II) levels in plasma (A) and kidney cortex (B) at the end of the experimental period in groups of Cyp1a1-Ren-2 rats exposed to I3C administration and c-AUCB treatment (n = 6–8).
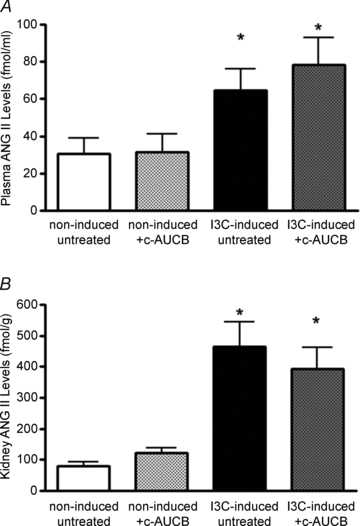
Data represent mean values ± s.e.m. *P < 0.05 vs. non-induced groups.
Although the administration of I3C did not alter the levels of EETs, treatment with the sEH inhibitor c-AUCB increased the concentration of EETs in renal cortex to similar levels in both non-induced and I3C-induced groups (Fig. 5A). DHETEs, products of the degradation of EETs, were significantly higher in the I3C-induced groups than those in non-induced groups of rats (Fig. 5B). Although inhibition of sEH did not change the concentration of DHETEs significantly in the non-induced group, there was a substantial reduction of DHETEs in I3C-induced rats treated with c-AUCB. Moreover, the EETs/DHETEs ratio reflected the efficiency of the sEH inhibitor to increase the levels of EETs (Fig. 5C).
Figure 5. Epoxyeicosatrienoic acids (EETs) (A) and dihydroxyeicosatrienoic acids (DHETEs) (B) in kidney cortex at the end of the experimental period in groups of Cyp1a1-Ren-2 rats exposed to I3C administration and c-AUCB treatment (n = 6–8) and EETs/DHETEs ratio (C), as an indicator of c-AUCB treatment efficiency.
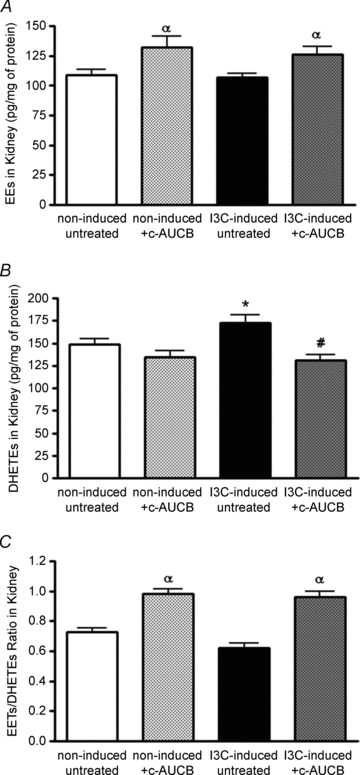
Data represent mean values ± s.e.m. αP < 0.05 vs. untreated groups; *P < 0.05 vs. non-induced groups; #P < 0.05 vs. I3C-induced untreated rats.
Immunoblot analysis showed no significant differences in protein expression of CYP2C23 and sEH in renal cortex and medulla between groups (Fig. 6). Data analysis is given in Table 2. Similar results were observed in protein expression of CYP2C11 epoxygenase (data are not shown). These data indicate that I3C administration as well as c-AUCB treatment did not alter the expression of epoxygenase enzymes in the present study.
Figure 6. Protein expression of CYP2C23 epoxygenase and soluble epoxide hydrolase (sEH) in kidney cortex and medulla in groups of Cyp1a1-Ren-2 rats at the end of the experimental protocol.
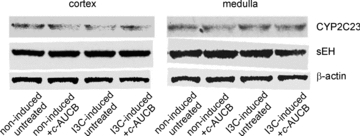
There were no significant differences between groups.
Table 2.
Protein expression of CYP2C23 epoxygenase and soluble epoxide hydrolase (sEH) in kidney cortex and medulla in groups of Cyp1a1-Ren-2 rats at the end of the experimental protocol
| Protein/β-actin ratio | |||||
|---|---|---|---|---|---|
| Group | n | CYP2C23 cortex | CYP2C11 medulla | sEH cortex | sEH medulla |
| Non-induced untreated | 8 | 1.13 ± 0.16 | 1.04 ± 0.11 | 0.99 ± 0.18 | 0.89 ± 0.14 |
| Non-induced + c-AUCB | 8 | 1.08 ± 0.19 | 1.05 ± 0.13 | 0.97 ± 0.20 | 0.90 ± 0.15 |
| I3C-induced untreated | 9 | 1.14 ± 0.23 | 1.04 ± 0.19 | 1.06 ± 0.22 | 0.99 ± 0.17 |
| I3C-induced + c-AUCB | 9 | 1.17 ± 0.19 | 1.07 ± 0.20 | 1.04 ± 0.19 | 1.01 ± 0.16 |
Values are means ± s.e.m. There are no significant differences between groups.
Renal haemodynamics
In a different series of experiments, renal clearance was performed under anaesthesia at the end of the same experimental protocol as described above. As both doses of c-AUCB exhibited similar responses during chronic studies, only the higher dose (26 mg l−1) has been used to determine mean arterial pressure (MAP) and renal functional parameters in non-induced and I3C-induced rats. Although treatment with c-AUCB did not significantly affect any renal parameters in non-induced normotensive rats (Figs 7 and 8), it significantly attenuated the rise in MAP (Fig. 7A) and normalized renal plasma flow (RPF), which was significantly reduced in I3C-induced rats (Fig. 7B). As shown in Fig. 8A, glomerular filtration rate (GFR) was not different between the groups. However, I3C-induced rats treated with c-AUCB exhibited higher sodium excretion than untreated rats (Fig. 8B).
Figure 7. Mean arterial pressure (A) and renal plasma flow (B) in groups of anaesthetized Cyp1a1-Ren-2 rats exposed to I3C administration and c-AUCB treatment (n = 8–9).
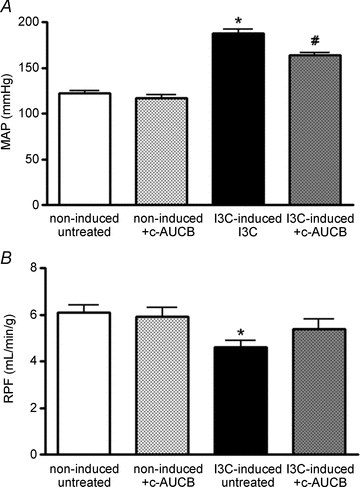
Data represent mean values ± s.e.m. *P < 0.05 vs. non-induced groups. #P < 0.05 vs. I3C-induced untreated rats.
Figure 8. Glomerular filtration rate (A) and absolute sodium excretion (B) in groups of anaesthetized Cyp1a1-Ren-2 rats exposed to I3C administration and c-AUCB treatment (n = 8–9).
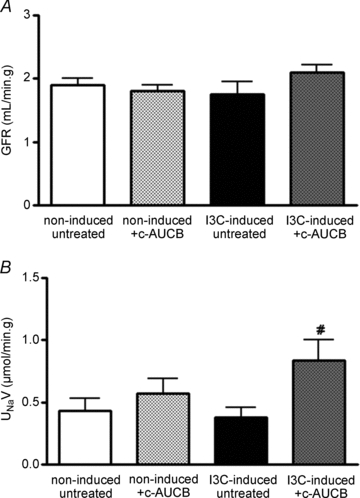
Data represent mean values ± s.e.m. #P < 0.05 vs. I3C-induced untreated rats.
Discussion
Treatment with inhibitors of sEH, the major enzyme that catalyses the degradation of EETs to biologically inactive DHETEs (Imig, 2005; Capdevila et al. 2007; Inceoglu et al. 2007), has been shown to lower BP in several models of hypertension including ANG II-induced hypertension (Imig et al. 2005; Jung et al. 2005; Huang et al. 2007; Loch et al. 2007). The present study demonstrates that oral administration of the novel sEH inhibitor c-AUCB significantly attenuates the development of malignant hypertension induced in Cyp1a1-Ren-2 transgenic rats. More importantly, this study clearly demonstrates an effect of sEH inhibition on renal haemodynamics and excretory function in this transgenic model of ANG II-dependent hypertension with endogenous RAS activation. The compound c-AUCB induced an improvement of renal function and furthermore it displayed significant renal protection by reducing proteinuria in these animals. These data strongly suggest that the antihypertensive effect of c-AUCB treatment in Cyp1a1-Ren-2 transgenic rats with induced malignant hypertension is likely to be mediated, at least in part, by these observed renal mechanisms. These results are in good agreement with the previous findings in ANG II infused hypertensive rats or mice (Zhao et al. 2004; Imig et al. 2005; Jung et al. 2005). The novel sEH inhibitor c-AUCB used in this study inhibits the sEH enzyme with a high potency due to its improved pharmacokinetic properties, particularly its oral bioavailability and water solubility (Hwang et al. 2007). This allows the compound to be administered at relatively low doses that are at or near the dose providing a maximum pharmacological efficacy. Accordingly, administration of the double dose of c-AUCB did not cause any further significant antihypertensive and renal functional effects in the present study. It is also important to notice that c-AUCB treatment exhibited no effects in BP or renal excretory parameters in normotensive non-induced control rats. Thus, our current results provide several new findings that expand our present knowledge with respect to a novel protective approach, namely that sEH inhibition by c-AUCB is associated with the attenuation of the characteristics of ANG II-dependent malignant hypertension in Cyp1a1-Ren-2 transgenic rats and the improvement of renal function in these animals.
We have shown in our previous study (Vanourková et al. 2006) that the rapid development of malignant hypertension in this model is associated with a prompt increase in plasma ANG II concentration during the first 24 h and a progressive rise in tissue ANG II content during the continuous administration of I3C in the diet. In the present study, I3C induces marked enhancement in RAS activity (renin and ANG II); however, it is important to note that treatment with c-AUCB did not alter plasma and kidney RAS activity. Thus, it is likely that the observed effects of the sEH inhibitor in our Cyp1a1-Ren-2 transgenic rats are primarily attributed to the substantial increases in the bioavailability of epoxy lipids, in particular EETs. Although the observed absolute changes in renal EET metabolism seem to be relatively small in the present study, it is conceivable that even moderate changes in the level of EETs or other epoxy lipids may significantly alter renal function (Imig et al. 2005; Jung et al. 2005; Chábová et al. 2007; Huang et al. 2007). Our data may also be explained by a high turnover of EETs, thus not allowing the accumulation of EETs in the kidney. In agreement with previous studies (Zhao et al. 2004; Imig et al. 2005; Chábová et al. 2007), significantly increased renal concentrations of DHETEs in I3C induced rats indicate a higher degradation of EETs in hypertensive animals than in non-induced rats. As the epoxide/diol ratio rather than the epoxide level itself is a better indicator of the sEH inhibitor's effectiveness (Zhao et al. 2004; Imig et al. 2005; Hwang et al. 2007), we also noticed that the EETs/DHETEs ratio indicates a significant efficacy of c-AUCB to inhibit, at least partially, the degradation of EETs by the sEH enzyme.
The inability to increase the level of EETs appropriately may contribute to the pathogenesis of hypertension (Yu et al. 2000; Zhao et al. 2004; Capdevila et al. 2007; Manhiani et al. 2009). In the present study, we also observed that the level of EETs in the renal cortex was not different in I3C-induced hypertensive rats compared to the non-induced normotensive group at the end of the experimental period indicating the inability to increase EETs as the expression of epoxygenases in the kidney remains unchanged. Thus, these data imply a relative lack of EETs during enhanced endogenous ANG II production (Zhao et al. 2004). The major statement is that EETs predominately act as paracrine or autocrine factors due to their lipophilicity and rapid incorporation into the cell membrane. As EETs can be extracted from several tissues or isolated platelets, the binding of EETs to proteins is likely to restrict their systemic distribution. The localization of the sEH enzyme in tissues shows the highest specific activity in the liver followed by the kidney, further supporting the local action of this system. Exogenous ANG II administration has been shown to up-regulate endothelial sEH (Zhao et al. 2004, Ai et al. 2007), which might further decrease the local availability of EETs. Although, renal expression of sEH protein was not significantly different in I3C-induced groups compared to non-induced groups in the present study, it is likely that the catalytic activity of the sEH enzyme is increased (Imig, 2006; Ai et al. 2007) and thus renal levels of DHETEs were higher in these I3C-induced hypertensive rats.
In recent studies, important interactions between nitric oxide (NO) and eicosanoids have been indicated (Oyekan et al. 1999; Jiang et al. 2007; Hercule et al. 2009). Therefore we determined urinary NOx excretion as an endogenous marker of NO metabolism and NO activity. In I3C-induced rats, we observed decreases in urinary NOx concentration indicating diminished NO levels during the development of malignant hypertension. c-AUCB at 13 and 26 mg l−1 did not significantly alter urinary NOx excretion in either non-induced or I3C-induced rats. These results suggest that the observed antihypertensive and natriuretic responses to c-AUCB were not attributable to an increase in NO levels. These data do not support a positive interaction between EETs and NO levels as recently indicated (Hercule et al. 2009); however, it is possible that NO release could be driven by DHETEs, which are lowered by c-AUCB in the present study. This present finding provides limited mechanistic insight to this interaction; nevertheless, more studies are required to address this issue more conclusively.
Severe hypertension in Cyp1a1-Ren-2 transgenic rats exposed to I3C is associated with extensive proteinuria (Kantachuvesiri et al. 2001; Mitchell et al. 2006; Ortiz et al. 2007). In the present study, we also observed that Cyp1a1-Ren-2 transgenic rats display progressive proteinuria during the exposure to I3C. Despite enhanced ANG II levels in these rats, both doses of c-AUCB treatment significantly attenuated proteinuria to a similar extent indicating substantial renoprotective properties of the compounds blocking sEH enzyme (Imig et al. 2005; Jung et al. 2005; Huang et al. 2007; Hwang et al. 2007). Moreover, c-AUCB prevented an initial sodium retention induced by I3C administration that was observed in untreated Cyp1a1-Ren-2 transgenic rats on day 2. Subsequently, c-AUCB promoted natriuretic responses during I3C exposure on days 7 and 11. Furthermore, our data strongly support the view that EETs are not only involved in the regulation of vascular tone but also directly influence tubular transport of sodium in the kidney (Capdevila et al. 2007; Imig, 2009). It is suggested that direct tubular effects of EETs are located in the proximal tubule where EETs regulate sodium tubular transport by inhibiting the translocation of the Na+/H+ exchanger to the apical membrane (Roman, 2002; Dos Santos et al. 2004, Imig, 2005). In the cortical collecting duct, EETs have also been shown to be potent inhibitors of Na+,K+-ATPase and amiloride-sensitive sodium transport (Satoh et al. 1993; Sakairi et al. 1995; Wei et al. 2004; Sun et al. 2006). Accordingly, we observed greater natriuresis and diuresis in I3C-induced Cyp1a1-Ren-2 transgenic rats treated with c-AUCB that may contribute to the antihypertensive effect. In addition, there were also slightly increased but non-significant natriuretic and diuretic responses to c-AUCB treatment in non-induced animals. However, this present study was not designed to delineate which tubular transport mechanisms are involved in observed natriuresis and diuresis. Furthermore, the enhanced tubuloglomerular feedback responsiveness mediated by the actions of ANG II has been reported in Cyp1a1-Ren-2 transgenic rats (Mitchell & Mullins, 2005), even though these hypertensive animals exhibit sustained GFR. The findings of the present study suggest that EETs induced direct tubular effects during c-AUCB treatment while having no appreciable effect on GFR. Thus we can only speculate that increasing EET levels during c-AUCB treatment may modulate the tubuloglomerular feedback in Cyp1a1-Ren-2 transgenic rats. This issue can be resolved by a comprehensive in vivo set of micropuncture experiments that are beyond the scope of this study. Despite limitations of this study, it is highly conceivable that the increase in biological availability of EETs in the kidney improves renal haemodynamic and tubular function in Cyp1a1-Ren-2 transgenic rats with malignant hypertension during the inhibition of sEH and thus may significantly reduce the development of malignant hypertension in this model.
In the present study renal haemodynamic parameters determined by renal clearance studies in anaesthetized rats at the end of the experimental period confirm that Cyp1a1-Ren-2 rats with malignant hypertension exhibit renal vasoconstriction (Mitchell et al. 2006). Interestingly, c-AUCB treatment that also reduced arterial pressure in the I3C induced group of rats normalized RPF, suggesting a significantly decreased renal vascular tone. Although there was no appreciable effect on GFR, sodium excretion was higher in c-AUCB treated rats exposed to I3C as compared to untreated animals. This further suggests that sEH inhibition displays a direct tubular action without much effect on the filtered load of sodium. As EETs have been postulated to directly inhibit tubular ion transport including sodium (Satoh et al. 1993; Sakairi et al. 1994; Wei et al. 2004; Sun et al. 2006), it is conceivable that the observed natriuretic effect is modulated by increased bioavailability of EETs in the kidney during sEH inhibition in the present study. Collectively, our data demonstrate that the observed renal mechanisms may significantly contribute to the antihypertensive effect of sEH inhibitor in Cyp1a1-Ren-2 transgenic rats with inducible malignant hypertension.
In summary, our present findings in Cyp1a1-Ren-2 transgenic rats extend previous observations that attributed to EETs an important role in the regulation of BP and renal function (Imig, 2005; Jung et al. 2005; Huang et al. 2007; Loch et al. 2007) is also involved in the pathophysiology of severe inducible hypertension. Our results demonstrate that substantial increases in the level of endogenous EETs in the presence of sEH inhibition significantly attenuate the development of malignant hypertension in Cyp1a1-Ren-2 transgenic rats particularly by the improvement of renal haemodynamics and sodium excretory function. These observed antihypertensive and renoprotective effects of the novel sEH inhibitor c-AUCB provide convincing evidence for a potentially new approach in antihypertensive therapy.
Acknowledgments
This study was supported by grant NS/9699-4 awarded to L.K. by the Internal Grant Agency of the Ministry of Health of the Czech Republic (IGA) and also by institutional finance support from the Institute for Clinical and Experimental Medicine (no. MZO 00023001) and financial support from the Center for Cardiovascular Research (no. 1M6798582302). L.K. and Z.Ho. are also partly supported by grant KJB 502030801 from the Grant Agency of the Academy of Science of the Czech Republic. Z.Hu. is supported by grant No. 305/08/P053 and A.S. is supported by grant P303/10/P170 awarded by the Czech Science Foundation (GAČR). H.J.K. is supported by the Deutsche Forschungsgemeinschaft (DFG) No. 436 TSE 113/57/0-1 and Deutscher Akademischer Austauschdienst (DAAD). J.D.I. is an Established Investigator of the American Heart Association and these studies were supported by NIH grants HL59699 and DK38226. H.-J.T. was supported in part by the Howard Hughes Foundation and a fellowship from the NIEHS Supported Basic Research Program. Partial support was provided by NIEHS Grant R01 ES02710 and NIEHS Superfund Basic Research Program P42 ES004699 awarded to B.D.H.. L.C. is currently supported by grant No. 305/08/J006 (GAČR) and grant NS/10499-3 (IGA). This study was also supported by the EU by the Operational Program Prague – Competitiveness; project ‘CEVKOON’ (no. CZ.2.16/3.1.00/22126).
Glossary
Abbreviations
- ANG II
angiotensin II
- BP
blood pressure
- BW
body weight
- c-AUCB
cis-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid
- Cyp
cytochrome P450
- DHETE
dihydroxyeicosatrienoic acid
- EET
epoxyeicosatrienoic acid
- GFR
glomerular filtration rate
- I3C
indole-3-carbinol
- RAS
renin–angiotensin system
- Ren
renin gene
- sEH
soluble epoxide hydrolase
- UV
urine volume
- UNaV
sodium excretion
- UNOxV
nitrate/nitrite excretion
- WI
water intake
Author contributions
Conception and design of the experiments : Z.Ho., Z.Hu., Z.V., A.S., S.H.H., B.D.H., J.D.I., L.Č., L.K. Collection, analysis and interpretation of data: Z.Ho., Z.Hu., Z.V., A.S., C.T., L.K. Drafting the article or revising it critically for important intellectual content : Z.Ho., H.J.K., S.H.H., C.T., B.D.H., J.D.I., L.Č., L.K. All authors approved the final version of the manuscript. The authors have no conflict of interest except B.D. H. and J.D.I. (advisory board of Arête Therapeutics, Hayward, CA, USA).
References
- Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, Hammock BD, Shyy JY, Zhu Y. Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:9018–9023. doi: 10.1073/pnas.0703229104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DJ, Karam H, Ménard J, Bruneval P, Mullins JJ. Prorenin contributes to angiotensin peptide formation in transgenic rats with rat prorenin expression targeted to the liver. Hypertension. 2009;54:1248–1253. doi: 10.1161/HYPERTENSIONAHA.109.138495. [DOI] [PubMed] [Google Scholar]
- Capdevila JH, Falck JR, Imig JD. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007;72:683–689. doi: 10.1038/sj.ki.5002394. [DOI] [PubMed] [Google Scholar]
- Cervenka L, Wang CT, Navar LG. Effects of acute AT1 receptor blockade by candesartan on arterial pressure and renal function in rats. Am J Physiol Renal Physiol. 1998;274:F940–945. doi: 10.1152/ajprenal.1998.274.5.F940. [DOI] [PubMed] [Google Scholar]
- Chábová VC, Kramer HJ, Vanecková I, Vernerová Z, Eis V, Tesar V, Skaroupková P, Thumová M, Schejbalová S, Husková Z, Vanourková Z, Kolský A, Imig JD, Cervenka L. Effects of chronic cytochrome P-450 inhibition on the course of hypertension and end-organ damage in Ren-2 transgenic rats. Vascul Pharmacol. 2007;47:145–159. doi: 10.1016/j.vph.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2005;46:842–848. doi: 10.1097/01.fjc.0000189600.74157.6d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dos Santos EA, Dahly-Vernon AJ, Hoagland KM, Roman RJ. Inhibition of the formation of EETs and 20-HETE with 1-aminobenzotriazole attenuates pressure-natriuresis. Am J Physiol Regul Integr Comp Physiol. 2004;287:R58–R68. doi: 10.1152/ajpregu.00713.2003. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbanová M, Thumová M, Husková Z, Vanecková I, Vanourková Z, Mullins JJ, Kramer HJ, Bürgelová M, Rakusan D, Cervenka L. Impairment of the autoregulation of renal hemodynamics and of the pressure–natriuresis relationship precedes the development of hypertension in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2009;27:575–586. doi: 10.1097/hjh.0b013e32831cbd5a. [DOI] [PubMed] [Google Scholar]
- Fleming I. Cytochrome P450 and vascular homeostasis. Circ Res. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- Graciano ML, Mouton CR, Patterson ME, Seth DM, Mullins JJ, Mitchell KD. Renal vascular and tubulointerstitial inflammation and proliferation in Cyp1a1-Ren2 transgenic rats with inducible ANG II-dependent malignant hypertension. Am J Physiol Renal Physiol. 2007;292:F1858–1866. doi: 10.1152/ajprenal.00469.2006. [DOI] [PubMed] [Google Scholar]
- Hall JE, Guyton AC, Smith MJ, Jr, Coleman TG. Blood pressure and renal function during chronic changes in sodium intake: role of angiotensin. Am J Physiol Renal Physiol. 1980;239:F271–F280. doi: 10.1152/ajprenal.1980.239.3.F271. [DOI] [PubMed] [Google Scholar]
- Hercule HC, Schunck WH, Gross V, Seringer J, Leung FP, Weldon SM, da Costa Goncalves ACh, Huang Y, Luft FC, Gollasch M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler Thromb Vasc Biol. 2009;29:54–60. doi: 10.1161/ATVBAHA.108.171298. [DOI] [PubMed] [Google Scholar]
- Huang H, Morisseau C, Wang J, Yang T, Falck JR, Hammock BD, Wang MH. Increasing or stabilizing renal epoxyeicosatrienoic acid production attenuates abnormal renal function and hypertension in obese rats. Am J Physiol Renal Physiol. 2007;293:F342–349. doi: 10.1152/ajprenal.00004.2007. [DOI] [PubMed] [Google Scholar]
- Husková Z, Vanourková Z, Erbanová M, Thumová M, Opocenský M, Mullins JJ, Kramer HJ, Bürgelová M, Cervenka L. Inappropriately high circulating and intrarenal angiotensin II levels during dietary salt loading exacerbate hypertension in Cyp1a1-Ren-2 transgenic rats. J Hypertens. 2010;28:495–509. doi: 10.1097/HJH.0b013e3283345d69. [DOI] [PubMed] [Google Scholar]
- Hwang SH, Tsai HJ, Liu JY, Morisseau C, Hammock BD. Orally bioavailable potent soluble epoxide hydrolase inhibitors. J Med Chem. 2007;50:3825–3840. doi: 10.1021/jm070270t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov. 2009;8:794–805. doi: 10.1038/nrd2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD, Zhao X, Zaharis CZ, Olearczyk JJ, Pollock DM, Newman JW, Kim IH, Watanabe T, Hammock BD. An orally active epoxide hydrolase inhibitor lowers blood pressure and provides renal protection in salt-sensitive hypertension. Hypertension. 2005;46:975–981. doi: 10.1161/01.HYP.0000176237.74820.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol. 2005;289:F496–503. doi: 10.1152/ajprenal.00350.2004. [DOI] [PubMed] [Google Scholar]
- Imig JD. Cardiovascular therapeutic aspects of soluble epoxide hydrolase inhibitors. Cardiovasc Drug Rev. 2006;24:169–188. doi: 10.1111/j.1527-3466.2006.00169.x. [DOI] [PubMed] [Google Scholar]
- Imig JD. Adenosine2A receptors and epoxyeicosatrienoic acids: a recipe for salt and blood pressure regulation. Hypertension. 2009;54:1223–1225. doi: 10.1161/HYPERTENSIONAHA.109.129981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inceoglu B, Schmelzer KR, Morisseau C, Jinks SL, Hammock BD. Soluble epoxide hydrolase inhibition reveals novel biological functions of epoxyeicosatrienoic acids (EETs) Prostaglandins Other Lipid Mediat. 2007;82:42–49. doi: 10.1016/j.prostaglandins.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinck PH, Forkert PG, Riddick DS, Okey AB, Michnovicz JJ, Bradlow HL. Ah receptor binding properties of indole carbinols and induction of hepatic estradiol hydroxylation. Biochem Pharmacol. 1993;45:1129–1136. doi: 10.1016/0006-2952(93)90258-x. [DOI] [PubMed] [Google Scholar]
- Jiang JG, Chen RJ, Xiao B, Yang S, Wang JN, Wang Y, Cowart LA, Xiao X, Wang DW, Xia Y. Regulation of endothelial nitric-oxide synthase activity through phosphorylation in response to epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat. 2007;82:162–174. doi: 10.1016/j.prostaglandins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension. 2005;45:759–765. doi: 10.1161/01.HYP.0000153792.29478.1d. [DOI] [PubMed] [Google Scholar]
- Kantachuvesiri S, Fleming S, Peters J, Peters B, Brooker G, Lammie AG, McGrath I, Kotelevtsev Y, Mullins JJ. Controlled hypertension, a transgenic toggle switch reveals differential mechanisms underlying vascular disease. J Biol Chem. 2001;276:36727–36733. doi: 10.1074/jbc.M103296200. [DOI] [PubMed] [Google Scholar]
- Kopkan L, Kramer HJ, Husková Z, Vanourková Z, Skaroupková P, Thurmová M, Cervenka L. The role of intrarenal angiotensin II in the development of hypertension in Ren-2 transgenic rats. J Hypertens. 2005;23:1531–1539. doi: 10.1097/01.hjh.0000174972.46663.5e. [DOI] [PubMed] [Google Scholar]
- Kopkan L, Majid DS. Superoxide contributes to development of salt sensitivity and hypertension induced by nitric oxide deficiency. Hypertension. 2005;46:1026–1031. doi: 10.1161/01.HYP.0000174989.39003.58. [DOI] [PubMed] [Google Scholar]
- Loch D, Hoey A, Morisseau C, Hammock BO, Brown L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem Biophys. 2007;47:87–98. doi: 10.1385/cbb:47:1:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol. 2009;297:F740–748. [Google Scholar]
- Mitchell KD, Bagatell SJ, Miller CS, Mouton CR, Seth DM, Mullins JJ. Genetic clamping of renin gene expression induces hypertension and elevation of intrarenal Ang II levels of graded severity in Cyp1a1-Ren2 transgenic rats. J Renin Angiotensin Aldosterone Syst. 2006;7:74–86. doi: 10.3317/jraas.2006.013. [DOI] [PubMed] [Google Scholar]
- Mitchell KD, Mullins JJ. Enhanced tubuloglomerular feedback in Cyp1a1-Ren2 transgenic rats with inducible ANG II-dependent malignant hypertension. Am J Physiol Renal Physiol. 2005;289:F1210–1216. doi: 10.1152/ajprenal.00461.2004. [DOI] [PubMed] [Google Scholar]
- Navar LG, Nishiyama A. Why are angiotensin concentrations so high in the kidney? Curr Opin Nephrol Hypertens. 2004;13:107–115. doi: 10.1097/00041552-200401000-00015. [DOI] [PubMed] [Google Scholar]
- Navar LG, Inscho EW, Majid SA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- Ortiz RM, Graciano ML, Mullins JJ, Mitchell KD. Aldosterone receptor antagonism alleviates proteinuria, but not malignant hypertension in Cyp1a1-Ren2 transgenic rats. Am J Physiol Renal Physiol. 2007;293:F1584–1591. doi: 10.1152/ajprenal.00124.2007. [DOI] [PubMed] [Google Scholar]
- Oyekan AO, Youseff T, Fulton D, Quilley J, McGiff JC. Renal cytochrome P450 ω-hydroxylase and epoxygenase activity are differentially modified by nitric oxide and sodium chloride. J Clin Invest. 1999;104:1131–1137. doi: 10.1172/JCI6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RJ. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002;82:131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- Sakairi Y, Jacobson HR, Noland TD, Capdevila JH, Falck JR, Breyer MD. 5,6-EET inhibits ion transport in collecting duct by stimulating endogenous prostaglandin synthesis. Am J Physiol Renal Physiol. 1995;268:F931–939. doi: 10.1152/ajprenal.1995.268.5.F931. [DOI] [PubMed] [Google Scholar]
- Satoh T, Cohen HT, Katz AI. Intracellular signaling in the regulation of renal Na-K-ATPase. II. Role of eicosanoids. J Clin Invest. 1993;91:409–415. doi: 10.1172/JCI116215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Lin DH, Wang T, Babilonia E, Wang Z, Jin Y, Kemp R, Nasjletti A, Wang WH. Low Na intake suppresses expression of CYP2C23 and arachidonic acid-induced inhibition of ENaC. Am J Physiol Renal Physiol. 2006;291:F1192–1200. doi: 10.1152/ajprenal.00112.2006. [DOI] [PubMed] [Google Scholar]
- Vanecková I, Kopkan L, Husková Z, Vanourková Z, Schejbalová S, Cervenka L, Kramer HJ. Long-term prevention of hypertension and end-organ damage in Ren-2 transgenic rats is achieved only with persistent but not transient AT1 receptor blockade. Kidney Blood Press Res. 2007;30:38–44. doi: 10.1159/000098869. [DOI] [PubMed] [Google Scholar]
- Vanourková Z, Kramer HJ, Husková Z, Vanecková I, Opocenský M, Chábová VC, Tesar V, Skaroupková P, Thumová M, Dohnalová M, Mullins JJ, Cervenka L. AT1 receptor blockade is superior to conventional triple therapy in protecting against end-organ damage in Cyp1a1-Ren-2 transgenic rats with inducible hypertension. J Hypertens. 2006;24:2465–2472. doi: 10.1097/01.hjh.0000251909.00923.22. [DOI] [PubMed] [Google Scholar]
- Véniant M, Whitworth CE, Ménard J, Sharp MGF, Gonzales MF, Bruneval P, Mullins JJ. Developmental studies demonstrate age-dependent elevation of renin activity in TGR(mRen2)27 rats. Am J Hypertens. 1995;8:1167–1176. doi: 10.1016/0895-7061(95)00254-5. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Morisseau C, Newman JW, Hammock BD. In vitro metabolism of the mammalian soluble epoxide hydrolase inhibitor, 1-cyclohexyl-3-dodecyl-urea. Drug Metab Dispos. 2003;31:846–853. doi: 10.1124/dmd.31.7.846. [DOI] [PubMed] [Google Scholar]
- Wei Y, Lin DH, Kemp R, Yaddanapudi GS, Nasjletti A, Falck JR, Wang WH. Arachidonic acid inhibits epithelial Na channel via cytochrome P450 (CYP) epoxygenase-dependent metabolic pathways. J Gen Physiol. 2004;124:719–727. doi: 10.1085/jgp.200409140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
- Zhao X, Yamamoto T, Newman JW, Kim IH, Watanabe T, Hammock BD, Stewart J, Pollock JS, Pollock DM, Imig JD. Soluble epoxide hydrolase inhibition protects the kidney from hypertension-induced damage. J Am Soc Nephrol. 2004;15:1244–1253. [PubMed] [Google Scholar]