

Abstract
Creatine kinase (CK) plays a key role both in energy provision and in signal transduction for the increase in skeletal muscle O2 uptake (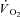 ) at exercise onset. The effects of acute CK inhibition by iodoacetamide (IA; 5 mm) on
) at exercise onset. The effects of acute CK inhibition by iodoacetamide (IA; 5 mm) on 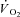 kinetics were studied in isolated canine gastrocnemius muscles in situ (n = 6) during transitions from rest to 3 min of electrically stimulated contractions eliciting ∼70% of muscle peak
kinetics were studied in isolated canine gastrocnemius muscles in situ (n = 6) during transitions from rest to 3 min of electrically stimulated contractions eliciting ∼70% of muscle peak 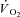 , and were compared to control (Ctrl) conditions. In both IA and Ctrl muscles were pump-perfused with constantly elevated blood flows. Arterial and venous [O2] were determined at rest and every 5–7 s during contractions.
, and were compared to control (Ctrl) conditions. In both IA and Ctrl muscles were pump-perfused with constantly elevated blood flows. Arterial and venous [O2] were determined at rest and every 5–7 s during contractions. 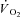 was calculated by Fick's principle. Muscle biopsies were obtained at rest and after ∼3 min of contractions. Muscle force was measured continuously. There was no fatigue in Ctrl (final force/initial force (fatigue index, FI) = 0.97 ± 0.06 (x ± s.d.)), whereas in IA force was significantly lower during the first contractions, slightly recovered at 15–20 s and then decreased (FI 0.67 ± 0.17). [Phosphocreatine] was not different in the two conditions at rest, and decreased during contractions in Ctrl, but not in IA.
was calculated by Fick's principle. Muscle biopsies were obtained at rest and after ∼3 min of contractions. Muscle force was measured continuously. There was no fatigue in Ctrl (final force/initial force (fatigue index, FI) = 0.97 ± 0.06 (x ± s.d.)), whereas in IA force was significantly lower during the first contractions, slightly recovered at 15–20 s and then decreased (FI 0.67 ± 0.17). [Phosphocreatine] was not different in the two conditions at rest, and decreased during contractions in Ctrl, but not in IA. 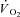 at 3 min was lower in IA (4.7 ± 2.9 ml 100 g−1 min−1) vs. Ctrl (16.6 ± 2.5 ml 100 g−1 min−1). The time constant (τ) of
at 3 min was lower in IA (4.7 ± 2.9 ml 100 g−1 min−1) vs. Ctrl (16.6 ± 2.5 ml 100 g−1 min−1). The time constant (τ) of 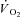 kinetics was faster in IA (8.1 ± 4.8 s) vs. Ctrl (16.6 ± 2.6 s). A second control condition (Ctrl-Mod) was produced by modelling a
kinetics was faster in IA (8.1 ± 4.8 s) vs. Ctrl (16.6 ± 2.6 s). A second control condition (Ctrl-Mod) was produced by modelling a 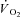 response that accounted for the ‘non-square’ force profile in IA, which by itself could have influenced
response that accounted for the ‘non-square’ force profile in IA, which by itself could have influenced 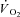 kinetics. However, τ in IA was faster than in Ctrl-Mod (13.8 ± 2.8 s). The faster
kinetics. However, τ in IA was faster than in Ctrl-Mod (13.8 ± 2.8 s). The faster 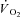 kinetics due to IA suggest that in mammalian skeletal muscle in situ, following contractions onset, temporal energy buffering by CK slows the kinetics of signal transduction for the activation of oxidative phosphorylation.
kinetics due to IA suggest that in mammalian skeletal muscle in situ, following contractions onset, temporal energy buffering by CK slows the kinetics of signal transduction for the activation of oxidative phosphorylation.
Non-technical summary
The ability to sustain skeletal muscle contractions is dependent on the conversion of chemical to mechanical energy – a process fueled by adenosine triphosphate (ATP). The link between two of the major mechanisms for ATP provision, phosphocreatine (PCr) breakdown and oxidative phosphorylation, was investigated in canine muscle. Infusion of a drug to prevent PCr breakdown (via inhibition of the enzyme creatine kinase; CK) caused (among other effects) a faster adjustment of energy provision from oxidation upon the onset of contractions. Thus, in mammalian skeletal muscle the CK enzyme slows the signal responsible for the activation of oxidative phosphorylation. Sudden increases in the demands for energy at the onset of exercise are met by PCr breakdown, but this process is functionally related, presumably through the levels of some of its metabolites, to the regulation of oxidative phosphorylation, the most important pathway for ATP resynthesis.
Introduction
In skeletal muscle, creatine kinase (CK) catalyses the reversible transfer of high energy phosphates between phosphocreatine (PCr) and ADP. The most abundant isoform of CK is located in the cytosol and is functionally coupled to the sites of energy usage. At exercise onset a rapid increase in ADP production through activation of ATPases is buffered by CK-catalysed rephosphorylation of ADP from PCr. An additional isoform, mitochondrial CK, is functionally associated with adenine translocases on the inner mitochondrial membrane. Together these two CK isoforms ‘shuttle’ PCr from mitochondria to the cytosol and Cr in the reverse direction (Mahler, 1980; Meyer et al. 1984), and are thought to act as a ‘damper’ in the control of cellular respiration (Mahler, 1980; Whipp & Mahler, 1980; Cerretelli & di Prampero, 1987; Meyer, 1988; Kaasik et al. 1999; Greenhaff, 2001; Rossiter et al. 2005). At exercise onset, substrate level phosphorylation (PCr breakdown and glycolysis: Cerretelli et al. 1979; Harrison et al. 2003) provides a temporal buffer for the initial demand for ATP turnover. This acts to slow the increase of oxidative phosphorylation by delaying fluctuations of the key energetic controlling signal(s) between sites of ATP hydrolysis and mitochondria, thereby controlling the finite kinetics of adjustment of O2 uptake (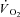 ) at the onset of exercise (Glancy et al. 2008).
) at the onset of exercise (Glancy et al. 2008).
In this scenario, CK inhibition should elicit a faster adjustment of oxidative phosphorylation at the onset of exercise, as also suggested by mathematical models (Roman et al. 2002; Korzeniewski & Zoladz, 2004). This suggestion has been corroborated by 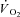 measurements across heart muscle of CK knockout (CK−/−) mice (Gustafson & Van Beek 2002). Inferences from experiments in knockout animals, however, are complicated by the compensatory adaptations incurred in skeletal muscles as a result of CK−/− (Van Deursen et al. 1993; Steeghs et al. 1997; Bruton et al. 2003), which could affect
measurements across heart muscle of CK knockout (CK−/−) mice (Gustafson & Van Beek 2002). Inferences from experiments in knockout animals, however, are complicated by the compensatory adaptations incurred in skeletal muscles as a result of CK−/− (Van Deursen et al. 1993; Steeghs et al. 1997; Bruton et al. 2003), which could affect 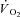 kinetics. A faster adjustment of mitochondrial respiration has also been described in isolated rabbit heart after acute CK inhibition obtained by the administration of iodoacetamide (IA) (Harrison et al. 1999, 2003). However,
kinetics. A faster adjustment of mitochondrial respiration has also been described in isolated rabbit heart after acute CK inhibition obtained by the administration of iodoacetamide (IA) (Harrison et al. 1999, 2003). However, 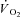 kinetics are much more rapid in heart muscle compared to skeletal muscle due to the much greater mitochondrial content and lower [PCr] (Balaban et al. 1986; Katz et al. 1989). Also, the magnitude of metabolic (oxidative) perturbation in heart muscle is much smaller than that which can be elicited in skeletal muscle. Therefore, the role of CK in damping respiratory control transduction is probably of much higher significance in skeletal muscle. The only study which examined the effects of acute CK inhibition on skeletal muscle bioenergetics was conducted on isolated amphibian muscle fibres (Kindig et al. 2005). After CK inhibition by IA these authors observed a faster decrease in intracellular
kinetics are much more rapid in heart muscle compared to skeletal muscle due to the much greater mitochondrial content and lower [PCr] (Balaban et al. 1986; Katz et al. 1989). Also, the magnitude of metabolic (oxidative) perturbation in heart muscle is much smaller than that which can be elicited in skeletal muscle. Therefore, the role of CK in damping respiratory control transduction is probably of much higher significance in skeletal muscle. The only study which examined the effects of acute CK inhibition on skeletal muscle bioenergetics was conducted on isolated amphibian muscle fibres (Kindig et al. 2005). After CK inhibition by IA these authors observed a faster decrease in intracellular 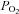 , which, in that preparation, is linearly related to
, which, in that preparation, is linearly related to 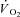 increase. The absence of myoglobin (Mb) in the preparation utilized by Kindig et al. (2005), however, could influence the adjustment of oxidative phosphorylation and its regulation.
increase. The absence of myoglobin (Mb) in the preparation utilized by Kindig et al. (2005), however, could influence the adjustment of oxidative phosphorylation and its regulation.
Thus, our aim was to investigate the role of CK-catalysed PCr breakdown at contraction onset in the control of oxidative phosphorylation in ‘whole’ mammalian skeletal muscle. We hypothesized that acute CK inhibition by IA administration would speed the adjustment of 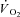 kinetics.
kinetics.
Methods
The study was conducted with approval of the Institutional Animal Care and Use Committee of Auburn University, Auburn, AL, USA, where the experiments were performed. The experiments comply with the policies and regulations described by Drummond (2009). Six adult mongrel dogs of either sex (body mass 19.6 ± 2.2 kg, mean ± s.d.) were anaesthetized with pentobarbital sodium (30 mg kg−1), given intravenously, with maintenance doses of 65–100 mg given as required to maintain a deep plane of surgical anaesthesia as indicated by a complete absence of pedal, palpebral and corneal reflexes. The initial injection of sodium pentobarbital was made into a prominent cephalic vein of the forelimb. Following this initial injection, which established surgical plane anaesthesia, a jugular vein was isolated and all subsequent booster doses were administered through the jugular. The dogs were intubated with an endotracheal tube and ventilated with a respirator (Model 613, Harvard). The rectal temperature was maintained at ∼37°C with a heating pad and a heating lamp. After surgical preparation the animals were treated with heparin (2000 U kg−1). Ventilation was maintained at a level that produced normal arterial 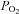 and
and 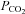 values.
values.
Surgical preparation
The gastrocnemius muscle complex (gastrocnemius + superficial digital flexor), for convenience referred to as ‘gastrocnemius’, was isolated as described previously (Stainsby & Welch, 1966). Briefly, a medial incision was made through the skin of the left hindlimb from midthigh to the ankle. The insertion tendons of the sartorius, gracilis, semitendinosus and semimembranosus muscles were cut to allow these muscles to be folded back to expose the gastrocnemius. To isolate the venous outflow from the gastrocnemius, all the vessels draining into the popliteal vein, except those from the gastrocnemius, were ligated. The popliteal vein was cannulated, and flow (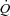 ) was measured with a flow-through-type transit-time ultrasound flow probe (6NRB440, Transonic Systems). Venous outflow was returned to the animal via a reservoir attached to a cannula in the left jugular vein. The arterial circulation to the gastrocnemius was isolated by ligating all vessels from the femoral and popliteal artery that did not enter the gastrocnemius. The right femoral artery was also isolated and cannulated. Blood from this artery was passed through tubing to a roller pump (Cole-Parmer Masterflex, Model No. 7520-25, Head Model No. 7016-20) and then through another cannula into the contralateral, isolated popliteal artery supplying the gastrocnemius. Y-connectors positioned before and after the pump allowed either spontaneous perfusion of the gastrocnemius, at the animal's own blood pressure, or controlled flow at any desired level by adjusting the pump setting. A T-connector in the tubing to the gastrocnemius was connected to a pressure transducer (Narco Biosystems, Model RP-1500) for measurement of muscle perfusion pressure. Arterial blood samples were taken from another T-connector in the cannula exiting the right femoral artery prior to the roller pump.
) was measured with a flow-through-type transit-time ultrasound flow probe (6NRB440, Transonic Systems). Venous outflow was returned to the animal via a reservoir attached to a cannula in the left jugular vein. The arterial circulation to the gastrocnemius was isolated by ligating all vessels from the femoral and popliteal artery that did not enter the gastrocnemius. The right femoral artery was also isolated and cannulated. Blood from this artery was passed through tubing to a roller pump (Cole-Parmer Masterflex, Model No. 7520-25, Head Model No. 7016-20) and then through another cannula into the contralateral, isolated popliteal artery supplying the gastrocnemius. Y-connectors positioned before and after the pump allowed either spontaneous perfusion of the gastrocnemius, at the animal's own blood pressure, or controlled flow at any desired level by adjusting the pump setting. A T-connector in the tubing to the gastrocnemius was connected to a pressure transducer (Narco Biosystems, Model RP-1500) for measurement of muscle perfusion pressure. Arterial blood samples were taken from another T-connector in the cannula exiting the right femoral artery prior to the roller pump.
A portion of the calcaneus, with the two tendons from the gastrocnemius attached, was cut away at the heel and clamped around a metal rod for connection to an isometric myograph via a load cell (Interface SM-250) and a universal joint coupler. The universal joint allowed the muscle to always pull directly in line with the load cell, and thus prevented the application of torque to the cell. The other end of the muscle was left attached to its origin; both the femur and the tibia were fixed to the base of the myograph by bone nails. A turnbuckle strut was placed parallel to the muscle between the tibial bone nail and the arm of the myograph to minimize flexing of the myograph.
The sciatic nerve, which innervates the gastrocnemius, was exposed and isolated near the muscle, doubly ligated and cut between the ties. The distal stump of the nerve, ∼1.5–3.0 cm in length, was pulled through a small epoxy electrode containing two wire loops for stimulation. The muscle was covered with saline-soaked gauze and a thin plastic sheet to prevent drying and cooling.
Experimental design
To evoke muscle contractions, the nerve was stimulated by supramaximal square pulses of 4.0–6.0 V amplitude and 0.2 ms duration (Grass S48 stimulator), isolated from ground by a stimulus isolator (Grass SIU8TB). Before each experiment, the muscle was set at optimal length by progressively lengthening the muscle as it was stimulated at a rate of 0.2 Hz, until a peak in developed tension (total tension minus resting tension) was obtained. For the experiments, isometric tetanic contractions were triggered by stimulation with trains of stimuli (4–6 V, 200 ms duration, 50 Hz frequency) at a rate of two contractions every 3 s for a 3 min period. Based on studies of peak 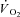 in this model (Kelley et al. 1996; Ameredes et al. 1998), this stimulation pattern elicited about 70% of peak metabolic rate for this muscle in the current experiments.
in this model (Kelley et al. 1996; Ameredes et al. 1998), this stimulation pattern elicited about 70% of peak metabolic rate for this muscle in the current experiments.
Tetanic contractions were chosen in order to allow a rapid attainment of a steady-state of developed force. Each isometric tetanic contraction lasted 200 ms, and was separated from the following by 1.3 s, during which the muscle was relaxing or relaxed.
For each dog, the experiment consisted of two contraction periods of 3 min duration, preceded by a resting baseline. The contraction periods were separated by at least 35 min of rest. The metabolic transition studied was therefore a rest-to-submaximal contractions transition. Two conditions were compared: (1) A control condition (Ctrl), in which the muscle was pump-perfused at a constant 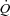 , adjusted 15–30 s before the start of the contraction to a level exceeding that attained in the steady-state of contractions, as determined in a preliminary trial with spontaneous adjustment of
, adjusted 15–30 s before the start of the contraction to a level exceeding that attained in the steady-state of contractions, as determined in a preliminary trial with spontaneous adjustment of 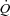 . When the blood supply to the gastrocnemius was switched from self-perfused to pump-perfused, at least 15 min were allowed for the haemodynamic parameters to stabilize. In Ctrl, 10 ml of saline solution were also infused intravenously at ∼1 ml min−1 for 10 min before contraction onset. (2) A ‘treatment’ condition, in which a 50 mm iodoacetamide (IA) solution in 10 ml of normal saline was infused into the muscle's arterial blood supply at a rate (∼1 ml min−1) to establish a 5 mm IA concentration (Harrison et al. 1999, 2003; Brault et al. 2003; Kindig et al. 2005) before contractions onset. In preliminary trials 2 mm IA was found to have little effect in this preparation. Also in IA the muscle was pump-perfused at the constant
. When the blood supply to the gastrocnemius was switched from self-perfused to pump-perfused, at least 15 min were allowed for the haemodynamic parameters to stabilize. In Ctrl, 10 ml of saline solution were also infused intravenously at ∼1 ml min−1 for 10 min before contraction onset. (2) A ‘treatment’ condition, in which a 50 mm iodoacetamide (IA) solution in 10 ml of normal saline was infused into the muscle's arterial blood supply at a rate (∼1 ml min−1) to establish a 5 mm IA concentration (Harrison et al. 1999, 2003; Brault et al. 2003; Kindig et al. 2005) before contractions onset. In preliminary trials 2 mm IA was found to have little effect in this preparation. Also in IA the muscle was pump-perfused at the constant 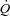 utilized in Ctrl. In both experimental conditions, to prevent vasoconstriction and inordinate pressure increases with the elevated
utilized in Ctrl. In both experimental conditions, to prevent vasoconstriction and inordinate pressure increases with the elevated 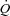 , 1–2 ml min−1 of a 10−2m adenosine solution (in normal saline) was also infused intra-arterially by a pump, beginning from 15–30 s before the onset of contractions. The adenosine infusion was then continued throughout the contraction period. This dosage of the drug was previously shown to be effective in obtaining a significant vasodilatation at the muscle level without causing significant metabolic effects (such as changes in resting
, 1–2 ml min−1 of a 10−2m adenosine solution (in normal saline) was also infused intra-arterially by a pump, beginning from 15–30 s before the onset of contractions. The adenosine infusion was then continued throughout the contraction period. This dosage of the drug was previously shown to be effective in obtaining a significant vasodilatation at the muscle level without causing significant metabolic effects (such as changes in resting 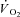 ,
, 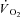 at the same submaximal level of contraction,
at the same submaximal level of contraction, 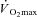 , acid–base status) (Kurdak et al. 1994; Grassi et al. 1998a,b, 2000, 2005). Since CK inhibition by IA is irreversible, the order of treatments could not be randomized, and Ctrl was always performed before IA. However, we have previously demonstrated in this in situ muscle model (Grassi et al. 1998a,b, 2002, 2005) that, given sufficient recovery, contractile protocols can be repeated with the same
, acid–base status) (Kurdak et al. 1994; Grassi et al. 1998a,b, 2000, 2005). Since CK inhibition by IA is irreversible, the order of treatments could not be randomized, and Ctrl was always performed before IA. However, we have previously demonstrated in this in situ muscle model (Grassi et al. 1998a,b, 2002, 2005) that, given sufficient recovery, contractile protocols can be repeated with the same 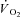 results.
results.
At the end of the experiments the dogs were killed with an overdose of pentobarbital. The gastrocnemius was excised, dissected free of connective tissue and weighed. The muscle was then dried to constant weight in an oven at 80°C to determine its percentage of water.
Measurements
Output from the pressure transducer was recorded on a strip chart recorder while outputs from the load cell and flowmeter (T206, Transonic Systems) were fed through strain gauge and transducer couplers, respectively, into a computerized (PowerComputing PowerBase 240 Macintosh clone) data acquisition system (GW Instruments Inc., SuperScope II and InstruNet Model 100B D/A input/output system). The load cell reached 90% of full response within 1 ms while the flowmeter was set to its highest pulsatile cut-off frequency of 100 Hz; both signals were sampled at a rate of 100 Hz by the computerized data acquisition system. The load cell was calibrated with known weights prior to each experiment. The flowmeter was calibrated with a graduated cylinder and clock during and after each experiment. Vascular resistance was calculated as muscle perfusion pressure (BPm) divided by 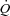 .
.
Samples of arterial blood entering the muscle and of venous blood from the popliteal vein were drawn anaerobically. Since the arterial values varied only slightly throughout each experiment, arterial samples were taken at rest, before the contractions and immediately after the contraction periods. A polyethylene tube (0.8 mm ID, 37 cm length, 0.25 ml total volume including luer hub) was threaded into the popliteal vein cannula to the point where the vein exited the gastrocnemius. This allowed collection of venous blood immediately draining from the muscle. Venous samples were taken at rest (∼10 s before the onset of contractions), every 5–7 s during the first 75 s of contractions, and every 30–45 s thereafter until the end of the contraction period. The precise time of each venous sample was recorded.
Blood samples were immediately stored in iced water and analysed within 30 min of collection. Both arterial and venous blood samples were analysed at 37°C for 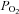 ,
, 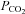 , plasma lactate concentration, and pH by a blood gas, pH, electrolytes, metabolites analyser (GEM Premier 3000, Instrumentation Laboratories, Lexington, MA, USA), and for haemoglobin concentration ([Hb]) and per cent saturation of Hb (
, plasma lactate concentration, and pH by a blood gas, pH, electrolytes, metabolites analyser (GEM Premier 3000, Instrumentation Laboratories, Lexington, MA, USA), and for haemoglobin concentration ([Hb]) and per cent saturation of Hb (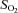 , %) with a CO-Oximeter (IL 682, Instrumentation Laboratories, Lexington, MA, USA), set for dog blood. These instruments were calibrated before and during each experiment. Dissolved O2 was also taken into account for the calculation of blood O2 concentration. Plasma lactate concentrations were corrected to whole blood values before use in exchange calculations. The difference between venous and arterial whole blood lactate concentration was multiplied by muscle blood flow in order to obtain blood lactate efflux from muscle.
, %) with a CO-Oximeter (IL 682, Instrumentation Laboratories, Lexington, MA, USA), set for dog blood. These instruments were calibrated before and during each experiment. Dissolved O2 was also taken into account for the calculation of blood O2 concentration. Plasma lactate concentrations were corrected to whole blood values before use in exchange calculations. The difference between venous and arterial whole blood lactate concentration was multiplied by muscle blood flow in order to obtain blood lactate efflux from muscle. 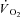 of the gastrocnemius was calculated by Fick's principle as
of the gastrocnemius was calculated by Fick's principle as 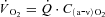 , where
, where 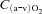 is the difference in O2 concentration between arterial blood (
is the difference in O2 concentration between arterial blood (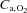 ) and venous blood (
) and venous blood (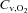 ).
). 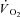 was calculated at discrete time intervals corresponding to the timing of the blood samples.
was calculated at discrete time intervals corresponding to the timing of the blood samples.
During both trials, muscle biopsies were obtained by superficial excision of muscle pieces with a scalpel, at rest and during the last 15 s of the contraction period. Biopsy samples were immediately frozen in liquid nitrogen. Subsequently, samples were freeze-dried and dissected from visible connective tissue and blood. The freeze-dried samples (4–8 mg) were placed into tubes and extracted with 0.5 m HClO4 (containing 1 mm EDTA) and neutralized with 2.2 m KHCO3. This extract was used for determination of adenosine triphosphate (ATP), phosphocreatine (PCr), creatine (Cr) and lactate concentrations by enzymatic spectrophotometric assays (Beckman DU 640B) (Bergmeyer, 1974). Muscle metabolite concentrations were expressed in mmol (kg dry mass)−1 (DM). Substrate level phosphorylation, that is total ‘anaerobic’ ATP yield, during contractions was estimated (in mmol kg DM−1 of ATP) as: Δ[PCr] + (1.5 Δ[lactate]) + 2 Δ[ATP] (Spriet et al. 1987), in which Δ indicates the difference between concentrations at rest and at 3 min during contractions. The net lactate efflux from muscle was calculated as 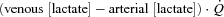 . The total amount of lactate coming out of the muscle during the contraction period was calculated as the area under the individual curves of net lactate efflux vs. time. This amount was taken into account in the calculation of substrate level phosphorylation.
. The total amount of lactate coming out of the muscle during the contraction period was calculated as the area under the individual curves of net lactate efflux vs. time. This amount was taken into account in the calculation of substrate level phosphorylation.
Analyses
Force
Peak force and tension–time integral (TTI) were calculated for each contraction. To calculate TTI, the pre-stimulation baseline was set as zero and the TTI was taken as the integral of force from the onset of force development in one contraction to the data sample before the beginning of force development in the next contraction. Any increase in baseline tension during contractions was accounted for by subtracting the area described by the straight line joining these two points and the pre-stimulation baseline. All these analyses were performed with commercial software (Origin, Microcal, USA).
Kinetics
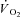 data were fitted by two equations, i.e. by eqn (1) and by eqn (2). Equation (1) was of the type:
data were fitted by two equations, i.e. by eqn (1) and by eqn (2). Equation (1) was of the type:
| (1) |
In this equation, yBAS indicates the baseline value obtained at rest before contraction onset, Af indicates the amplitude between yBAS and the steady-state value at the end of the contraction period, TDf the time delay and τf the time constant of the function. The suffix f indicates that these parameters relate to the ‘fundamental’ component of the 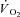 kinetics (Whipp et al. 2002).
kinetics (Whipp et al. 2002).
Equation (2) was of the type:
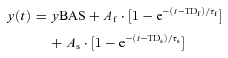 |
(2) |
In this equation, As, TDs and τs indicate, respectively, the amplitude, the time delay and the time constant of the ‘slow’ component of the kinetics (Whipp et al. 2002). The equation that best fitted the experimental data was determined by F test (see below). That is to say, when eqn (2) provided a better fit of data, a slow component of the  kinetics was present, superimposed on the fundamental component.
kinetics was present, superimposed on the fundamental component.
Peak force and TTI were constant during Ctrl (see below), but not in IA. This has potentially significant implications for parameter estimation because time-dependent alterations in the ‘error signal’ (assumed here to be linearly related to TTI) could alter the time-dependent response in  . Thus, a second control condition was established, in which a
. Thus, a second control condition was established, in which a 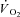 response was modelled using the TTI calculated for each contraction in IA and kinetics parameter estimates obtained in Ctrl. The
response was modelled using the TTI calculated for each contraction in IA and kinetics parameter estimates obtained in Ctrl. The 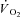 response for this ‘modelled control’ condition (Ctrl-Mod) was generated using the following equation:
response for this ‘modelled control’ condition (Ctrl-Mod) was generated using the following equation:
| (3) |
In this equation, G is a ‘gain’ factor and was established from A/TTI (see eqn 1) in the Ctrl condition, as were values for yBAS, TDf and τf. The TTIi was the instantaneous tension–time integral measured during IA. This allowed a Ctrl-Mod data set to be produced, which was reduced to a sampling frequency similar to Ctrl. Parameter values for Ctrl-Mod were then calculated by fitting to eqns 1 and (2) (i.e. using the same criteria as measured Ctrl and IA data). Therefore, this approach generated the 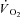 kinetic parameter values that would be expected during a non-constant TTI profile, assuming that IA had no effect on the
kinetic parameter values that would be expected during a non-constant TTI profile, assuming that IA had no effect on the 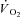 kinetics. Parameter estimates from Ctrl-Mod response profiles thereby allowed the influence of dynamic alterations in TTI during the IA condition on
kinetics. Parameter estimates from Ctrl-Mod response profiles thereby allowed the influence of dynamic alterations in TTI during the IA condition on  kinetics to be taken into account.
kinetics to be taken into account.
Statistical analysis
Values were expressed as means ± standard deviations (s.d.). To determine the statistical significance of differences between two means, a Student's paired t test (two-tailed) was performed. To determine the statistical significance of differences among more than two means, a repeated-measures analysis of variance was performed. A Tukey's post hoc test was utilized to discriminate where significant differences occurred. Data fitting by exponential functions was performed by an iterative least-squares approach. Comparisons between fittings with different exponential models were carried out by F test. The level of significance was set at P < 0.05. Data fitting and statistical analyses were carried out by using a commercially available software package (GraphPad Prism, GraphPad Software Inc.).
Results
The mass of the gastrocnemius muscles at the end of the experiments was 86 ± 13 g. The electrical stimulation and combined IA treatment caused some oedema, resulting in a percentage of water that was 80 ± 2%. Due to this oedema, the dry mass was used to calculate a corrected muscle mass for normalization of appropriate values (e.g.  ); the correction was to 76.5% water, a value based on the average of 65 gastrocnemius muscles in previous studies in this lab in which no oedema was observed. The corrected mass was 74 ± 11 g.
); the correction was to 76.5% water, a value based on the average of 65 gastrocnemius muscles in previous studies in this lab in which no oedema was observed. The corrected mass was 74 ± 11 g.
Mean (± s.d.) values of peak force are shown in Fig. 1. Peak force was lower in IA vs. Ctrl throughout the contraction period. There was no fatigue in Ctrl (final peak force/initial peak force (fatigue index, FI) = 0.97 ± 0.06). The high  utilized in the present study (see below) presumably minimized muscle fatigue in Ctrl. On the other hand, in IA force dramatically declined (by ∼30%) during the 2nd–4th contractions vs. the first, slightly recovered at 15–20 s and then slightly decreased during the ensuing contraction period (FI at 3 min = 0.67 ± 0.17). When TTI was considered, the FI at 3 min in IA was slightly lower (0.60 ± 0.11), as a consequence of an increase in baseline tension.
utilized in the present study (see below) presumably minimized muscle fatigue in Ctrl. On the other hand, in IA force dramatically declined (by ∼30%) during the 2nd–4th contractions vs. the first, slightly recovered at 15–20 s and then slightly decreased during the ensuing contraction period (FI at 3 min = 0.67 ± 0.17). When TTI was considered, the FI at 3 min in IA was slightly lower (0.60 ± 0.11), as a consequence of an increase in baseline tension.
Figure 1. Mean (± s.d.) values of peak force in the two experimental conditions (control, Ctrl and iodoacetamide infusion, IA).
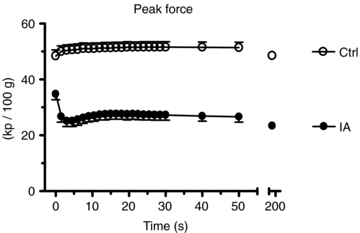
The x axis is broken in order to allow the reader to better notice the time-course of the variable during the first 50 s of contractions. During the first 30 s peak force values obtained during each contraction are shown, whereas after 30 s of contractions mean values calculated every 10 s are presented. Throughout the contraction period peak force was lower in IA vs. Ctrl. There was substantially no fatigue in Ctrl, whereas in IA peak force declined by about 30% during the 2nd–4th contractions vs. the first, slightly recovered at 15–20 s and then slightly decreased during the ensuing contraction period. See text for further details.
Values at rest and at ∼3 min of contractions for the main variables pertinent to O2 transport and utilization, acid–base status and haemodynamics are shown in Table 1. Arterial pH, perfusion pressure and vascular resistance at rest were lower in IA vs. Ctrl. No other resting variables were different between Ctrl and IA. CK inhibition was associated with a lower arterial pH also at ∼3 min of contractions, whereas all other variables related to convective and diffusive O2 delivery (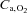 ,
, 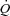 ,
, 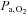 ) were not different between Ctrl and IA.
) were not different between Ctrl and IA. 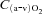 , fractional O2 extraction and
, fractional O2 extraction and 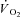 were significantly lower (by about 70%) in IA.
were significantly lower (by about 70%) in IA.
Table 1.
Values at rest and at ∼3 min of contractions for the main variables pertinent to O2 transport and utilization, acid-base status and haemodynamics, in the two experimental conditions (Ctrl and IA)
| Rest | 3 min of contractions | |||
|---|---|---|---|---|
| Ctrl | IA | Ctrl | IA | |
| [Hb]a (g 100 ml−1) | 15.1 ± 0.9 | 16.3 ± 2.2 | 15.3 ± 1.4 | 16.4 ± 2.2 |
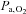 (Torr) (Torr) |
95.7 ± 12.9 | 97.5 ± 20.9 | 103.3 ± 20.1 | 97.3 ± 29.2 |
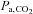 (Torr) (Torr) |
33.0 ± 3.2 | 33.8 ± 6.5 | 32.5 ± 2.8 | 30.7 ± 8.7 |
| pHa | 7.39 ± 0.06 | 7.32 ± 0.05* | 7.39 ± 0.05 | 7.32 ± 0.08* |
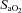 (%) (%) |
94.0 ± 5.1 | 92.3 ± 6.6 | 94.4 ± 4.7 | 90.9 ± 7.2 |
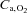 (ml 100 ml−1) (ml 100 ml−1) |
20.0 ± 1.5 | 21.3 ± 3.2 | 20.4 ± 1.7 | 21.1 ± 3.2 |
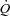 (ml 100 g−1 min−1) (ml 100 g−1 min−1) |
172.8 ± 40.9 | 171.2 ± 45.5 | 172.8 ± 40.9 | 171.2 ± 45.5 |
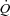 . .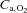 (ml 100 g−1 min−1) (ml 100 g−1 min−1) |
34.3 ± 6.4 | 35.2 ± 2.4 | 34.9 ± 6.4 | 34.9 ± 2.4 |
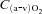 (ml 100 ml−1) (ml 100 ml−1) |
0.4 ± 1.0 | 0.1 ± 0.7 | 10.0 ± 2.6§ | 3.0 ± 2.0*§ |
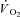 (ml 100 g−1 min−1) (ml 100 g−1 min−1) |
0.6 ± 1.7 | 0.1 ± 1.0 | 16.6 ± 2.5§ | 4.7 ± 2.9*§ |
| Fractional O2 extraction | 0.017 ± 0.048 | 0.002 ± 0.033 | 0.49 ± 0.10 | 0.14 ± 0.09*§ |
| BPm (mmHg) | 180 ± 20 | 135 ± 22* | 156 ± 17§ | 155 ± 28§ |
| Vascular resistance (mmHg ml−1 100 g min) | 1.07 ± 0.18 | 0.82 ± 0.21* | 0.93 ± 0.18§ | 0.94 ± 0.24§ |
[Hb]a, arterial haemoglobin concentration; 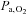 , oxygen partial pressure in arterial blood;
, oxygen partial pressure in arterial blood; 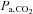 , carbon dioxide partial pressure in arterial blood; pHa, arterial pH;
, carbon dioxide partial pressure in arterial blood; pHa, arterial pH; 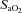 , haemoglobin saturation in arterial blood;
, haemoglobin saturation in arterial blood; 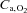 , arterial oxygen concentration;
, arterial oxygen concentration; 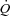 , muscle blood flow;
, muscle blood flow; 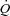 .
. , muscle O2 delivery;
, muscle O2 delivery;  , artero-venous oxygen concentration difference;
, artero-venous oxygen concentration difference;  , muscle O2 consumption; BPm, muscle perfusion pressure. Values are means ± s.d.; n = 6. Ctrl, control condition; IA, iodoactetamide infusion condition. *P < 0.05 vs. Ctrl. §P < 0.05 vs. rest in the same experimental condition (IA or Ctrl).
, muscle O2 consumption; BPm, muscle perfusion pressure. Values are means ± s.d.; n = 6. Ctrl, control condition; IA, iodoactetamide infusion condition. *P < 0.05 vs. Ctrl. §P < 0.05 vs. rest in the same experimental condition (IA or Ctrl).
Mean (± s.d.) values of muscle [ATP] and [PCr] at rest and at ∼3 min of contractions are shown in Fig. 2. [ATP] was lower in IA vs. Ctrl, both at rest and during contractions. Resting [PCr] was not different in the two conditions. [PCr] decrease during contractions in Ctrl (−30.8 ± 8.8 mmol kg−1 DM) was significantly higher than that observed in IA (−6.5 ± 10.5), confirming an effective CK inhibition obtained by IA administration.
Figure 2. Mean (± s.d.) values of muscle [ATP] and [PCr] at rest and after ∼3 min of contractions in the two experimental conditions.
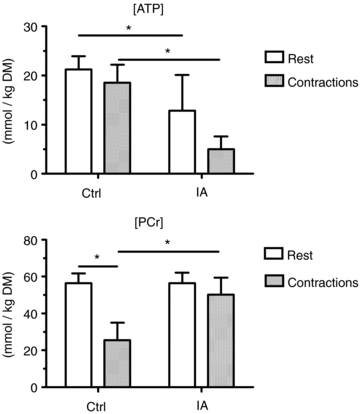
[ATP] was lower in IA vs. Ctrl, both at rest and during contractions. Resting [PCr] was not different in the two conditions. [PCr] decreased during contractions in Ctrl, but not in IA. See text for further details. *P < 0.05.
Mean (± s.d.) values of muscle [lactate] at rest and at ∼3 min of contractions are shown in the upper panel of Fig. 3. In IA muscle [lactate] tended to be higher at rest, but it did not increase during contractions, whereas it significantly increased during contractions in Ctrl. Mean (± s.d.) values of net lactate efflux from muscle are shown as a function of time of contractions in the lower panel of Fig. 3. Net lactate efflux was higher in IA vs. Ctrl at rest and early in the contraction period, whereas the opposite was true at the end of the contraction period. The total amount of lactate coming out of the muscle during the contraction period was not significantly different in IA (76 ± 104 μmol (100 g of ‘wet’ muscle)−1) vs. Ctrl (100 ± 86 μmol (100 g of ‘wet’ muscle)−1). Substrate level phosphorylation was lower in IA (25.0 ± 35.8 mmol (kg of ‘dry’ muscle)−1vs. Ctrl (72.1 ± 24.9 mmol (kg of ‘dry’ muscle)−1).
Figure 3. Mean (± s.d.) values of muscle [lactate] and net lactate efflux from muscle in the two experimental conditions.
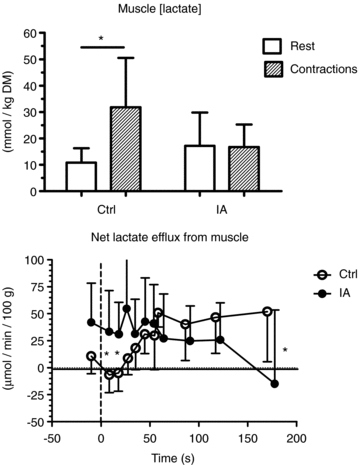
In IA muscle [lactate] tended to be higher at rest, but it did not increase during contractions, whereas it significantly increased during contractions in Ctrl. Net lactate efflux was higher in IA vs. Ctrl at rest and early in the contraction period, whereas the opposite was true at the end of the contraction period. See text for further details. *P < 0.05.
Individual 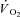 kinetics are shown for a typical experiment in Fig. 4. Time 0 indicates contractions onset. In all experiments
kinetics are shown for a typical experiment in Fig. 4. Time 0 indicates contractions onset. In all experiments 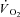 data were fitted by a monoexponential function (see Methods), which is also shown in Fig. 4. Thus, a ‘slow component’ of
data were fitted by a monoexponential function (see Methods), which is also shown in Fig. 4. Thus, a ‘slow component’ of 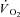 kinetics was not detected in any experiment. As discussed above (see Table 1) steady-state
kinetics was not detected in any experiment. As discussed above (see Table 1) steady-state 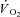 values during contractions were markedly lower in IA vs. Ctrl. As also discussed above (see Methods), for the analysis of
values during contractions were markedly lower in IA vs. Ctrl. As also discussed above (see Methods), for the analysis of 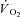 kinetics a second control condition (Ctrl-Mod) was introduced, which accounted for the effects on
kinetics a second control condition (Ctrl-Mod) was introduced, which accounted for the effects on 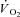 kinetics deriving from the non-square-wave error signal in IA. Calculated baseline (BAS), amplitude (Af), time delay (TDf) and time constant (τf) values are presented for individual dogs in Table 2. Mean (± s.d.) values of TDf and τf are also shown in Fig. 5. TDf in Ctrl was almost identical compared to Ctrl-Mod; in both conditions TDf was slightly lower than in IA (no significant difference). τf in Ctrl-Mod was not different compared to Ctrl; τf values in IA were lower (indicating a faster
kinetics deriving from the non-square-wave error signal in IA. Calculated baseline (BAS), amplitude (Af), time delay (TDf) and time constant (τf) values are presented for individual dogs in Table 2. Mean (± s.d.) values of TDf and τf are also shown in Fig. 5. TDf in Ctrl was almost identical compared to Ctrl-Mod; in both conditions TDf was slightly lower than in IA (no significant difference). τf in Ctrl-Mod was not different compared to Ctrl; τf values in IA were lower (indicating a faster 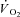 kinetics) compared to both Ctrl and Ctrl-Mod. In all conditions, the 95% confidence intervals for the τf estimates were narrow (2.0 ± 0.8, 1.0 ± 0.6 and 4.0 ± 0.5 s in Ctrl, Ctrl-Mod and IA, respectively).
kinetics) compared to both Ctrl and Ctrl-Mod. In all conditions, the 95% confidence intervals for the τf estimates were narrow (2.0 ± 0.8, 1.0 ± 0.6 and 4.0 ± 0.5 s in Ctrl, Ctrl-Mod and IA, respectively).
Figure 4. Individual kinetics from a typical experiment.
kinetics from a typical experiment.
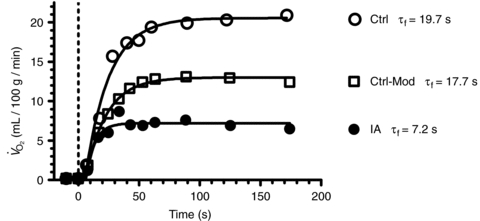
Time = 0 and the vertical hatched line indicates contractions onset.  data were fitted by a mono-exponential function, which is also shown in the figure. A second control condition (Ctrl-Mod) was introduced, which accounted for the effects on
data were fitted by a mono-exponential function, which is also shown in the figure. A second control condition (Ctrl-Mod) was introduced, which accounted for the effects on  kinetics deriving from the non-square wave error signal in IA. Calculated time constants (τf) are also presented. See text for further details.
kinetics deriving from the non-square wave error signal in IA. Calculated time constants (τf) are also presented. See text for further details.
Table 2.
Calculated baselines (BAS), amplitudes (Af), time delay (TDf) and time constant (τf) for the ‘fundamental’ component of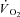 kinetics in Ctrl, Ctrl-Mod and in IA
kinetics in Ctrl, Ctrl-Mod and in IA
| Ctrl | Ctrl-Mod | IA | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dog | BAS (ml/100 g min−1) | Af (ml/100 g min−1) | TDf (s) | τf (s) | BAS (ml/100 g min−1) | Af (ml/100 g min−1) | TDf (s) | τf (s) | BAS (ml/100 g min−1) | Af (ml/100 g min−1) | TDf (s) | τf (s) |
| 1 | 0.4 | 16.4 | 3.8 | 15.3 | 0.4 | 13.0 | 4.1 | 13.8 | 0.3 | 8.3 | 5.6 | 11.4 |
| 2 | 0.4 | 13.9 | 6.5 | 19.8 | 0.4 | 8.2 | 6.5 | 16.4 | 0.3 | 5.5 | 3.7 | 6.4 |
| 3 | 0.2 | 20.3 | 5.6 | 19.7 | 0.2 | 12.8 | 5.6 | 17.6 | 0.1 | 7.1 | 6.3 | 7.2 |
| 4 | 0.5 | 13.3 | 10.1 | 16.2 | 0.5 | 5.9 | 9.9 | 10.2 | 0.1 | 3.2 | 24.6 | 5.2 |
| 5 | 0.3 | 14.4 | 9.3 | 15.3 | 0.3 | 5.8 | 9.2 | 12.6 | 0.5 | 4.0 | 6.5 | 13.9 |
| 6 | 0.3 | 17.8 | 4.3 | 13.5 | 0.3 | 10.7 | 4.5 | 12.0 | 0.2 | 1.3 | 7.9 | 4.8 |
| x | 0.4 | 16.0 | 6.6 | 16.6 | 0.4 | 9.4* | 6.6 | 13.8 | 0.2 | 4.9*§ | 9.1 | 8.1*§ |
| s.d. | 0.1 | 2.7 | 2.6 | 2.6 | 0.1 | 3.2 | 2.4 | 2.8 | 0.2 | 2.6 | 7.7 | 4.8 |
Values are means ± s.d.; n = 6. Ctrl, control condition; Ctrl-Mod, ‘modelled control’ condition; IA, iodoactetamide infusion condition.
Significantly different from the corresponding value obtained in Ctrl;
significantly different from the corresponding value obtained in Ctrl-Mod. See text for further details.
Figure 5. Mean (± s.d.) values of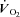 kinetics parameters (TDf, time delay; τf, time constant) in Ctrl, Ctrl-Mod and IA.
kinetics parameters (TDf, time delay; τf, time constant) in Ctrl, Ctrl-Mod and IA.
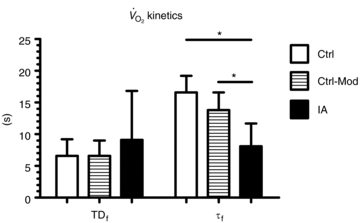
TDf in Ctrl was almost identical compared to Ctrl-Mod; in both conditions TDf was slightly lower than in IA (no significant difference). τf in Ctrl-Mod was not different compared to Ctrl; τf values in IA were lower (indicating a faster 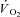 kinetics) compared to both Ctrl and Ctrl-Mod. See text for further details. *P < 0.05.
kinetics) compared to both Ctrl and Ctrl-Mod. See text for further details. *P < 0.05.
Discussion
Following CK inhibition by IA we measured faster 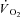 kinetics in an isolated dog gastrocnemius preparation in situ. This observation confirmed our hypothesis and suggests that, in normal conditions, CK-catalysed breakdown of PCr at contractions onset slows the rate of increase of oxidative phosphorylation. By acting as a high-capacitance energy buffer (Meyer, 1988), PCr breakdown appears to delay or attenuate the increase in [ADP] within mitochondria following rapid increases in ATP demand, thereby ‘damping’ a more rapid activation of oxidative phosphorylation. Our findings support the ‘dual’ role of PCr breakdown at exercise onset (Greenhaff, 2001): an almost instantaneous increase in energy yield (di Prampero, 1981) and, due to the PCr–Cr ‘shuttle’ from the sites of ATP utilization and the sites of oxidative phosphorylation in mitochondria (Mahler, 1980; Whipp & Mahler, 1980; Meyer et al. 1984; Cerretelli & di Prampero, 1987; Meyer, 1988; Kaasik et al. 1999; Greenhaff, 2001; Rossiter et al. 2005), an important role in
kinetics in an isolated dog gastrocnemius preparation in situ. This observation confirmed our hypothesis and suggests that, in normal conditions, CK-catalysed breakdown of PCr at contractions onset slows the rate of increase of oxidative phosphorylation. By acting as a high-capacitance energy buffer (Meyer, 1988), PCr breakdown appears to delay or attenuate the increase in [ADP] within mitochondria following rapid increases in ATP demand, thereby ‘damping’ a more rapid activation of oxidative phosphorylation. Our findings support the ‘dual’ role of PCr breakdown at exercise onset (Greenhaff, 2001): an almost instantaneous increase in energy yield (di Prampero, 1981) and, due to the PCr–Cr ‘shuttle’ from the sites of ATP utilization and the sites of oxidative phosphorylation in mitochondria (Mahler, 1980; Whipp & Mahler, 1980; Meyer et al. 1984; Cerretelli & di Prampero, 1987; Meyer, 1988; Kaasik et al. 1999; Greenhaff, 2001; Rossiter et al. 2005), an important role in 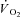 regulation. In other words, the bioenergetic mechanism which is fast enough to meet sudden increases in metabolic demand, that is PCr breakdown, is functionally related, through the levels of some of its metabolites, to the regulation of oxidative phosphorylation, the most important mechanism for ATP resynthesis.
regulation. In other words, the bioenergetic mechanism which is fast enough to meet sudden increases in metabolic demand, that is PCr breakdown, is functionally related, through the levels of some of its metabolites, to the regulation of oxidative phosphorylation, the most important mechanism for ATP resynthesis.
Following the pioneering work by Piiper et al. (1968), the isolated muscle preparation in situ utilized in the present study has been previously used by our group in numerous studies investigating the limiting and controlling factors for 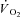 kinetics. Whereas in humans interventions such as training, ‘priming’ exercise, nitric oxide synthesis inhibition and others have obtained faster pulmonary
kinetics. Whereas in humans interventions such as training, ‘priming’ exercise, nitric oxide synthesis inhibition and others have obtained faster pulmonary 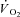 kinetics, CK inhibition is the first intervention in this preparation to have resulted in faster than normal
kinetics, CK inhibition is the first intervention in this preparation to have resulted in faster than normal 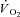 kinetics during a transition from rest to a submaximal metabolic rate (with the exception of a study in which a priming exercise was employed: Hernández et al. 2010). Previous attempts to speed
kinetics during a transition from rest to a submaximal metabolic rate (with the exception of a study in which a priming exercise was employed: Hernández et al. 2010). Previous attempts to speed 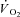 kinetics by enhancing convective (Grassi et al. 1998a) and diffusive (Grassi et al. 1998b) O2 delivery, by activating pyruvate dehydrogenase activity (dichloroacetate administration) (Grassi et al. 2002) or by relieving NO inhibition of cytochrome oxidase (l-NAME administration) (Grassi et al. 2005) were not successful. From those studies we concluded that (see e.g. Grassi, 2005): (a) in normal conditions the main limiting factor for
kinetics by enhancing convective (Grassi et al. 1998a) and diffusive (Grassi et al. 1998b) O2 delivery, by activating pyruvate dehydrogenase activity (dichloroacetate administration) (Grassi et al. 2002) or by relieving NO inhibition of cytochrome oxidase (l-NAME administration) (Grassi et al. 2005) were not successful. From those studies we concluded that (see e.g. Grassi, 2005): (a) in normal conditions the main limiting factor for  kinetics resides in a delayed metabolic activation of intracellular oxidative metabolism to adjust to the augmented metabolic needs; (b) this delayed activation is presumably functionally related to cytoplasmic PCr breakdown. A regulatory role of PCr breakdown in
kinetics resides in a delayed metabolic activation of intracellular oxidative metabolism to adjust to the augmented metabolic needs; (b) this delayed activation is presumably functionally related to cytoplasmic PCr breakdown. A regulatory role of PCr breakdown in 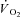 kinetics was hypothesized decades ago by Margaria et al. (1965): ‘…the oxidative processes in muscles are dictated by the concentration at a given time of the high energy compounds when split’, as well as by di Prampero & Margaria (1968), who described a linear relationship between muscle [PCr] decrease and
kinetics was hypothesized decades ago by Margaria et al. (1965): ‘…the oxidative processes in muscles are dictated by the concentration at a given time of the high energy compounds when split’, as well as by di Prampero & Margaria (1968), who described a linear relationship between muscle [PCr] decrease and 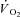 increase at steady state in the same preparation utilized in the present study. More recently, experimental evidence in support of this concept has been provided both from amphibian (Mahler, 1985) and murine muscles (Meyer, 1988). Subsequently, Rossiter et al. (1999) demonstrated that in humans pulmonary
increase at steady state in the same preparation utilized in the present study. More recently, experimental evidence in support of this concept has been provided both from amphibian (Mahler, 1985) and murine muscles (Meyer, 1988). Subsequently, Rossiter et al. (1999) demonstrated that in humans pulmonary 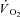 kinetics during the ‘metabolically relevant’ phase 2 (Grassi et al. 1996; Whipp et al. 2002) were not different from the kinetics of PCr breakdown, as determined by magnetic resonance spectroscopy. The same concept was confirmed also for the so-called ‘slow component’ of pulmonary
kinetics during the ‘metabolically relevant’ phase 2 (Grassi et al. 1996; Whipp et al. 2002) were not different from the kinetics of PCr breakdown, as determined by magnetic resonance spectroscopy. The same concept was confirmed also for the so-called ‘slow component’ of pulmonary 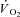 kinetics (Rossiter et al. 2002). A linear relationship between [PCr] decrease and
kinetics (Rossiter et al. 2002). A linear relationship between [PCr] decrease and 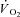 kinetics has been proposed in a model by Korzeniewski and Zoladz (2004): relatively small [PCr] decreases during rest-to-work transitions would be associated with relatively fast
kinetics has been proposed in a model by Korzeniewski and Zoladz (2004): relatively small [PCr] decreases during rest-to-work transitions would be associated with relatively fast 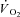 kinetics, whereas relatively large [PCr] decreases would be associated with relatively slower
kinetics, whereas relatively large [PCr] decreases would be associated with relatively slower 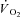 kinetics. In a study carried out on isolated mitochondria in vitro, Glancy et al. (2008) observed a linear relationship between the total creatine pool (PCr and Cr) and the τ of mitochondrial respiration adjustment following increases of ATP turnover elicited by hexokinase addition. A linear relationship between resting [PCr] and the τ of pulmonary
kinetics. In a study carried out on isolated mitochondria in vitro, Glancy et al. (2008) observed a linear relationship between the total creatine pool (PCr and Cr) and the τ of mitochondrial respiration adjustment following increases of ATP turnover elicited by hexokinase addition. A linear relationship between resting [PCr] and the τ of pulmonary 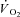 kinetics has been described by Francescato et al. (2008). An increase in resting [PCr] obtained by a dietary creatine supplementation was associated with slower [PCr] breakdown kinetics (Jones et al. 2009), although, rather surprisingly, with no changes in pulmonary
kinetics has been described by Francescato et al. (2008). An increase in resting [PCr] obtained by a dietary creatine supplementation was associated with slower [PCr] breakdown kinetics (Jones et al. 2009), although, rather surprisingly, with no changes in pulmonary 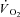 kinetics (Jones et al. 2002). Utilizing skinned human muscle, Walsh et al. (2001) demonstrated that mitochondrial ADP-stimulated respiration was attenuated or sensitised by [PCr] and [Cr], respectively, highlighting the central role of the CK system in the transduction of respiratory control. In none of these studies, however, was CK directly stimulated or inhibited, with a subsequent analysis of PCr and/or
kinetics (Jones et al. 2002). Utilizing skinned human muscle, Walsh et al. (2001) demonstrated that mitochondrial ADP-stimulated respiration was attenuated or sensitised by [PCr] and [Cr], respectively, highlighting the central role of the CK system in the transduction of respiratory control. In none of these studies, however, was CK directly stimulated or inhibited, with a subsequent analysis of PCr and/or 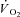 kinetics.
kinetics.
Faster 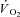 kinetics were modelled on the basis of experimentally determined PCr kinetics by Roman et al. (2002) in skeletal muscle of CK−/− mice. Gustafson & Van Beek (2002) observed faster
kinetics were modelled on the basis of experimentally determined PCr kinetics by Roman et al. (2002) in skeletal muscle of CK−/− mice. Gustafson & Van Beek (2002) observed faster 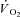 kinetics across heart muscle in CK−/− mice. However, as mentioned in the Introduction, there are limitations associated with knockout models, mainly related to the compensatory adaptations that occur in skeletal muscles as a result of CK−/−, such as increased mitochondrial volume density and oxidative capacity, structural reorganization of mitochondria and altered Ca2+ responses (Van Deursen et al. 1993; Steeghs et al. 1997; Bruton et al. 2003). All these changes could have a significant effect on
kinetics across heart muscle in CK−/− mice. However, as mentioned in the Introduction, there are limitations associated with knockout models, mainly related to the compensatory adaptations that occur in skeletal muscles as a result of CK−/−, such as increased mitochondrial volume density and oxidative capacity, structural reorganization of mitochondria and altered Ca2+ responses (Van Deursen et al. 1993; Steeghs et al. 1997; Bruton et al. 2003). All these changes could have a significant effect on 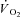 kinetics. Moreover, signs of histological damage and skeletal muscle wasting have been described in CK−/− mice (Momken et al. 2005).
kinetics. Moreover, signs of histological damage and skeletal muscle wasting have been described in CK−/− mice (Momken et al. 2005).
A faster adjustment of mitochondrial respiration after acute CK inhibition by IA was described in heart muscle by Harrison et al. (1999, 2003). However, CK is suggested to play a much more important role in damping respiratory control transduction in skeletal muscle tissue in comparison to cardiac muscle, in which 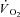 kinetics are much more rapid due to greater mitochondrial content and lower [PCr] (Balaban et al. 1986; Katz et al. 1989). In heart muscle a 5-fold increase in
kinetics are much more rapid due to greater mitochondrial content and lower [PCr] (Balaban et al. 1986; Katz et al. 1989). In heart muscle a 5-fold increase in 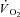 following a work increase is associated with essentially no change in [ATP], [PCr], [Pi] and [ADP]free (Balaban et al. 1986; Katz et al. 1989). It should also be considered that the magnitude of metabolic (oxidative) perturbation in heart muscle is much lower than what is found in skeletal muscle.
following a work increase is associated with essentially no change in [ATP], [PCr], [Pi] and [ADP]free (Balaban et al. 1986; Katz et al. 1989). It should also be considered that the magnitude of metabolic (oxidative) perturbation in heart muscle is much lower than what is found in skeletal muscle.
A faster rate of fall in intracellular 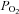 has been described in amphibian myoglobin-free single fibres following IA administration (Kindig et al. 2005). This is the only previous study in which acute CK inhibition was investigated in skeletal muscle fibres. In that study, however, intracellular the
has been described in amphibian myoglobin-free single fibres following IA administration (Kindig et al. 2005). This is the only previous study in which acute CK inhibition was investigated in skeletal muscle fibres. In that study, however, intracellular the 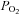 decrease did not reach a clear steady state, so that the faster decrease observed after IA administration could derive either from faster
decrease did not reach a clear steady state, so that the faster decrease observed after IA administration could derive either from faster 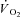 kinetics, a decreased efficiency of muscle contraction or from a combination of the two. Additionally, the absence of myoglobin (Mb) in the preparation utilized by Kindig et al. (2005) could influence the adjustment of oxidative phosphorylation and its regulation during metabolic transitions. The roles of Mb as a ‘buffer’ for intracellular
kinetics, a decreased efficiency of muscle contraction or from a combination of the two. Additionally, the absence of myoglobin (Mb) in the preparation utilized by Kindig et al. (2005) could influence the adjustment of oxidative phosphorylation and its regulation during metabolic transitions. The roles of Mb as a ‘buffer’ for intracellular 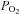 changes during contraction, and as a facilitator for peripheral O2 diffusion are well established (see e.g. Ordway & Garry, 2004). Moreover, it has been demonstrated that Mb rapidly desaturates (within 20 s) after the onset of submaximal exercise in human muscle (Richardson et al. 1995).
changes during contraction, and as a facilitator for peripheral O2 diffusion are well established (see e.g. Ordway & Garry, 2004). Moreover, it has been demonstrated that Mb rapidly desaturates (within 20 s) after the onset of submaximal exercise in human muscle (Richardson et al. 1995).
A confirmation of the results by Kindig et al. (2005) in ‘whole’ mammalian skeletal muscle was therefore necessary. In the present study we have directly demonstrated, for the first time, that the pharmacological ‘removal’ of the CK system markedly speeds the rate of adjustment of oxidative phosphorylation following an increase in ATP demand in a mammalian, blood-perfused skeletal muscle preparation in situ. In our study we did not determine PCr kinetics, but effective inhibition of CK, and therefore of PCr breakdown, was clearly demonstrated (see Fig. 2). With the experimental intervention we employed, however, this faster adjustment was achieved at the cost of a lower force production.
Force production
In rabbit hearts IA has significant effects on contractile function, with loss of contractile reserve (Harrison et al. 1999, 2003). In the present mammalian skeletal muscle preparation, IA infusion had profound negative consequences on force production (as well as on other variables). It is possible that muscle pH was lower at rest in IA due to an enhanced glycolysis, as suggested by the trends towards higher muscle [lactate] and net lactate efflux from muscle in IA vs. Ctrl. The lower resting muscle [ATP] in IA vs. Ctrl suggests that basal ATPase activity is normally buffered by PCr breakdown.
The lower arterial (and presumably muscle) pH and the lower [ATP] at rest were probably responsible for the lower peak force observed in IA vs. Ctrl during the first contraction. The time-course of peak force observed during the ensuing contraction period was similar to those described by previous authors in isolated amphibian skeletal muscle fibres (Kindig et al. 2005) and in skeletal muscles of CK−/− mice (Steeghs et al. 1997; Roman et al. 2002). Following a marked force decrease occurring with the second contraction, presumably directly related to the lack of ATP replacement by PCr breakdown, peak force only partially recovered during the ensuing ∼20 s. This partial recovery could in theory be attributed to the intervention of the other mechanisms of ATP resynthesis, namely glycolysis, oxidative phosphorylation and possibly adenylate kinase (AK). However, calculation of TTI in IA reveals that the partial recovery of force was accompanied by a rise in the baseline tension due to incomplete tension recovery between contractions. Therefore, the small peak force recovery in IA was not associated with a recovery in TTI. In IA, TTI decrease at the end of the contraction period was ∼40% of the initial force, whereas in terms of peak force the decrease was only ∼33% (see FI data). Muscle [ATP], already low at rest, manifested a further significant decrease during contractions, whereas (as expected) no decrease was observed in Ctrl. This supports the idea that CK function is essential not only for the temporal buffering of ATP, but also for the facilitation of transport of ATP/ADP equivalents between mitochondria and ATP utilization sites. The lower muscle [ATP], besides directly impairing force output, was probably associated with increased muscle [ADP] and [ADP]free. Muscle [ADP]free is typically too low to be directly determined, and it was not measured in the present experiments. However, during CK inhibition any reduction in [ATP] should be directly reflected by a near-reciprocal increase in [ADP]free. The reduced phosphorylation potential deriving from [ATP] decrease and [ADP]free increase would lower the Gibbs free energy made available from ATP hydrolysis, and contribute to the impairment of force production. On the other hand, high [ADP] is known to increase TTI through the inhibition of the release of the power stroke of the crossbridge cycle, or via Ca2+-related mechanisms (Mcdonald & Stephenson, 2004). The results are a slowed relaxation and an increased resting tension, which were both observed in the IA condition of the present study. According to Kindig et al. (2005) the decrease in peak tension described in their single-fibre model after IA administration was associated with a decreased peak cytosolic [Ca2+]. A lower muscle pH in IA, probably present in our study considering the lower arterial pH throughout the contraction period, could negatively affect sarcoplasmic reticulum function and muscle contractility (Allen et al. 2008).
Whatever the mechanism, the markedly altered peak force and TTI profiles in IA, particularly during the early phase of the contractile period, had the potential to alter 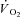 kinetics independently from the specific effects of the drug on muscle respiration. Thus, a second control condition (Ctrl-Mod) was introduced (see Methods), in which the influence of the ‘non-square’ change in the TTI profile (error signal) in IA was taken into account. Ctrl-Mod showed that the variable TTI profile throughout the contraction period would predict slightly faster
kinetics independently from the specific effects of the drug on muscle respiration. Thus, a second control condition (Ctrl-Mod) was introduced (see Methods), in which the influence of the ‘non-square’ change in the TTI profile (error signal) in IA was taken into account. Ctrl-Mod showed that the variable TTI profile throughout the contraction period would predict slightly faster 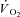 kinetics (τf ∼ 14 s) than those seen in Ctrl (τf ∼ 17 s). Nevertheless, these ‘modelled control’ kinetics were still significantly slower than the
kinetics (τf ∼ 14 s) than those seen in Ctrl (τf ∼ 17 s). Nevertheless, these ‘modelled control’ kinetics were still significantly slower than the 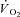 kinetics measured in IA (τf ∼ 8 s). This confirmed a direct effect of CK inhibition on respiratory control signal transduction, and that the fast
kinetics measured in IA (τf ∼ 8 s). This confirmed a direct effect of CK inhibition on respiratory control signal transduction, and that the fast 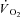 kinetics in IA were not simply due to an indirect effect of reduced tension development.
kinetics in IA were not simply due to an indirect effect of reduced tension development.
Energy provision during CK blockade
With energy provision unavailable from PCr breakdown, rephosphorylation of ADP produced by the Ca2+ and acto-mysin ATPases during contractile activity would need to be provided by oxidative phosphorylation, glycolysis and possibly adenylate kinase (AK). In rabbit heart, IA administration partially inhibited the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Harrison et al. 1999). In rat heart, IA did not affect the activities of phosphofructokinase and lactate dehydrogenase, whereas it did determine a dose-dependent inhibition of GAPDH (Tian et al. 1997). In that model, however, GAPDH activity normally exceeds glycolytic rate >50-fold; therefore, the glycolytic rate was not affected by the drug (Tian et al. 1997). Although no specific data are available, something similar could occur also in mammalian skeletal muscle. In the present study, resting muscle [lactate] and net lactate efflux from muscle at rest were greater in IA compared to Ctrl, consistent with a high rate of glycolytic flux. However, net lactate efflux gradually (but significantly) decreased throughout the contractile period in IA, compared to the increase efflux seen in Ctrl. Thus, muscle lactate production was actually attenuated during contractions in IA. Indeed, in IA after 3 min of contractions, muscle [lactate] was the same as at rest, whereas in Ctrl it increased by ∼3-fold. Calculated substrate level phosphorylation during contractions was lower in IA than in Ctrl. Therefore, it can be hypothesized that the decrease in resting [ATP] by IA may result in an increased glycolytic flux at rest, whereas the rapid increase in [ADP] following the onset of contractions, in the absence of CK, could determine a rapid activation of oxidative phosphorylation, leading to a rapid utilization of pyruvate. The resulting fall in lactate accumulation and/or production during contractions in IA would then be compatible with the faster 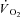 kinetics that was observed.
kinetics that was observed.
The energy output of the AK mechanism, which is normally low, was enhanced in studies in which CK inhibition was obtained by the administration of 1-fluoro-2,4-dinitrobenzene (Dzeja et al. 1996). ATP production from AK is usually enhanced under conditions of low [ATP] and high [ADP], which were observed in the present experiments. However, in isolated rabbit heart, CK inhibition by IA did not affect AK activity (Harrison et al. 1999). No inferences on the AK mechanism can be obtained from the data of the present study, in which AK activity was not determined. This applies also to other variables which could have allowed a deeper insight into the effects of IA, such as, for example, CK activity, mitochondrial density and fibre typing.
In the present study steady-state 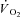 was markedly (by ∼70%) lower in IA vs. Ctrl, whereas peak force was reduced by only ∼33% (or ∼40% when TTI was considered). No clear-cut explanation can be forwarded for this discrepancy. As mentioned above, substrate level phosphorylation was lower in IA vs. Ctrl. The contribution of the AK mechanism, which could in theory be increased in IA (see above), could not be evaluated in the present study. At first sight the scenario might suggest a lower energy cost of contractions in IA. Gustafson and Van Beek (2002) observed indeed, in isolated hearts of CK−/− mice, higher levels of pressure development at similar levels of myocardial
was markedly (by ∼70%) lower in IA vs. Ctrl, whereas peak force was reduced by only ∼33% (or ∼40% when TTI was considered). No clear-cut explanation can be forwarded for this discrepancy. As mentioned above, substrate level phosphorylation was lower in IA vs. Ctrl. The contribution of the AK mechanism, which could in theory be increased in IA (see above), could not be evaluated in the present study. At first sight the scenario might suggest a lower energy cost of contractions in IA. Gustafson and Van Beek (2002) observed indeed, in isolated hearts of CK−/− mice, higher levels of pressure development at similar levels of myocardial 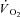 , pointing to an enhanced myocardial efficiency. No similar data are available for IA administration in mammalian skeletal muscle. IA did not affect oxidative phosphorylation in rabbit heart mitochondria (Harrison et al. 1999). The mechanism of the apparent improvement of contractile efficiency in the present study remains to be determined.
, pointing to an enhanced myocardial efficiency. No similar data are available for IA administration in mammalian skeletal muscle. IA did not affect oxidative phosphorylation in rabbit heart mitochondria (Harrison et al. 1999). The mechanism of the apparent improvement of contractile efficiency in the present study remains to be determined.
Conclusions
Following CK inhibition by IA, we observed faster 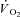 kinetics in an isolated dog gastrocnemius preparation in situ. This observation confirms that in mammalian skeletal muscle, under normal conditions, CK-catalysed PCr breakdown at contractions onset slows the rate of increase of oxidative phosphorylation. By acting as a high-capacitance energy buffer (Meyer, 1988), PCr breakdown appears to delay or attenuate the increase in [Cr] and [ADP] following rapid increases in ATP demand, thereby ‘damping’ a more rapid activation of oxidative phosphorylation.
kinetics in an isolated dog gastrocnemius preparation in situ. This observation confirms that in mammalian skeletal muscle, under normal conditions, CK-catalysed PCr breakdown at contractions onset slows the rate of increase of oxidative phosphorylation. By acting as a high-capacitance energy buffer (Meyer, 1988), PCr breakdown appears to delay or attenuate the increase in [Cr] and [ADP] following rapid increases in ATP demand, thereby ‘damping’ a more rapid activation of oxidative phosphorylation.
Acknowledgments
Financial support by the Agenzia Spaziale Italiana (ASI-OSMA Contract I/007/06/0 – Workpackage 1B-32-1) (Italy); by Telethon - UILDM GUP08007) (Italy); by BBSRC (UK) Project Grant BB/F019521/1; by National Institutes of Health (USA) grant AR40155 is acknowledged.
Glossary
Abbreviations
- A
amplitude
- AK
adenylate kinase
- BAS
baseline
- BPm
muscle perfusion pressure
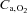
arterial blood O2 concentration
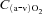
arterio-venous O2 concentration difference
- CK
creatine kinase
- Cr
creatine
- Ctrl
control
- Ctrl-Mod
modelled control
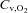
venous blood O2 concentration
- DM
dry mass
- FI
fatigue index
- G
gain
- Hb
haemoglobin
- IA
iodoacetamide
- Mb
myoglobin
- PCr
phosphocreatine
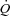
blood flow
- TD
time delay
- TTI
tension–time integral
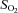
per cent saturation of Hb
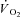
oxygen uptake
Author contributions
The experiments were carried out in the laboratory of the Department of Kinesiology, Auburn University, Auburn, AL, USA. B.G., H.B.R., M.C.H. and L.B.G. contributed to the conception and design of the experiment, drafting the article and to its critical revision. All authors contributed to the collection, analysis and interpretation of data. All authors approved the final version of the manuscript.
Author's present addresses
Matthew L. Goodwin: Weill Cornell Medical College, Cornell University, New York, NY, USA.
John L. Dobson: Department of Applied Physiology and Kinesiology, University of Florida, Gainesville, FL, USA.
James E. Harris: Aptos, CA, USA.
References
- Allen DG, Lamb GD, Westerblad H. Skeletal muscle fatigue: cellular mechanisms. Physiol Rev. 2008;88:287–332. doi: 10.1152/physrev.00015.2007. [DOI] [PubMed] [Google Scholar]
-
Ameredes BT, Brechue WF, Stainsby WN. Mechanical and metabolic determination of
 and fatigue during repetitive isometric contractions in situ. J Appl Physiol. 1998;84:1909–1916. doi: 10.1152/jappl.1998.84.6.1909. [DOI] [PubMed] [Google Scholar]
and fatigue during repetitive isometric contractions in situ. J Appl Physiol. 1998;84:1909–1916. doi: 10.1152/jappl.1998.84.6.1909. [DOI] [PubMed] [Google Scholar] - Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science. 1986;232:1121–1123. doi: 10.1126/science.3704638. [DOI] [PubMed] [Google Scholar]
- Bergmeyer HU. Methods of Enzymatic Analysis. New York: Academic; 1974. [Google Scholar]
- Brault JJ, Abraham KA, Terjung RL. Phosphocreatine content of freeze-clamped muscle: influence of creatine kinase inhibition. J Appl Physiol. 2003;94:1751–1756. doi: 10.1152/japplphysiol.01070.2002. [DOI] [PubMed] [Google Scholar]
- Bruton JD, Dahlstedt AJ, Abbate F, Westerblad H. Mitochondrial function in intact skeletal muscle fibres of creatine kinase deficient mice. J Physiol. 2003;552:393–342. doi: 10.1113/jphysiol.2003.050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerretelli P, di Prampero PE. Gas exchange in exercise. In: Fahri LE, Tenney SM, editors. Handbook of Physiology, Section 3, The Respiratory System, vol. IV, Gas Exchange. Bethesda: American Physiological Society; 1987. pp. 297–339. [Google Scholar]
-
Cerretelli P, Pendergast D, Paganelli WC, Rennie DW. Effects of specific muscle training on
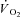 on-response and early blood lactate. J Appl Physiol. 1979;47:761–769. doi: 10.1152/jappl.1979.47.4.761. [DOI] [PubMed] [Google Scholar]
on-response and early blood lactate. J Appl Physiol. 1979;47:761–769. doi: 10.1152/jappl.1979.47.4.761. [DOI] [PubMed] [Google Scholar] - di Prampero PE. Energetics of muscular exercise. Rev Physiol Biochem Pharmacol. 1981;89:143–222. doi: 10.1007/BFb0035266. [DOI] [PubMed] [Google Scholar]
- di Prampero PE, Margaria R. Relationship between O2 consumption, high energy phosphates and the kinetics of the O2 debt in exercise. Pflügers Arch. 1968;304:11–19. doi: 10.1007/BF00586714. [DOI] [PubMed] [Google Scholar]
- Drummond GB. Reporting ethical matters in The Journal of Physiology: standards and advice. J Physiol. 2009;587:713–719. doi: 10.1113/jphysiol.2008.167387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzeja PP, Zeleznikar RJ, Goldberg ND. Suppression of creatine kinase-catalyzed phosphotransfer results in increased phosphoryl transfer by adenylate kinase in intact skeletal muscle. J Biol Chem. 1996;271:12847–12851. doi: 10.1074/jbc.271.22.12847. [DOI] [PubMed] [Google Scholar]
- Francescato MP, Cettolo V, di Prampero PE. Influence of phosphagen concentration on phosphocreatine breakdown kinetics. Data from human gastrocnemius muscle. J Appl Physiol. 2008;105:158–164. doi: 10.1152/japplphysiol.00007.2008. [DOI] [PubMed] [Google Scholar]
- Glancy B, Barstow TB, Willis WT. Linear relation between time constant of oxygen uptake kinetics, total creatine, and mitochondrial content in vitro. Am J Physiol Cell Physiol. 2008;294:C79–C87. doi: 10.1152/ajpcell.00138.2007. [DOI] [PubMed] [Google Scholar]
-
Grassi B. Limitation of skeletal muscle
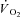 on-kinetics by inertia of cellular respiration. In: Jones AM, Poole DC, editors. Oxygen Uptake Kinetics in Sport, Exercise and Medicine. London, UK: Routledge; 2005. pp. 212–229. [Google Scholar]
on-kinetics by inertia of cellular respiration. In: Jones AM, Poole DC, editors. Oxygen Uptake Kinetics in Sport, Exercise and Medicine. London, UK: Routledge; 2005. pp. 212–229. [Google Scholar] -
Grassi B, Gladden LB, Samaja M, Stary CM, Hogan MC. Faster adjustment of O2 delivery does not affect
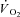 on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998a;85:1394–1403. doi: 10.1152/jappl.1998.85.4.1394. [DOI] [PubMed] [Google Scholar]
on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998a;85:1394–1403. doi: 10.1152/jappl.1998.85.4.1394. [DOI] [PubMed] [Google Scholar] -
Grassi B, Gladden LB, Stary CM, Wagner PD, Hogan MC. Peripheral O2 diffusion does not affect
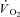 on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998b;85:1404–1412. doi: 10.1152/jappl.1998.85.4.1404. [DOI] [PubMed] [Google Scholar]
on-kinetics in isolated in situ canine muscle. J Appl Physiol. 1998b;85:1404–1412. doi: 10.1152/jappl.1998.85.4.1404. [DOI] [PubMed] [Google Scholar] -
Grassi B, Hogan MC, Greenhaff PL, Hamann JJ, Kelley KM, Aschenbach WG, Constantin-Teodosiu D, Gladden LB.
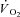 on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. J Physiol. 2002;538:195–207. doi: 10.1113/jphysiol.2001.012984. [DOI] [PMC free article] [PubMed] [Google Scholar]
on-kinetics in dog gastrocnemius in situ following activation of pyruvate dehydrogenase by dichloroacetate. J Physiol. 2002;538:195–207. doi: 10.1113/jphysiol.2001.012984. [DOI] [PMC free article] [PubMed] [Google Scholar] -
Grassi B, Hogan MC, Kelley KM, Aschenbach WG, Hamann JJ, Evans RK, Patillo RE, Gladden LB. Role of convective O2 delivery in determining
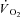 on-kinetics in canine muscle contracting at peak
on-kinetics in canine muscle contracting at peak 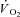 . J Appl Physiol. 2000;89:1293–1301. doi: 10.1152/jappl.2000.89.4.1293. [DOI] [PubMed] [Google Scholar]
. J Appl Physiol. 2000;89:1293–1301. doi: 10.1152/jappl.2000.89.4.1293. [DOI] [PubMed] [Google Scholar] - Grassi B, Hogan MC, Kelley KM, Howlett RA, Gladden LB. Effects of NOS inhibition by l-NAME on oxygen uptake kinetics in isolated canine muscle in situ. J Physiol. 2005;568:1021–1033. doi: 10.1113/jphysiol.2005.090068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi B, Poole DC, Richardson RS, Knight DR, Erickson BK, Wagner PD. Muscle O2 uptake kinetics in humans: implications for metabolic control. J Appl Physiol. 1996;80:988–998. doi: 10.1152/jappl.1996.80.3.988. [DOI] [PubMed] [Google Scholar]
- Greenhaff PL. The creatine–phosphocreatine system: there's more than one song in its repertoire. J Physiol. 2001;537:657. doi: 10.1111/j.1469-7793.2001.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson LA, Van Beek JHGM. Activation time of myocardial oxidative phosphorylation in creatine kinase and adenylate kinase knockout mice. Am J Physiol Heart Circ Physiol. 2002;282:H2259–H2264. doi: 10.1152/ajpheart.00264.2001. [DOI] [PubMed] [Google Scholar]
- Harrison GJ, van Wijhe MH, de Groot B, Dijk FJ, Gustafson LA, van Beek JHGM. Glycolytic buffering affects cardiac bioenergetic signalling and contractile reserve similar to creatine kinase. Am J Physiol Heart Circ Physiol. 2003;285:H883–H890. doi: 10.1152/ajpheart.00725.2002. [DOI] [PubMed] [Google Scholar]
- Harrison GJ, van Wijhe MH, de Groot B, Dijk FJ, van Beek JH. CK inhibition accelerates transcytosolic energy signalling during rapid workload steps in isolated rabbit heart. Am J Physiol Heart Circ Physiol. 1999;276:H134–H140. doi: 10.1152/ajpheart.1999.276.1.H134. [DOI] [PubMed] [Google Scholar]
-
Hernández A, McDonald JR, Lai N, Gladden LB. A prior bout of contractions speeds
 and blood flow on-kinetics and reduces the
and blood flow on-kinetics and reduces the 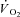 slow-component amplitude in canine skeletal muscle contracting in situ. J Appl Physiol. 2010;108:1169–1176. doi: 10.1152/japplphysiol.01318.2009. [DOI] [PubMed] [Google Scholar]
slow-component amplitude in canine skeletal muscle contracting in situ. J Appl Physiol. 2010;108:1169–1176. doi: 10.1152/japplphysiol.01318.2009. [DOI] [PubMed] [Google Scholar] - Jones AM, Carter H, Pringle JSM, Campbell IT. Effect of creatine supplementation on oxygen uptake kinetics during submaximal cycle exercise. J Appl Physiol. 2002;92:2571–2577. doi: 10.1152/japplphysiol.01065.2001. [DOI] [PubMed] [Google Scholar]
- Jones AM, Wilkerson DP, Fulford J. Influence of dietary creatine supplementation on muscle phosphocreatine kinetics during knee-extensor exercise in humans. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1078–R1087. doi: 10.1152/ajpregu.90896.2008. [DOI] [PubMed] [Google Scholar]
- Kaasik A, Minajeva A, De Sousa E, Ventura-Clapier R, Veksler V. Nitric oxide inhibits cardiac energy production via inhibition of mitochondrial creatine kinase. FEBS Lett. 1999;444:75–77. doi: 10.1016/s0014-5793(99)00033-2. [DOI] [PubMed] [Google Scholar]
- Katz LA, Swain JA, Portman MA, Balaban RS. Relation between phosphate metabolites and oxygen consumption of heart in vivo. Am J Physiol Heart Circ Physiol. 1989;256:H265–H274. doi: 10.1152/ajpheart.1989.256.1.H265. [DOI] [PubMed] [Google Scholar]
-
Kelley KM, Hamann JJ, Aschenbach WG, Gladden LB. Canine gastrocnemius muscle in situ:
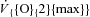 . Med Sci Sports Exerc. 1996;28:S62. Abstract. [Google Scholar]
. Med Sci Sports Exerc. 1996;28:S62. Abstract. [Google Scholar] - Kindig CA, Howlett RA, Stary CM, Walsh B, Hogan MC. Effects of acute creatine kinase inhibition on metabolism and tension development in isolated single myocytes. J Appl Physiol. 2005;98:541–549. doi: 10.1152/japplphysiol.00354.2004. [DOI] [PubMed] [Google Scholar]
-
Korzeniewski B, Zoladz JA. Factors determining the oxygen consumption rate (
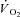 ) on-kinetics in skeletal muscles. Biochem J. 2004;379:703–710. doi: 10.1042/BJ20031740. [DOI] [PMC free article] [PubMed] [Google Scholar]
) on-kinetics in skeletal muscles. Biochem J. 2004;379:703–710. doi: 10.1042/BJ20031740. [DOI] [PMC free article] [PubMed] [Google Scholar] - Kurdak SS, Hogan MC, Wagner PD. Adenosine infusion does not improve maximal O2 uptake in isolated working dog muscle. J Appl Physiol. 1994;76:2820–2824. doi: 10.1152/jappl.1994.76.6.2820. [DOI] [PubMed] [Google Scholar]
- Macdonald WA, Stephenson DG. Effects of ADP on action potential-induced force responses in mechanically skinned rat fast-twitch fibres. J Physiol. 2004;559:433–447. doi: 10.1113/jphysiol.2004.067603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler M. Kinetics and control of oxygen consumption in skeletal muscle. In: Cerretelli P, Whipp BJ, editors. Exercise Bioenergetics and Gas Exchange. Amsterdam: Elsevier; 1980. pp. 53–66. [Google Scholar]
-
Mahler M. First-order kinetics of muscle oxygen consumption, and equivalent proportionality between
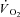 and phosphorylcreatine level. Implications for the control of respiration. J Gen Physiol. 1985;86:135–165. doi: 10.1085/jgp.86.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
and phosphorylcreatine level. Implications for the control of respiration. J Gen Physiol. 1985;86:135–165. doi: 10.1085/jgp.86.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar] - Margaria R, Mangili F, Cuttica F, Cerretelli P. The kinetics of the oxygen consumption at the onset of muscular exercise in man. Ergonomics. 1965;8:49–54. [Google Scholar]
- Meyer RA. A linear model of muscle respiration explains monoexponential phosphocreatine changes. Am J Physiol Cell Physiol. 1988;254:C548–C553. doi: 10.1152/ajpcell.1988.254.4.C548. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Sweeney HL, Kushmerick MJ. A simple analysis of the “phosphocreatine shuttle”. Am J Physiol Cell Physiol. 1984;246:C365–C377. doi: 10.1152/ajpcell.1984.246.5.C365. [DOI] [PubMed] [Google Scholar]
- Momken I, Lechêne P, Koulmann N, Fortin D, Mateo P, Doan BT, Hoerter J, Bigard X, Veksler V, Ventura-Clapier R. Impaired voluntary running capacity of creatine kinase-deficient mice. J Physiol. 2005;565:951–964. doi: 10.1113/jphysiol.2005.086397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway GA, Garry DJ. Myoglobin: an essential hemoprotein in skeletal muscle. J Exp Biol. 2004;207:3341–3446. doi: 10.1242/jeb.01172. [DOI] [PubMed] [Google Scholar]
- Piiper J, di Prampero PE, Cerretelli P. Oxygen debt and high-energy phosphates in gastrocnemius muscle of the dog. Am J Physiol. 1968;215:523–531. doi: 10.1152/ajplegacy.1968.215.3.523. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Noyszewski EA, Kendrick KF, Leigh JS, Wagner PD. Myoglobin O2 desaturation during exercise. Evidence of limited O2 transport. J Clin Invest. 1995;96:1916–1926. doi: 10.1172/JCI118237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman BB, Meyer RA, Wiseman RW. Phosphocreatine kinetics at the onset of contractions in skeletal muscle of MM creatine kinase knockout mice. Am J Physiol Cell Physiol. 2002;283:C1776–C1783. doi: 10.1152/ajpcell.00210.2002. [DOI] [PubMed] [Google Scholar]
-
Rossiter HB, Howe FA, Ward SA. Intramuscular phosphate and pulmonary
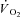 kinetics during exercise: Implications for control of skeletal muscle O2 consumption. In: Jones AM, Poole DC, editors. Oxygen Uptake Kinetics in Sport, Exercise and Medicine. London: Routledge; 2005. pp. 154–184. [Google Scholar]
kinetics during exercise: Implications for control of skeletal muscle O2 consumption. In: Jones AM, Poole DC, editors. Oxygen Uptake Kinetics in Sport, Exercise and Medicine. London: Routledge; 2005. pp. 154–184. [Google Scholar] - Rossiter HB, Ward SA, Doyle VL, Howe FA, Griffiths JR, Whipp BJ. Inferences from pulmonary O2 uptake with respect to intramuscular [phosphocreatine] kinetics during moderate exercise in humans. J Physiol. 1999;518:921–932. doi: 10.1111/j.1469-7793.1999.0921p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossiter HB, Ward SA, Kowalchuck JM, Howe FA, Griffiths JR, Whipp BJ. Dynamic asymmetry of phosphocreatine concentration and O2 uptake between the on- and off-transients of moderate- and high-intensity exercise in humans. J Physiol. 2002;541:991–1002. doi: 10.1113/jphysiol.2001.012910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spriet LL, Söderlund K, Bergström M, Hultman E. Anaerobic energy release in skeletal muscle during electrical stimulation in men. J Appl Physiol. 1987;62:611–615. doi: 10.1152/jappl.1987.62.2.611. [DOI] [PubMed] [Google Scholar]
- Stainsby WN, Welch HG. Lactate metabolism of contracting skeletal muscle in situ. Am J Physiol. 1966;211:177–183. doi: 10.1152/ajplegacy.1966.211.1.177. [DOI] [PubMed] [Google Scholar]
- Steeghs K, Benders A, Oerlemans F, de Haan A, Heerschep A, Ruitenbeek W, Jost C, Van Deursen J, Perryman B, Pette D, BrUckwilder M, Kondijs J, Jap P, Veerkamp J, Wieringa B. Altered Ca2+ responses in muscles with combined mitochondrial and cytosolic creatine kinase deficiencies. Cell. 1997;89:93–103. doi: 10.1016/s0092-8674(00)80186-5. [DOI] [PubMed] [Google Scholar]
- Tian R, Christe ME, Spindler M, Hopkins JCA, Halow JM, Camacho SA, Ingwall JS. Role of MgADP in the development of diastolic dysfunction in the intact beating rat heart. J Clin Invest. 1997;99:745–751. doi: 10.1172/JCI119220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deursen J, Heerschap A, Oerlemans F, Ruitenbeek W, Jap P, ter Laak H, Wieringa B. Skeletal muscle of mice deficient in muscle creatine kinase lack burst activity. Cell. 1993;74:621–631. doi: 10.1016/0092-8674(93)90510-w. [DOI] [PubMed] [Google Scholar]
- Walsh B, Tonkonogi M, Söderlund K, Hultman E, Saks V, Sahlin K. The role of phosphorylcreatine and creatine in the regulation of mitochondrial respiration in human skeletal muscle. J Physiol. 2001;537:971–978. doi: 10.1111/j.1469-7793.2001.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whipp BJ, Mahler M. Dynamics of pulmonary gas exchange during exercise. In: West JB, editor. Pulmonary Gas Exchange. II. New York: Academic; 1980. pp. 33–96. [Google Scholar]
- Whipp BJ, Rossiter HB, Ward SA. Exertional oxygen uptake kinetics: a stamen of stamina? Biochem Soc Trans. 2002;30:237–247. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]