

Abstract
Brain abscesses are mainly caused by either direct or indirect inoculation of gram positive bacteria including Stapylococcus aureus (S. aureus) or Streptococcus species into the central nervous system. In the present study, we aimed to compare potential changes in brain abscess pathogenesis induced by two different strains of S. aureus, namely the laboratory strain RN6390 and the clinical isolate Reynolds. Although the Reynolds strain was expected to be more resistant to eradication by the host, due to the existence of a polysaccharide capsule, and subsequently to be more virulent, instead we found parenchymal damage and mortality rates to be more prominent following RN6390 infection. In contrast, the Reynolds strain proliferated faster and induced early expression of the chemokine CXCL2, matrix metalloproteinase-9 (MMP-9), and complement 3a and C5. Furthermore, there were early and more abundant infiltration of PMNs, T cells and erythrocyte extravasation in brain abscesses induced by the Reynolds strain. However, several immune parameters were not different between the two strains during the later stages of the disease. These results suggest that capsular S. aureus can modulate innate immunity and complement system activation differently than the acapsular strain RN6390, and the early changes induced by Reynolds strain may have an important impact on survival.
Keywords: S. aureus, Brain abscess, Mast cells, Immune cells, Complement 3a, Complement 5
1. Introduction
Staphylococcus aureus (S. aureus) is an opportunistic gram positive bacterium that causes a wide spectrum of clinical manifestations, ranging from localized soft-tissue infections to life-threatening bacteremia, endocarditis, and abscesses within multiple organs including the brain (Archer, 1998; Lowy, 1998; Townsend and Scheld, 1998). Many virulence factors contribute to the pathogenesis of staphylococcal infections including both bacterial structural components and secreted products such as surface-associated adhesion molecules, exoenzymes, exotoxins, and capsular polysaccharides (Projan and Novick, 1997). Capsular polysaccharides (CP) are produced by more than 90% of all S. aureus isolates (O’Riordan and Lee, 2004). In addition, serotyping studies reveal that CP5 and CP8 are the predominant polysaccharides of those isolates recovered from humans with virulent S. aureus infections (25% and 50%, respectively) (Hochkeppel et al., 1987; Sompolinsky et al., 1985).
Abscesses in brain parenchyma develop as a consequence of local spread of pyogenic bacteria from the paranasal sinuses, middle ear, or oral cavity, via hematogenous dissemination from a systemic infection (e.g. endocarditis), or by direct penetrating trauma to the head. The most common etiologies of brain abscess are the streptococcal species and S. aureus (Mathisen and Johnson, 1997; Townsend and Scheld, 1998).
During the last decade, an experimental brain abscess model in rats and mice has been established by direct intra-cerebral injection of live S. aureus (Flaris and Hickey, 1992; Kielian and Hickey, 2000). This system has been investigated intensely to understand the mechanisms in disease pathogenesis and the possible treatment modalities (Flaris and Hickey, 1992; Kielian and Hickey, 2000). It mimics accurately the natural course of brain abscess development in humans. Thus, during the early stage of brain abscess development (days 1–3), microglial and astrocyte activation, as well as neutrophil accumulation become evident, and are accompanied by subsequent tissue necrosis, and edema. Microglial and astrocyte activation is observed in all stages throughout abscess development. Predominant infiltration of macrophages and lymphocytes is observed from days 4–9 and characterizes the intermediate or late cerebritis stage. At the final or capsule stage (days 10 onward), a well-vascularized abscess wall is formed, sequestering the lesion and protecting the surrounding normal brain parenchyma from additional damage. Although RN6390, derived from the strain NCTC 8325, is widely used in several disease models due to the fact it is amenable to genetic manipulation, it is a laboratory strain and is quite different than the strains isolated from clinical cases. One important difference is the absence of capsular structure outside the bacteria. In addition, RN6390 has known mutations which affect its virulence (Blevins et al., 2002; Cassat et al., 2006). Moreover, accumulating data have suggested that regulatory models based on these strains are not entirely representative of the situation observed in clinical isolates.
Therefore, we aimed to evaluate S. aureus strain differences in the pathogenesis of brain abscess. One of the common clinical isolates, strain Reynolds, which has a capsule and expresses CP5 was directly compared with the uncapsulated strain RN6390. Our results show that although the abscess size was smaller and caused less damage to the surrounding tissue, the capsular strain Reynolds proliferated faster and caused early induction of chemoattractant mediator CXCL2, resulting in more polymorphonuclear leukocyte infiltration into the abscess area. Furthermore, the strain Reynolds caused early induction of MMP-9 and more local T cell infiltration, more erythrocyte extravasation and red blood cell lysis, as well as, greater elaboration of C3a and C5b complement components. Therefore, we suggest that the early induction of several host mechanisms may benefit the host in terms of disease control.
2. Materials and methods
2.1. Bacterial strains and generation of experimental brain abscesses
The live S. aureus strains RN6390 and Reynolds (generously provided by Dr. Kielian and Dr. Lee, respectively, at University of Arkansas for Medical Sciences), were laden with agarose prior to implantation in the brain as previously described (Kielian et al., 2001b; Kielian et al., 2001a). The animals injected with sterile agarose beads (i.e. PBS-encapsulated agarose) were used as controls. Brain abscesses were induced in 6 to 8 week old C57BL/6 mice. For all studies described here, equal numbers of age-matched male and female animals were used; previous work has established that both genders exhibit qualitatively similar inflammatory profiles following bacterial challenge (Baldwin and Kielian, 2004; Kielian et al., 2001b; Kielian et al., 2001a). Briefly, mice were anesthetized with 2.5% avertin (Sigma) intra-peritoneally and a 1-cm longitudinal incision was made along the vertex of the skull. A rodent stereotaxic apparatus for mouse (Stoelting, Kiel, WI) was used to implant S. aureus-, or PBS-encapsulated beads into the caudate putamen region of left hemisphere using the following coordinates relative to bregma: +1.0 mm rostral, +2.0 mm lateral, and −3.0 mm deep from the surface of the brain. A burr hole was made and a 5 μl Hamilton syringe fitted with a 26-gauge needle was used to slowly deliver 2 μl beads (104CFU) into the brain parenchyma. To minimize bead efflux and potential leakage into the meninges, the needle kept in place for 2.5 min following injection. The animal protocol under which these experiments were performed has been approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee and is in accord with the National Institutes of Health guidelines for the use of rodents.
2.2. Simultaneous collection of RNA and protein from brain abscesses
To collect brain abscess extracts for analysis, lesion sites were localized by the stab wound created during the injections and dissected within 1 to 2 mm on all sides. Tissues were homogenized in 500 μl of PBS supplemented with a Complete™ protease inhibitor cocktail tablet (Roche, Indianapolis, IN) and 160 U/ml of RNAse inhibitor (Promega, Madison, WI) using a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). At this point, a 20 μl aliquot of abscess homogenate was removed for quantitative culture of viable bacteria as described below. The remaining homogenate was divided into two aliquots and centrifuged at 14,000 rpm for 15 min at 4 °C to pellet material. Supernatants were removed and stored at −70 °C until ELISA analysis could be performed, as described below. One of the pellet fractions was used for extraction of total RNA and qRT-PCR, while the other was used for protein analysis through Western blotting.
2.3. Quantitation of viable bacteria from brain abscesses
The numbers of viable bacteria associated with brain abscesses in vivo were quantified by plating the serial dilutions of brain abscess homogenates onto blood agar plates (Becton Dickinson). Titers were calculated by enumerating colony growth and are expressed as colony forming units (CFU) per milliliter of homogenate.
2.4. Quantitation of brain abscess size
At the indicated time points post-infection, RN6390- or Reynolds-injected mice were perfused transcardially to eliminate leukocytes from the vasculature, whereupon brains were removed and immediately flash frozen on dry ice. Brain tissues were embedded in optimal cutting temperature (OCT) medium and serial 10 μm cryo-sections were made through the entire lesioned tissue and stained with hematoxylin and eosin (H & E) to identify abscess margins. Evaluation of serial sections throughout the affected brain region ensured that the largest abscess cross-sectional area was identified for comparisons of lesion size. The sections were visualized under 1.25× objective to cover the whole section, and abscess area (reported as mm2) was calculated using the MetaMorph image analysis program (Universal Imaging Corporation, Downingtown, PA).
2.5. Quantitation of T cells, microglia, macrophages, and PMNs in brain abscesses
To determine whether the presence or absence of a capsular structure on bacteria influenced the influx of T cells, PMNs, macrophages, and/or microglia into and around lesions, abscess-associated cells were quantified by FACS analysis as previously described (Carson et al., 1998; Ford et al., 1995; Kielian et al., 2007; Renno et al., 1995). Briefly, mice were perfused with PBS to eliminate leukocytes from the vasculature, whereupon the entire infected hemisphere was collected to recover abscess-associated cells. This approach ensured that equivalent tissue regions were procured from each group for comparison purposes. Following vascular perfusion, tissues were minced in HBSS (Mediatech, Herndon, VA) supplemented with 10% FBS (HyClone, Logan, UT) and filtered through a 70 μm nylon mesh cell strainer using a rubber policeman. The resulting slurry was digested for 30 min at 37 °C in HBSS supplemented with 2 mg/ml collagenase type I (Sigma, St. Louis, MO) and 5000 U/ml DNAse I (Invitrogen, Carlsbad, CA) to obtain a single-cell suspension. Following enzyme neutralization, cells were layered onto a discontinuous Percoll gradient (1.122 to 1.088 g/ml) and centrifuged at 2400 rpm for 20 min at room temperature in a swinging bucket rotor. After centrifugation, myelin debris was carefully aspirated and the cell interface collected. Following extensive washes and incubation in Fc Block™ (BD Biosciences, San Diego, CA) to minimize non-specific antibody binding to Fc receptors, cells were stained with directly-conjugated antibodies for 4-color FACS to detect neutrophils (Gr-1+, CD11b+, CD45hi), macrophages (Gr-1−, CD11b+, CD45hi), and microglia (Gr-1−, CD11b+, CD45lo-intermediate). To enumerate abscess-associated T cell infiltrates, cells were analyzed by 3-color FACS using anti-CD3, -CD4, and -CD8 antibodies. All antibodies were purchased from BD Biosciences. Cells were analyzed using a BD FACSAria with compensation set based on the staining of each individual fluorochrome alone (CD11b-AlexaFluor488, Gr1-APC, CD45-FITC, CD3-PE, CD4-PECy7, or CD8-APC) and correction for autofluorescence with unstained cells. Controls included cells stained with directly-conjugated isotype control antibodies to assess the degree of non-specific staining.
The differences in immune cell influx between RN6390- and Reynolds-infected mice were reported as the percentage of positive cells for each innate immune cell population and pooled data from a minimum of three independent experiments was shown.
2.6. Quantitative real-time RT-PCR (qRT-PCR)
Total RNA from brain abscesses of RN6390- or Reynolds-infected as well as sterile bead injected and naïve control mice was isolated using the TriZol reagent and treated with DNAse1 (both from Invitrogen, Carlsbad, CA) prior to use in qRT-PCR studies. The overall experimental procedure was performed as previously described (Kielian et al., 2005). Briefly, GAPDH primers and TAMRA TaqMan probes were designed as previously described (Esen et al., 2004; Tanga et al., 2005) and synthesized by Applied Biosystems (ABI, Foster City, CA). An ABI Assays-on-Dema™ Taqman kit was utilized to investigate MMP-9 and MMP-2 mRNA levels. Gene expression differences between abscesses from RN6390-, and Reynolds-infected mice as well as sterile bead injected controls were calculated after normalizing cycle thresholds against the “housekeeping” gene GAPDH and are presented as the fold-induction or fold-reduction (2−ΔΔCt) value relative to naïve mice.
2.7. Enzyme linked immunosorbent assay (ELISA)
Protein levels of murine TNF-α, IL-12p40, IL-10 (OptiEIA, BD PharMingen, Carpenteria, CA), CXCL2/MIP-2 (DuoSet, R&D Systems, Minneapolis, MN), and IgG (Betty Laboratories, Montgomery, TX) were quantified in brain abscess homogenates using ELISA kits according to the manufacturer’s instructions. All results were normalized to the amount of total protein extracted from tissues to correct for differences in lesion size as previously described (Baldwin and Kielian, 2004; Kielian et al., 2004a).
2.8. Quantitation of hemoglobin
Hemoglobin (Hb) quantitation was performed indirectly, which has been shown to be more sensitive than direct measurement (Montano and Morrison, 1999). The assay is based on the pseudoperoxidase activity of Hb on 2–7 diaminofluorene (DAF, Sigma) which is oxidized in the presence of Hb yielding the blue colored compound, fluorine. As described previously (Montano and Morrison, 1999), the working solution of DAF was prepared by mixing 10 volumes of diluent (0.2 M Tris–HCL, pH 7.2) with one volume of the DAF stock (1% w/v DAF dissolved in 90% glacial acetic acid), and 0.02 volumes of 30% hydrogen peroxide. Then, 50 μl of tissue homogenates was mixed with 100 μl of the working solution in a 96-well plate and incubated 20 min at room temperature, whereupon the optical density values were obtained at 630 nm wavelength.
2.9. Immunofluorescence staining and confocal microscopy
The effects of the two different strains of S. aureus on fibronectin deposition and mast cell accumulation during brain abscess development were evaluated by immunofluorescence staining for fibronectin and tryptase, respectively. In addition, the presence of any B cell infiltration into the brain following bacterial infection was investigated by immunofluorescence staining for CD19. Brain abscess tissues were collected at the indicated time points following bacterial challenge and 10 μm cryostat sections were mounted onto SuperFrost Plus slides (Fisher Scientific), air dried, and stored at −80 °C until use. Staining was initiated by equilibrating slides at room temperature for 15 min followed by incubation in ice-cold methanol. Following numerous rinses in PBS, tissues were incubated with PBS/10% normal donkey serum to block non-specific binding of antibodies. Tissues were then reacted with either a rabbit anti-mouse fibronectin (Affinity Bioreagents, Golden, CO), a goat anti-mouse mast cell tryptase (Santa Cruz Biotechnology, Santa Cruz, CA), or a rat anti-mouse CD19 (BD Biosciences, San Jose, CA) antibodies overnight at 4 °C in a humidified chamber. Following several rinses in PBS, tissues were incubated with either a biotinylated donkey anti-rabbit IgG antibody (for fibronectin), a biotinylated donkey anti-goat IgG antibody (for tryptase), or a FITC conjugated donkey anti-rat antibody (for CD19) for 1 h at room temperature. All secondary antibodies were purchased from Jackson Immunoresearch (West Grove, PA). Fibronectin, and tryptase expression were visualized by the addition of a streptavidin-Alexa Fluor 568 conjugate (Molecular Probes, Eugene, OR), and Hoechst 33342 (2 μM; Molecular Probes) was added for labeling of nuclei. Slides were then coverslipped using the Prolong anti-fade reagent (Invitrogen, Carlsbad, CA) and sealed using nail polish. Slides were imaged using a Zeiss laser scanning confocal microscope (LSM 510, Carl Zeiss Microimaging Inc, Thornwood, NY) with the confocal pinhole set to obtain an optical section thickness of 1.6 μm. CD19-stained slides were imaged under an immunofluorescence microscope (Nikon Eclipse Ti, Nikon Instruments Inc., Melville, NY). Specific staining was confirmed by the absence of fluorescence signal following incubation of brain abscess tissues with secondary antibodies alone (data not shown).
2.10. Western blotting
Differences in matrix metalloproteinase 9 (MMP-9), complement 3 (C3), and C3a receptor (C3aR) expression in brain abscesses of RN6390- or Reynolds-infected mice were evaluated by Western blot analysis as previously described (Kielian et al., 2004b). Blots were probed using rabbit anti-rat MMP-9 (Millipore, Billerica, MA), rat anti-mouse C3, rabbit anti-mouse C3aR, goat anti-mouse C5α (all three from Santa Cruz Biotechnology, Santa Cruz, CA) antibody, followed by horseradish peroxidase-conjugated donkey anti-rabbit, anti-rat, or anti-goat immunoglobulin G (Jackson Immunoresearch, West Grove, PA). Blots were stripped and reprobed with a rabbit anti-actin polyclonal antibody (Sigma, St. Louis, MO) to verify uniformity in gel loading and developed using the ChemiGlow West substrate (Alpha Innotech, San Leandro, CA) followed by exposure to X-ray film.
2.11. Statistics
Significant differences between experimental groups were determined using the unpaired Student’s t-test at the 95% confidence interval with Sigma Stat (SPSS Science, Chicago, IL). In addition, significant differences between the various time points in the same experimental group were determined using one-way ANOVA followed by the Holm-Sidak method for pair-wise multiple comparisons with Sigma Stat.
3. Results
3.1. Bacterial burdens following infection with the capsular strain of S. aureus were higher during the early time period of infection
It has been well established that within 24 h following brain infection, bacterial burdens exceed 2–3 logs of the number of bacteria injected (Baldwin and Kielian, 2004; Kielian et al., 2001a). When we compared the bacterial titers following either Reynolds or RN6390 infection, we found that bacterial burdens were significantly higher at day 1 following challenge with the capsular S. aureus strain Reynolds (Fig. 1A). However, this difference had disappeared at later time points of infection (Fig. 1A). On the other hand, we found the mortality rates one day after the infection around 50–60% in RN6390 injected mice compared to 5–10% in Reynolds-injected group (Fig. 1B). Although the lower doses of RN6390 would cause less mortality, for scientific reasons we prepared the dose, which was optimal for the Reynolds strain, from both strains. The mortality rates or disease profiles did not show any gender bias among the groups. Moreover, the finding that bacterial burdens were still pronounced in brain abscesses of RN6390- infected mice, which survived to the later time points, excluded the possibility that these late survivors were weakly infected.
Fig. 1.

S. aureus Reynolds is proliferated faster. Mice (n=4–6 mice per group/time point) were injected with either agarose-laden S. aureus strain RN6390- or S. aureus strain Reynolds-beads (104CFU) as described in the Materials and methods. Animals were euthanized at the indicated time points and the number of viable organisms associated with brain abscesses was determined by quantitative culture. Titers are expressed as the mean log10 CFU per milliliter of brain abscess homogenate (A). (B) mortality rates are determined as the percentages of dead mice to surviving mice at the indicated time points (n=20–25 mice per group). Significant differences in mortality rates between S. aureus RN6390- and Reynolds-infected mice are denoted with asterisks (**, p<0.001). Results are the mean (±SEM) of three independent experiments.
Measurement of abscess sizes, however, indicated that the Reynolds strain generated significantly smaller abscess area at day 1 post-infection, although differences at later time points were not significant (Fig. 2A and B). As shown in Fig. 2A, tissue damage typified by necrotic and edematous areas around the abscess were more prominent following infection with strain RN6390. These findings demonstrate that bacterial burdens, per se, are not the sole determinant that dictates the extent of tissue injury and/or mortality during brain abscess development.
Fig. 2.

Following Reynolds infection the early damage to surrounding tissue is controlled better. Mice (n=4–6 mice per group/time point) were injected with either agarose-laden S. aureus strain RN6390- or S. aureus strain Reynolds-beads (104CFU) as described in the Materials and methods. Animals were euthanized at the indicated time points, whereupon brain tissues were flash frozen on dry ice for subsequent cryostat sectioning. Serial sections were prepared throughout the entire abscess to ensure that the maximal cross-sectional area was identified and stained with H & E to demarcate the histological appearance of lesions. (A) Stained slices were imaged under 1.25× objective to visualize the whole section. Lesions from 3 individual animals/group are presented to demonstrate the extent of abscess formation. (B) Abscess area (mm2; mean±SEM) was quantitated using the MetaMorph image analysis program by measuring the two largest lesion sizes for each tissue specimen. Significant differences in brain abscess size between S. aureus RN6390- and Reynolds-infected mice are denoted with asterisks (*, p<0.05). Results are mean of three independent experiments.
3.2. The neutrophil chemokine CXCL2 is more robustly induced by S. aureus Reynolds during the early period of infection
We have shown that in the experimental brain abscess model that RN6390 induced significant amounts of proinflammatory mediators including TNF-α and CXCL2/MIP-2 in brain extracts (Baldwin and Kielian, 2004; Kielian et al., 2004a). Similar induction of these mediators was observed in Reynolds-induced brain abscesses (Fig. 3A and B). Although both S. aureus strains induced significant TNF-α and CXCL2 release compared to sterile beads, the only significant difference found between the two strains was elevated CXCL2 levels at day 1 following Reynolds infection compared to RN6390 challenge (Fig. 3B). The latter pathogen induced a significant and steady rise in CXCL2 protein expression through the later time points evaluated, although the differences at later time points did not reach significant levels. Since CXCL2 is an important chemoattractant for neutrophils, early induction may have an influence on the recruitment of neutrophils into the brain.
Fig. 3.

Strain Reynolds induces more proinflammatory, but less anti-inflammatory mediator earlier than RN6390. Brain abscess homogenates from S. aureus strain RN6390- or Reynolds-infected animals (n=4 to 6 per group for each time point), as well as sterile bead injected mice (n=2 per group for each time point) were prepared at the indicated time points and analyzed for TNF-α (A), CXCL2 (B), IL-10 (C), and IL-12p40 (D) protein expression by ELISA. Abscess-associated cytokine levels were normalized to the amount of total protein recovered to correct for differences in tissue sampling size and reported as the mean values of cytokine (picograms) per milligram of protein. The results are the mean (±SEM) of 3 independent experiments. Significant differences between S. aureus strain RN6390- and Reynolds-infected mice are denoted with asterisks (*, p<0.05), while the pound sign represents the differences in the same group among different time points (#, p<0.05).
In addition to proinflammatory mediators, we also evaluated the effects of the two strains of S. aureus on induction of anti-inflammatory mediators or molecules which have a role in the interaction between innate and adaptive immunity, namely IL-10 and IL-12, respectively. The major physiological function of IL-10 is to attenuate macrophage activation in response to pathogens or their byproducts to minimize chronic inflammation (Murray, 2006). IL-10 can also regulate IL-12 production which is a key cytokine in the induction of cell mediated immunity following exposure to pathogens (Trinchieri, 2003) through its ability to induce IFN-γ production and Th1 differentiation (Moser and Murphy, 2000). Interestingly, we found that IL-10 expression was significantly higher at day 1 following RN6390 infection and remained elevated compared to both Reynolds-infected or sterile bead injected mice (Fig. 3C). On the other hand, IL-12p40, which is a subunit of either IL-12p70 or IL-23, was similarly expressed between all experimental groups until day 7 post-infection where it was significantly higher in RN6390 infected animals (Fig. 3D). Since the levels of IL-12p40 at early time points following bacteria injection were not significantly different compared to the sterile bead injected group, we suggest that anti-inflammatory mediators are controlled in a steady state following Reynolds infection, whereas RN630 infection induced more of an anti-inflammatory mediator profile.
3.3. Dynamics of peripheral immune cell infiltration into experimental brain abscesses
Previous studies have shown that S. aureus strain RN6390 led to the initial accumulation of neutrophils at the site of infection, which were evident at 24 h following bacterial exposure, followed by macrophage infiltration and microglial activation (Baldwin and Kielian, 2004; Kielian et al., 2001b). Since we found that the neutrophil chemoattractant, CXCL2, was significantly induced at day 1 following Reynolds infection, we investigated the relative kinetics of immune cell infiltration into the brain following infection with the two different strains of S. aureus. In accordance with the elevated levels of CXCL2 expression, the proportion of PMNs associated with brain abscessed induced by the Reynolds strain were significantly higher at day 1 compared to the acapsular strain RN6390 (Fig. 4A). Conversely, the differences in macrophage infiltration did not reach statistical significance while microglial population was significantly more prominent in RN6390 infected brains (Fig. 4B and C). These findings suggests that there is a significant recruitment of PMNs at the very beginning following Reynolds exposure which may be compensated with microglial proliferation in RN6390 infected brains. Following the early cerebritis stage, the intermediate cerebritis stage of brain abscess is associated with lymphocyte infiltration first evident at 4 days following infection (Flaris and Hickey, 1992; Kielian and Hickey, 2000). In agreement with these earlier studies, we also observed the accumulation of both CD3+CD4+ and CD3+CD8+ cells beginning at day 3 post-infection, which continued to increase out to day 7 in brain abscesses of both RN6390- and Reynolds-infected animals (Fig. 5A and B). However, the Reynolds strain induced significantly greater infiltration of both Th and Tc cells at all time points evaluated (Fig. 5). These data suggest that the encapsulated S. aureus strain Reynolds is more efficient at inducing local T cell recruitment compared to uncapsulated S. aureus RN6390. However, at this point we cannot determine whether these differences in T cell populations arise from differential retention and/or proliferation or differential recruitment.
Fig. 4.

Reynolds induces early infiltration of PMNs into brain abscesses. Abscess-associated cells were recovered from mice (3–4 animal/group/time point) at days 1, 3 and 7 following S. aureus strain RN6390 or Reynolds infection using a Percoll gradient method and analyzed by FACS. Results are presented as the percent positive cells for each population pooled from three independent experiments (mean±SEM). Significant differences between S. aureus strain RN6390- and Reynolds-infected mice are denoted with asterisks (*, p<0.05).
Fig. 5.
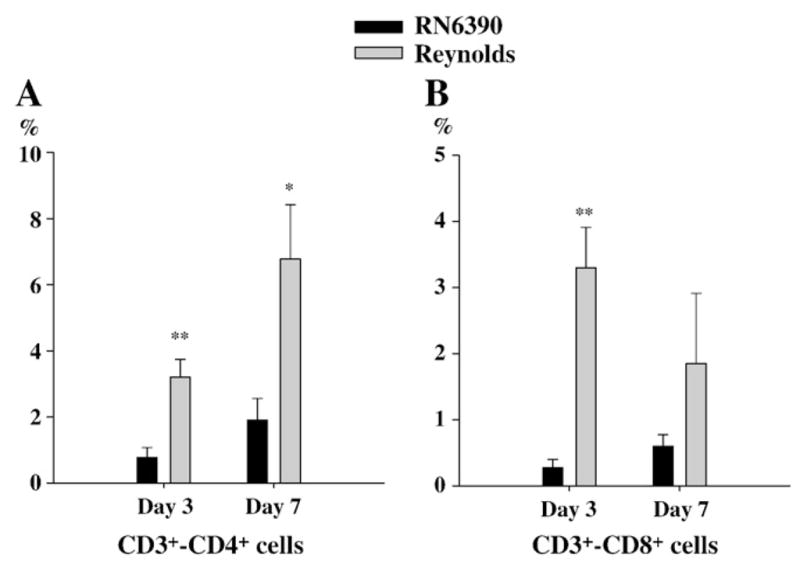
More T cells subsets infiltrate into brain abscesses following Reynolds infection. Abscess-associated cells were recovered from mice (3–4 animal/group/time point) at days 1, 3 and 7 following S. aureus strain RN6390 or Reynolds infection using a Percoll gradient method and analyzed by FACS. Results are presented as the percent positive cells for each population pooled from three independent experiments (mean±SEM). Significant differences between S. aureus strain RN6390- and Reynolds-infected mice are denoted with asterisks (*, p<0.05, **, p<0.001).
3.4. Early induction of MMP-9 expression was evident following Reynolds exposure
It has been shown that increased MMP proteolytic activity mediates acute neuronal injury (Thornton et al., 2008) and induces leukocyte invasion into the CNS (Sellebjerg and Sorensen, 2003), thus contributing to the pathogenesis of many neuroinflammatory and neurodegenerative conditions. However, it remains unclear whether these proteases are induced by the primary insult within CNS or by the inflammation associated with injury. Recently, cell wall components of S. aureus have been reported to induce MMP-9 production by neutrophils (Wang et al., 2005) and macrophages (Souza et al., 2008). In the present study, we found more prominent MMP-9 expression at both the mRNA and protein levels at day 1 following Reynolds exposure compared to RN6390 infection (Fig. 6A and B). However, MMP-9 expression levels steadily increased in RN6390- infected animals at later time points to exceed that of Reynolds-infected mice at day 7 (Fig. 6A). On the other hand, MMP-2 mRNA expression was not detectable in the brain following infection with either strain (data not shown). These data demonstrate that capsular structures of S. aureus cause an early induction of MMP-9, which may in turn, along with CXCL2, account for the significant neutrophil infiltrates observed at day 1 following Reynolds exposure.
Fig. 6.
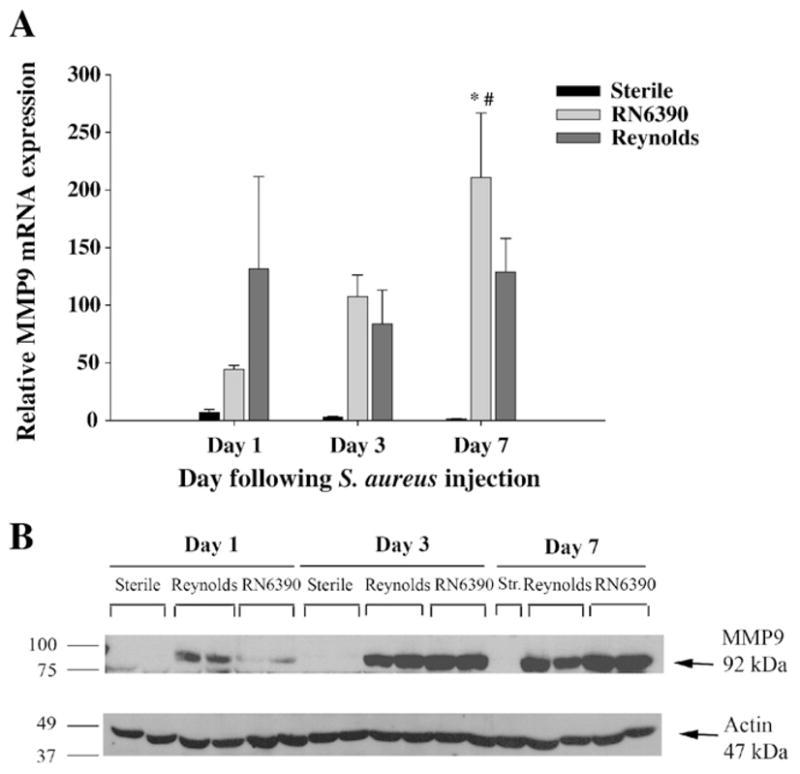
The expression of MMP-9 is augmented earlier in Reynolds-infected mice. Mice infected with either S. aureus strain RN6390- or S. aureus strain Reynolds- encapsulated agarose beads (104CFU) as well as sterile beads as described in Materials and methods, whereupon brain abscess homogenates were prepared (n=4 to 6 animals per group for each time point) at the indicated intervals and analyzed for MMP-9 mRNA and protein expression by qRT-PCR (A) and Western blotting (B), respectively. (A) MMP-9 gene expression was normalized to the house keeping gene GAPDH expression for each tissue and then normalized to that of naïve brain mRNA. The results represent the mean (±SEM) of three independent experiments. Significant differences between S. aureus strain RN6390- and Reynolds-infected mice are denoted with asterisks (*, p<0.05), while the pound sign represents the difference in the same group from other time points (#, p<0.05). (B) Western blots were stripped and reprobed with an antibody specific for actin to verify the uniformity in gel loading. Results are presented from two or three individual mice per group for each time point and representative of three independent experiments.
3.5. Effects of capsular structures on local complement activation
The complement system promotes phagocytosis and local inflammation and as such, represents an irreplaceable key component of innate immune defense against microbial pathogens. Several microbial components are able to activate the complement cascade. Previously, Kielian and co-workers have reported that IgG levels were significantly induced in brain abscesses following RN6390 infection (Kielian et al., 2001b). In the classical complement activation pathway, formation of antibody antigen complexes is the first step necessary to eradicate the bacteria. However, capsular structures prevent the formation of those complexes to some degree (Chavakis et al., 2007; Karakawa et al., 1988). Therefore, we wanted to compare whether the local accumulation of antibody was different between capsular and acapsular strains of S. aureus. As shown in Fig. 7A, there was significant accumulation of IgG in brain abscesses elicited by either S. aureus strain at all time points examined when compared to brains injected with sterile beads. However, total IgG levels were not significantly different between the two infected groups. Since the stab wound induced IgG levels were negligible, it was clear that bacterial infection induced antibody accumulation significantly. In addition, due to the fact that S. aureus is a commensal bacterium, it was not surprising to see very high levels of IgG even at day 1 post-infection suggesting a memory B cell response. Although the source of high IgG levels is likely due to plasma leakage based on previous data showing that the blood–brain barrier (BBB) is disrupted for at least two weeks following intra-cerebral injections RN6390 in the brain abscess model (Baldwin and Kielian, 2004; Kielian et al., 2001a, Kielian et al., 2001b), we wanted to evaluate the possibility of local antibody production by examining the presence of CD19+ B cells in the CNS. However, we were unable to detect any significant CD19+ B cell infiltration into brain abscesses following either RN6390 or Reynolds infection at any time point evaluated (data not shown), supporting the possibility that IgG accumulation is likely due to leakage from the circulation. Still, further studies are needed to examine B cell infiltration using more sensitive methods.
Fig. 7.

Reynolds infection induces significant hemolysis in brain tissue. Brain abscess homogenates from S. aureus strain RN6390- or Reynolds-infected animals (n=4 to 6 per group for each time point) as well as sterile bead injected mice (n=2 per group for each time point) were prepared at the indicated time points and analyzed for IgG (A) protein expression by ELISA and colorimetric hemoglobin (Hb) quantitation (B). Abscess-associated IgG levels were normalized to the amount of total protein recovered to correct for differences in tissue sampling size and reported as the mean values of IgG (picograms) per milligram of protein. Raw OD readings at 630 nm are used directly without any further normalization since all tissues were perfused prior to homogenization. The results represent the mean (±SEM) of three independent experiments. Significant differences between S. aureus strain RN6390- and Reynolds-infected mice are denoted with asterisks (*, p<0.05, **, p<0.001).
During the course of our studies, we noticed the presence of more petechial hemorrhages in brain abscesses of Reynolds compared to RN6390- infected mice. Erythrocytes via their complement receptor (CR1) specific for C3b, which is a key component of both the classical and alternative pathways, can play an important role in innate immunity (Birmingham, 1995; Birmingham and Hebert, 2001; Takemura et al., 1984). We measured local tissue hemoglobin (Hb) levels as a reflection of erythrocyte content and found significantly higher amounts in Reynolds-infected brains at both days 3 and 7 post-infection compared to sterile bead and RN6390- injected animals (Fig. 7B). These results imply that capsular S. aureus through unknown mechanisms augments erythrocyte extravasation which, in turn, might have a role in preventing bacterial spread. Next, we evaluated the levels of C3 and C5 in infected brain tissue homogenates to further investigate the effect of capsular and acapsular S. aureus on complement activation. C3b, along with C3a is cleaved from C3 by C3 convertase, which is the central reaction in both the classical and alternative complement pathways. C3a (9 kDa) is a strong chemoattractant for neutrophils, T cells and mast cells and mediates the local inflammatory process (Boos et al., 2004). We could not detect either the whole C3 protein or the complex forming component C3b (104 kDa) in abscess homogenates by Western blot, but interestingly, there was induction of C3a (Fig. 8). C3a expression was first detected at day 3, and peaked at day 7, where it was more pronounced in the Reynolds-infected brains (Fig. 8). Faint bands were observed around 49 kDa; however, since this size does not match any known possible cleavage component of C3, we considered them non-specific. In Fig. 8, we elected to present whole entire blot to emphasize that no C3 product, with the exception of C3a was expressed in the brain following S. aureus infection. On the other hand, the expression of the C3a receptor (C3aR) did not significantly change following infection at anytime point evaluated (data not shown). C5 (190 kDa) levels were examined by using an antibody against the C5 precursor, C5α chain and C5α′ chain. Interestingly, its levels were induced prominently at day 7 in the Reynolds-infected brains compared to RN6390- infected brains (Fig. 9). Taken together, these results suggest that S. aureus strain Reynolds was a more potent inducer of the key components of the complement system.
Fig. 8.

S. aureus regulates C3a induction in experimental brain abscess. Mice infected with either agarose-laden S. aureus strain RN6390- or S. aureus strain Reynolds-beads (104CFU) as described in Materials and methods, whereupon brain abscess protein extracts (40 μg per sample) were prepared at the indicated days post-infection and analyzed for C3 expression by Western blotting as described in Materials and methods. To compare with the baseline expression of C3 in the brain, naïve brain tissue was also included. Blots were stripped and reprobed with an antibody specific for actin to verify uniformity in gel loading. Results are presented from three individual animals per group and are representative of three independent experiments.
Fig. 9.
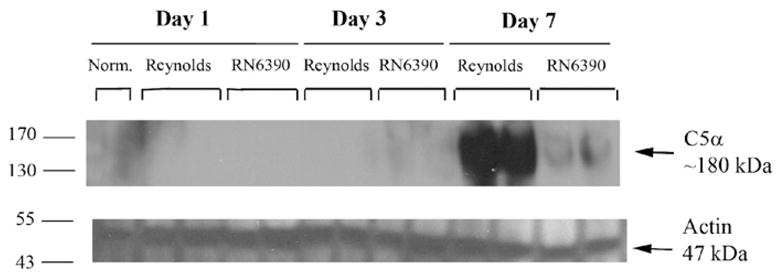
S. aureus strain Reynolds induce C5 levels prominently in experimental brain abscess. Mice infected with either agarose-laden S. aureus strain RN6390- or S. aureus strain Reynolds-beads (104CFU) as described in Materials and methods, whereupon brain abscess protein extracts (40 μg per sample) were prepared at the indicated days post-infection and analyzed for C5 expression by Western blotting as described in Materials and methods. To compare with the baseline expression of C5 in the brain, naïve brain tissue was also included. Blots were stripped and reprobed with an antibody specific for actin to verify uniformity in gel loading. Results are presented from two individual animals per group and are representative of three independent experiments.
3.6. S. aureus infection elicits mast cell infiltration into brain abscesses
Brain abscess is one of the few CNS insults that caused fibrosis instead of gliosis (Flaris and Hickey, 1992; Kielian and Hickey, 2000). At the final stage of abscess formation, a well-vascularized capsule forms around the necrotic tissue to protect the surrounding normal brain parenchyma. The inflammatory and necrotic area in the capsule is eventually replaced with connective tissue a process called fibrosis (Kielian, 2004; Wynn, 2008). Myofibroblasts can contribute to the formation of abscess wall, although the origin of these cells in the brain is not well understood. Recently, several lines of evidence have suggested the participation of mast cells in fibroblast proliferation in various organs. Increased numbers of mast cells are observed in close proximity to proliferating fibroblasts in healing wounds as well as in fibrotic lesions of lungs, vessel walls, and skin (Claman, 1989; Kawanami et al., 1979; Leroy et al., 1989; Trabucchi et al., 1988). Tryptase, a member of the serine protease enzyme family, is produced almost exclusively by mast cells, and impacts fibrosis by inducing the synthesis of fibronectin and collagen type I (Kondo et al., 2001). Therefore, we aimed to determine what effects different S. aureus strains had on abscess wall evolution and mast cell infiltration into the brain. Similar to our recent report (Kielian et al., 2008), fibronectin immunoreactivity appeared starting at day 3 and became prominent at day 7 following bacterial inoculation (Fig. 10). In addition, both strains of S. aureus were able to induce fibronectin expression to similar extents. To the best of our knowledge, for the first time we have demonstrated mast cell infiltration into the abscess area which was evident with tryptase immunoreactivity starting at day 3 and became prominent at day 7 (Fig. 11 and data not shown). Similar to fibronectin staining, mast cell infiltration was equivalent between both strains of S. aureus examined.
Fig. 10.
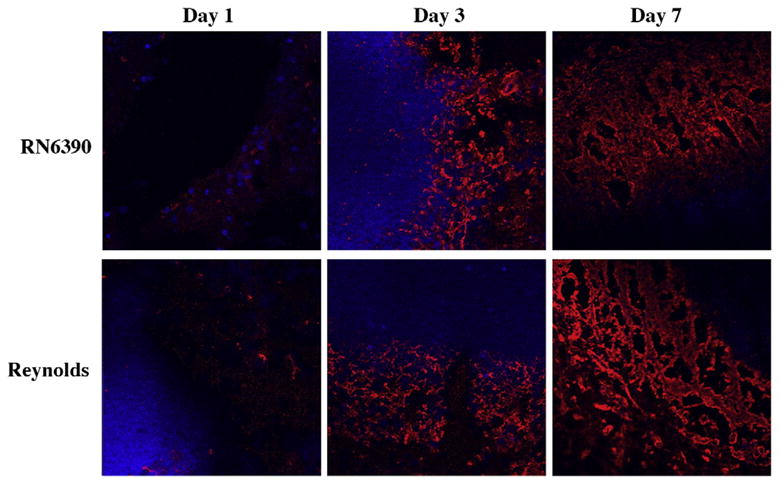
Fibronectin deposition along the developing brain abscess wall become prominent at day 7. Mice (n=4–6 mice per group/time point) were injected with either S. aureus strain RN6390- or S. aureus strain Reynolds- encapsulated agarose beads (104CFU) as described in the Materials and methods. Animals were euthanized at the indicated time points, whereupon brain tissues were flash frozen on dry ice for subsequent cryostat sectioning. Serial 10 μm thick sections were prepared throughout the entire abscess, subjected to immunofluorescence staining for fibronectin (red), and imaged using a 40× oil immersion objective lens by confocal microscopy. Nuclei were visualized using Hoechst 33342 (blue). Fibronectin immunoreactivity is shown along the peri-abscess area in the vicinity of the developing wall, where the dark areas represent necrotic regions. Results are representative of two independent experiments.
Fig. 11.

Mice (n=4–6 mice per group/time point) were injected with either S. aureus strain RN6390- or S. aureus strain Reynolds-beads (104CFU) as described in the Materials and methods. Animals were euthanized at the indicated time points, whereupon brain tissues were flash frozen on dry ice for subsequent cryostat sectioning. Serial 10 μm thick sections were prepared throughout the entire abscess, subjected to immunofluorescence staining for tryptase (red), and imaged by confocal microscopy (magnification 63×). Nuclei were visualized using Hoechst 33342 (blue). Tryptase immunoreactivity was prominent at day 7 in the abscess area. Results are representative of two independent experiments.
4. Discussion
In the present study, we compared brain abscess pathogenesis using two different strains of S. aureus; one a widely used, uncapsulated laboratory strain (RN6390) and the other a clinical isolate (Reynolds), which expresses capsular polysaccharide type 5. The main differences we observed following Reynolds infection in the experimental brain abscess model were earlier induction of CXCL2 production, neutrophil infiltration, MMP-9 expression and C3a as well as C5b induction compared to RN6390 infection. Furthermore, bacterial burdens were higher during the early stages of Reynolds infection. In contrast, damage to surrounding brain parenchyma and overall mortality rates were higher following RN6390 infection (Figs. 1B and 2A). These data were opposite to what we had hypothesized, as the polysaccharide capsule of the Reynolds strain should have made the bacteria relatively resistant to phagocytosis and more virulent as previously reported in bacteremia, as well as abdominal and renal abscess models (Nilsson et al., 1997; Tzianabos et al., 2001). However, consistent with what we observed in the brain abscess model, the acapsular strain RN6390 was found to be more virulent in several other disease models (Abdelnour et al., 1993; Beenken et al., 2003; Booth et al., 1997; Cheung et al., 1994; Nilsson et al., 1996). Studies which have compared the bacterial regulatory events among RN6390 and several clinical isolates have suggested that RN6390 may not be representative of most clinical isolates due to the differences in its production of proteases and exotoxins, and in its capacity to bind to host proteins (Blevins et al., 2002; Cassat et al., 2006). In addition, data from other studies showing that another S. aureus clinical isolate (ATCC 25923) did not cause mortality in their brain abscess model (Stenzel et al., 2005; Stenzel et al., 2008) also support the idea that RN6390 behaves differently than clinical isolates. These differences might explain how RN6390 induced a larger necrotic area and surrounding edema during brain abscess development compared to Reynolds. Conversely, it may be that differences in the early host response to infection have an important impact on the outcome from infection. Ongoing studies aim to discriminate between bacterial and host factors in disease pathogenesis.
Interestingly, we found that the CP5-producing Reynolds strain induced the early expression of CXCL2 and neutrophil infiltration into brain abscesses. In addition, greater infiltration of T cells into brain abscesses in response to Reynolds infection correlated with higher local levels of C3a. However, we were not able to detect other cleavage products of C3 within brain abscesses, including C3b. On the other hand, expression of the receptor for C3a (C3aR) was found unchanged during infection (data not shown), even though several studies have reported increased C3aR expression in various experimental models of CNS diseases (Akiyama et al., 2000; Barnum et al., 2002; Davoust et al., 1999; Gasque et al., 1998; Van Beek et al., 2000). Similar to C3a, C5 α was prominently induced in Reynolds-infected animals at day 7 post-infection (Fig. 9). Since a commercially available antibody against mouse C5a could not be located, we used an anti-C5 precursor C5α chain antibody to indirectly query C5a levels. C5 convertase cleaves C5 with one proteolytic cleavage at an arginine 75 residues downstream from the C5α chain N terminus (Sandoval et al., 2000), releasing C5a and C5b. The band we detected was smaller than intact C5 (i.e. 190 kDa), therefore, we propose it represents the larger fragment C5b, which interacts with other complement proteins to form the membrane attack complex (Guo and Ward, 2005). Induction of this larger fragment following Reynolds infection also indirectly suggests that larger amounts of C5a had been released compared to RN6390 infection. C5a is one of the most potent inflammatory peptides, with a broad spectrum of functions. In addition to its involvement in chemotaxis it stimulates the production of several cytokines and chemokines (Guo and Ward, 2005; van Beek et al., 2003). Therefore it likely contributes to the exacerbation of tissue damage during CNS infiammation. However, recent data have shown that C5a can be also protective for neurons against neurotoxic insults (Mukherjee and Pasinetti, 2001; van Beek et al., 2003). Similarly, some potential anti-inflammatory properties of C3a were also suggested in certain inflammatory conditions (Kildsgaard et al., 2000). To our knowledge this is the first study suggesting an association between brain abscess and activation of key components of the complement system, namely C3a and C5b. Therefore, controlling or interfering with C3a and/or C5a signaling might represent a potential target for therapeutic treatment modalities in various inflammatory disorders, and quite possibly a potential application in brain abscesses induced by S. aureus strain Reynolds.
During our studies, we noticed gross histopathological differences in brain abscesses of RN6390- and Reynolds-infected mice with obvious hemorrhagic changes visible only around the former. While minor local bleeding is typical in both RN6390-induced brain abscesses and following the injection of sterile beads as a consequence of trauma, there were very obvious hemorrhagic lesions that persisted until day 7 in Reynolds- inoculated tissues. Indeed, Hb levels, which correlate with the number of erythrocytes in tissue, were significantly elevated in the Reynolds-infected brains (Fig. 7B). Tissue extravasation of erythrocytes in several clinical Staphylococcal infections including skin and kidney infections is not uncommon (Galimberti et al., 2003; von Kockritz-Blickwede et al., 2008). This can be explained by the effects of infiammatory mediators such as chemokines and MMP-9 on endothelial barrier permeability (Engelhardt, 2008; Hoffman et al., 2009; Yamaguchi et al., 2007; Yamaguchi et al., 2008). Although we found such mediators were induced earlier in Reynolds-infected brain tissue, this may not explain the persistent extravasation of erythrocytes, since these mediators were also induced in RN6390- infected brains. It can also not be explained by blood–brain barrier disruption, since S. aureus injection leads to a chronic disruption of the barrier (Baldwin and Kielian, 2004; Kielian et al., 2001b). Recently, it has been demonstrated that erythrocytes via surface expression of complement receptor 1(CR1, CD35, C3b/C4b receptor) participate in host defense by binding complement opsonized foreign antigens and deliver them to liver and spleen for destruction and immune response (Lindorfer et al., 2001; Krych-Goldberg et al., 2005). Therefore, the accumulation of erythrocytes around the abscess may represent a compensatory mechanism to augment the function of phagocytic cells or to remove C3b complexes. In addition, locally produced C3a could have an effect on persistent erythrocyte recruitment. These possibilities need to be clarified in future studies with the use of C3 knockout animals to investigate the role of erythrocytes and complement in the abscess microenviroment. However, further studies need to be conducted to clarify the functional role of complement activation in brain abscess pathogenesis.
Although the roles played by immune cells infiltrating brain abscesses have been extensively characterized (Baldwin and Kielian, 2004; Kielian and Hickey, 2000), this is the first study that demonstrates the presence of mast cells with brain abscesses. However, mast cell infiltrates were present at equivalent numbers in both RN6390 and Reynolds lesions, suggesting that they are recruited during infection independent of the presence of bacterial capsule. Still the possibility exists that the techniques we used were not sensitive enough to detect potential differences among the groups and needs to be clarified in future studies. The presence of mast cells in the brain also correlated with prominent fibronectin levels at day 7 and these cells may participate in fibrotic wall formation based on previous data demonstrating that mast cell proteases are elevated at sites of excess scarring, and that they promote fibroblast proliferation and collagen synthesis (Asano-Kato et al., 2005; Gruber et al., 1997; Matsushima et al., 2006). In addition, tryptase, one of the most abundant mast cell proteins induces fibroblast proliferation through the newly synthesized products of cyclooxygenase 2 activity, prostaglandin (PG)J2 and its metabolite 15 day-PGJ2 (Frungieri et al., 2002; Levi-Schaffer and Piliponsky, 2003). However, further studies utilizing more sensitive techniques are needed to clarify the association of mast cell infiltration with the fibrotic abscess wall.
In conclusion, our study demonstrates that the capsular Reynolds strain of S. aureus replicates faster in the brain compared to the acapsular laboratory RN6390 strain, inducing earlier recruitment of immune cells including neutrophils, T cells, and erythrocytes, and immune response mediators such as CXCL2, MMP-9, C3a, and C5b. However, many of these changes were transient and only detected at day 1 following bacterial infection, revealing capsule-independent effects induced by the pathogen. Furthermore, the presence of mast cells is a novel finding in S. aureus induced brain abscesses. Although the expression of several immune mediators were not significantly different in brain abscesses elicited by RN6390 and Reynolds strains, we believe that it is more relevant to use clinical isolates of bacteria in animal models due to the fact that laboratory strain RN6390 causes more tissue destruction and subsequent mortality at the dose we used. In addition, use of capsular S. aureus strains in the brain abscess model may shed light on the role of complement sytem activation and erythrocytes in the immunologic responses to S. aureus. Further studies are needed to clarify the individual contributions of these immune responses during brain abscess pathogenesis.
Acknowledgments
The authors would like to thank Remzi T. Bilgin for excellent technical assistance. The authors also thank Dr. Tammy Kielian and Dr. David Irani for their critical review of the manuscript. This work was supported by University of Arkansas for Medical Sciences, Medical Research Endowment grant to N.E., T. Kielian Laboratory at UAMS, and the NINDS-supported Core Facility at UAMS (P30 NS047546). Support for the Digital and Confocal Microscopy Laboratory at the University of Arkansas for Medical Sciences was provided by NIH/INBRE P20 RR6460 and NIH/NCRR S10 RR19395.
References
- Abdelnour A, Arvidson S, Bremell T, Ryden C, Tarkowski A. The accessory gene regulator (agr) controls Staphylococcus aureus virulence in a murine arthritis model. Infect Immun. 1993;61:3879–3885. doi: 10.1128/iai.61.9.3879-3885.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer GL. Staphylococcus aureus: a well-armed pathogen. Clin Infect Dis. 1998;26:1179–1181. doi: 10.1086/520289. [DOI] [PubMed] [Google Scholar]
- Asano-Kato N, Fukagawa K, Okada N, Dogru M, Tsubota K, Fujishima H. Tryptase increases proliferative activity of human conjunctival fibroblasts through protease-activated receptor-2. Invest Ophthalmol Vis Sci. 2005;46:4622–4626. doi: 10.1167/iovs.05-0388. [DOI] [PubMed] [Google Scholar]
- Baldwin AC, Kielian T. Persistent immune activation associated with a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuroimmunol. 2004;151:24–32. doi: 10.1016/j.jneuroim.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Barnum SR, Ames RS, Maycox PR, Hadingham SJ, Meakin J, Harrison D, Parsons AA. Expression of the complement C3a and C5a receptors after permanent focal ischemia: an alternative interpretation. Glia. 2002;38:169–173. doi: 10.1002/glia.10069. [DOI] [PubMed] [Google Scholar]
- Beenken KE, Blevins JS, Smeltzer MS. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun. 2003;71:4206–4211. doi: 10.1128/IAI.71.7.4206-4211.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham DJ. Erythrocyte complement receptors. Crit Rev Immunol. 1995;15:133–154. doi: 10.1615/critrevimmunol.v15.i2.20. [DOI] [PubMed] [Google Scholar]
- Birmingham DJ, Hebert LA. CR1 and CR1-like: the primate immune adherence receptors. Immunol Rev. 2001;180:100–111. doi: 10.1034/j.1600-065x.2001.1800109.x. [DOI] [PubMed] [Google Scholar]
- Blevins JS, Beenken KE, Elasri MO, Hurlburt BK, Smeltzer MS. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect Immun. 2002;70:470–480. doi: 10.1128/IAI.70.2.470-480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173:4708–4714. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- Booth MC, Cheung AL, Hatter KL, Jett BD, Callegan MC, Gilmore MS. Staphylococcal accessory regulator (sar) in conjunction with agr contributes to Staphylococcus aureus virulence in endophthalmitis. Infect Immun. 1997;65:1550–1556. doi: 10.1128/iai.65.4.1550-1556.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson MJ, Reilly CR, Sutcliffe JG, Lo D. Mature microglia resemble immature antigen-presenting cells. Glia. 1998;22:72–85. doi: 10.1002/(sici)1098-1136(199801)22:1<72::aid-glia7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Cassat J, Dunman PM, Murphy E, Projan SJ, Beenken KE, Palm KJ, Yang SJ, Rice KC, Bayles KW, Smeltzer MS. Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiology. 2006;152:3075–3090. doi: 10.1099/mic.0.29033-0. [DOI] [PubMed] [Google Scholar]
- Chavakis T, Preissner KT, Herrmann M. The anti-inflammatory activities of Staphylococcus aureus. Trends Immunol. 2007;28:408–418. doi: 10.1016/j.it.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Cheung AL, Yeaman MR, Sullam PM, Witt MD, Bayer AS. Role of the sar locus of Staphylococcus aureus in induction of endocarditis in rabbits. Infect Immun. 1994;62:1719–1725. doi: 10.1128/iai.62.5.1719-1725.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claman HN. On scleroderma. Mast cells, endothelial cells, and fibroblasts. Jama. 1989;262:1206–1209. doi: 10.1001/jama.262.9.1206. [DOI] [PubMed] [Google Scholar]
- Davoust N, Jones J, Stahel PF, Ames RS, Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. 1999;26:201–211. doi: 10.1002/(sici)1098-1136(199905)26:3<201::aid-glia2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274:23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- Esen N, Tanga FY, DeLeo JA, Kielian T. Toll-like receptor 2 (TLR2) mediates astrocyte activation in response to the Gram-positive bacterium Staphylococcus aureus. J Neurochem. 2004;88:746–758. doi: 10.1046/j.1471-4159.2003.02202.x. [DOI] [PubMed] [Google Scholar]
- Flaris NA, Hickey WF. Development and characterization of an experimental model of brain abscess in the rat. Am J Pathol. 1992;141:1299–1307. [PMC free article] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Frungieri MB, Weidinger S, Meineke V, Kohn FM, Mayerhofer A. Proliferative action of mast-cell tryptase is mediated by PAR2, COX2, prostaglandins, and PPARgamma: possible relevance to human fibrotic disorders. Proc Natl Acad Sci U S A. 2002;99:15072–15077. doi: 10.1073/pnas.232422999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti R, Pietropaolo N, Galimberti G, Kowalczuk A. Adult purpura fulminans associated with staphylococcal infection and administration of colony-stimulating factors. Eur J Dermatol. 2003;13:95–97. [PubMed] [Google Scholar]
- Gasque P, Singhrao SK, Neal JW, Wang P, Sayah S, Fontaine M, Morgan BP. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol. 1998;160:3543–3554. [PubMed] [Google Scholar]
- Gruber BL, Kew RR, Jelaska A, Marchese MJ, Garlick J, Ren S, Schwartz LB, Korn JH. Human mast cells activate fibroblasts: tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J Immunol. 1997;158:2310–2317. [PubMed] [Google Scholar]
- Guo RF, Ward PA. Role of C5a in inflammatory responses. Annu Rev Immunol. 2005;23:821–852. doi: 10.1146/annurev.immunol.23.021704.115835. [DOI] [PubMed] [Google Scholar]
- Hochkeppel HK, Braun DG, Vischer W, Imm A, Sutter S, Staeubli U, Guggenheim R, Kaplan EL, Boutonnier A, Fournier JM. Serotyping and electron microscopy studies of Staphylococcus aureus clinical isolates with monoclonal antibodies to capsular polysaccharide types 5 and 8. J Clin Microbiol. 1987;25:526–530. doi: 10.1128/jcm.25.3.526-530.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman WH, Stamatovic SM, Andjelkovic AV. Inflammatory mediators and blood brain barrier disruption in fatal brain edema of diabetic ketoacidosis. Brain Res. 2009;1254:138–148. doi: 10.1016/j.brainres.2008.11.100. [DOI] [PubMed] [Google Scholar]
- Karakawa WW, Sutton A, Schneerson R, Karpas A, Vann WF. Capsular antibodies induce type-specific phagocytosis of capsulated Staphylococcus aureus by human polymorphonuclear leukocytes. Infect Immun. 1988;56:1090–1095. doi: 10.1128/iai.56.5.1090-1095.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanami O, Ferrans VJ, Fulmer JD, Crystal RG. Ultrastructure of pulmonary mast cells in patients with fibrotic lung disorders. Lab Invest. 1979;40:717–734. [PubMed] [Google Scholar]
- Kielian T. Immunopathogenesis of brain abscess. J Neuroinflammation. 2004;1:16. doi: 10.1186/1742-2094-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Hickey WF. Proinflammatory cytokine, chemokine, and cellular adhesion molecule expression during the acute phase of experimental brain abscess development. Am J Pathol. 2000;157:647–658. doi: 10.1016/S0002-9440(10)64575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Cheung A, Hickey WF. Diminished virulence of an alphatoxin mutant of Staphylococcus aureus in experimental brain abscesses. Infect Immun. 2001a;69:6902–6911. doi: 10.1128/IAI.69.11.6902-6911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Barry B, Hickey WF. CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J Immunol. 2001b;166:4634–4643. doi: 10.4049/jimmunol.166.7.4634. [DOI] [PubMed] [Google Scholar]
- Kielian T, Bearden ED, Baldwin AC, Esen N. IL-1 and TNF-alpha play a pivotal role in the host immune response in a mouse model of Staphylococcus aureus-induced experimental brain abscess. J Neuropathol Exp Neurol. 2004a;63:381–396. doi: 10.1093/jnen/63.4.381. [DOI] [PubMed] [Google Scholar]
- Kielian T, McMahon M, Bearden ED, Baldwin AC, Drew PD, Esen N. S. aureus-dependent microglial activation is selectively attenuated by the cyclopentenone prostaglandin 15-deoxy-Delta12, 14- prostaglandin J2 (15d-PGJ2) J Neurochem. 2004b;90:1163–1172. doi: 10.1111/j.1471-4159.2004.02579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Haney A, Mayes PM, Garg S, Esen N. Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun. 2005;73:7428–7435. doi: 10.1128/IAI.73.11.7428-7435.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Phulwani NK, Esen N, Syed MM, Haney AC, McCastlain K, Johnson J. MyD88-dependent signals are essential for the host immune response in experimental brain abscess. J Immunol. 2007;178:4528–4537. doi: 10.4049/jimmunol.178.7.4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielian T, Syed MM, Liu S, Phulwani NK, Phillips N, Wagoner G, Drew PD, Esen N. The synthetic peroxisome proliferator-activated receptor-gamma agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. J Immunol. 2008;180:5004–5016. doi: 10.4049/jimmunol.180.7.5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Immunol. 2000;165:5406–5409. doi: 10.4049/jimmunol.165.10.5406. [DOI] [PubMed] [Google Scholar]
- Kondo S, Kagami S, Kido H, Strutz F, Muller GA, Kuroda Y. Role of mast cell tryptase in renal interstitial fibrosis. J Am Soc Nephrol. 2001;12:1668–1676. doi: 10.1681/ASN.V1281668. [DOI] [PubMed] [Google Scholar]
- Krych-Goldberg M, Barlow Paul N, Mallin Rosie L, Atkinson JP. C3b/C4b binding site of complement receptor type 1 (CR1, CD35) In: Lambris JD, Morikis D, editors. Structural Biology of Complement System. CRC Press, Boca Raton; Florida, USA: 2005. pp. 179–232. [Google Scholar]
- Leroy EC, Smith EA, Kahaleh MB, Trojanowska M, Silver RM. A strategy for determining the pathogenesis of systemic sclerosis. Is transforming growth factor beta the answer? Arthritis Rheum. 1989;32:817–825. [PubMed] [Google Scholar]
- Levi-Schaffer F, Piliponsky AM. Tryptase, a novel link between allergic inflammation and fibrosis. Trends Immunol. 2003;24:158–161. doi: 10.1016/s1471-4906(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Lindorfer MA, Hahn CS, Foley PL, Taylor RP. Heteropolymer-mediated clearance of immune complexes via erythrocyte CR1: mechanisms and applications. Immunol Rev. 2001;183:10–24. doi: 10.1034/j.1600-065x.2001.1830102.x. [DOI] [PubMed] [Google Scholar]
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- Mathisen GE, Johnson JP. Brain abscess. Clin Infect Dis. 1997;25:763–779. doi: 10.1086/515541. quiz 780–761. [DOI] [PubMed] [Google Scholar]
- Matsushima R, Takahashi A, Nakaya Y, Maezawa H, Miki M, Nakamura Y, Ohgushi F, Yasuoka S. Human airway trypsin-like protease stimulates human bronchial fibroblast proliferation in a protease-activated receptor-2-dependent pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290:L385–395. doi: 10.1152/ajplung.00098.2005. [DOI] [PubMed] [Google Scholar]
- Montano RF, Morrison SL. A colorimetric-enzymatic microassay for the quantitation of antibody-dependent complement activation. J Immunol Methods. 1999;222:73–82. doi: 10.1016/s0022-1759(98)00181-1. [DOI] [PubMed] [Google Scholar]
- Moser M, Murphy KM. Dendritic cell regulation of TH1–TH2 development. Nat Immunol. 2000;1:199–205. doi: 10.1038/79734. [DOI] [PubMed] [Google Scholar]
- Mukherjee P, Pasinetti GM. Complement anaphylatoxin C5a neuroprotects through mitogen-activated protein kinase-dependent inhibition of caspase 3. J Neurochem. 2001;77:43–49. doi: 10.1046/j.1471-4159.2001.00167.x. [DOI] [PubMed] [Google Scholar]
- Murray PJ. Understanding and exploiting the endogenous interleukin-10/STAT3-mediated anti-inflammatory response. Curr Opin Pharmacol. 2006;6:379–386. doi: 10.1016/j.coph.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Nilsson IM, Bremell T, Ryden C, Cheung AL, Tarkowski A. Role of the staphylococcal accessory gene regulator (sar) in septic arthritis. Infect Immun. 1996;64:4438–4443. doi: 10.1128/iai.64.11.4438-4443.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson IM, Lee JC, Bremell T, Ryden C, Tarkowski A. The role of staphylococcal polysaccharide microcapsule expression in septicemia and septic arthritis. Infect Immun. 1997;65:4216–4221. doi: 10.1128/iai.65.10.4216-4221.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev. 2004;17:218–234. doi: 10.1128/CMR.17.1.218-234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Projan SJ, Novick RP. The molecular basis of pathogenicity. In: Crossley GL, Archer KB, editors. The Staphyloccci in Human Disease. Churchill Livingstone; New York, NY: 1997. pp. 55–81. [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T. TNF-alpha expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- Sandoval A, Ai R, Ostresh JM, Ogata RT. Distal recognition site for classical pathway convertase located in the C345C/netrin module of complement component C5. J Immunol. 2000;165:1066–1073. doi: 10.4049/jimmunol.165.2.1066. [DOI] [PubMed] [Google Scholar]
- Sellebjerg F, Sorensen TL. Chemokines and matrix metalloproteinase-9 in leukocyte recruitment to the central nervous system. Brain Res Bull. 2003;61:347–355. doi: 10.1016/s0361-9230(03)00097-2. [DOI] [PubMed] [Google Scholar]
- Sompolinsky D, Samra Z, Karakawa WW, Vann WF, Schneerson R, Malik Z. Encapsulation and capsular types in isolates of Staphylococcus aureus from different sources and relationship to phage types. J Clin Microbiol. 1985;22:828–834. doi: 10.1128/jcm.22.5.828-834.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza LF, Jardim FR, Sauter IP, Souza MM, Barreto F, Margis R, Bernard EA. Lipoteichoic acid from Staphylococcus aureus increases matrix metalloproteinase 9 expression in RAW 264.7 macrophages: modulation by A2A and A2B adenosine receptors. Mol Immunol. 2008 doi: 10.1016/j.molimm.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Stenzel W, Soltek S, Miletic H, Hermann MM, Korner H, Sedgwick JD, Schluter D, Deckert M. An essential role for tumor necrosis factor in the formation of experimental murine Staphylococcus aureus-induced brain abscess and clearance. J Neuropathol Exp Neurol. 2005;64:27–36. doi: 10.1093/jnen/64.1.27. [DOI] [PubMed] [Google Scholar]
- Stenzel W, Soltek S, Sanchez-Ruiz M, Akira S, Miletic H, Schluter D, Deckert M. Both TLR2 and TLR4 are required for the effective immune response in Staphylococcus aureus-induced experimental murine brain abscess. Am J Pathol. 2008;172:132–145. doi: 10.2353/ajpath.2008.070567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura S, Deguchi M, Ueda M, Yoshida N, Kato H, Yoshikawa T, Sugino S, Kondo M. C3b receptor (CR1) on erythrocytes in various diseases. Immunol Lett. 1984;7:325–328. doi: 10.1016/0165-2478(84)90089-0. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton P, Pinteaux E, Allan SM, Rothwell NJ. Matrix metalloproteinase-9 and urokinase plasminogen activator mediate interleukin-1-induced neurotoxicity. Mol Cell Neurosci. 2008;37:135–142. doi: 10.1016/j.mcn.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Townsend GC, Scheld WM. Infections of the central nervous system. Adv Intern Med. 1998;43:403–447. [PubMed] [Google Scholar]
- Trabucchi E, Radaelli E, Marazzi M, Foschi D, Musazzi M, Veronesi AM, Montorsi W. The role of mast cells in wound healing. Int J Tissue React. 1988;10:367–372. [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Tzianabos AO, Wang JY, Lee JC. Structural rationale for the modulation of abscess formation by Staphylococcus aureus capsular polysaccharides. Proc Natl Acad Sci U S A. 2001;98:9365–9370. doi: 10.1073/pnas.161175598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Beek J, Bernaudin M, Petit E, Gasque P, Nouvelot A, MacKenzie ET, Fontaine M. Expression of receptors for complement anaphylatoxins C3a and C5a following permanent focal cerebral ischemia in the mouse. Exp Neurol. 2000;161:373–382. doi: 10.1006/exnr.1999.7273. [DOI] [PubMed] [Google Scholar]
- van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- von Kockritz-Blickwede M, Rohde M, Oehmcke S, Miller LS, Cheung AL, Herwald H, Foster S, Medina E. Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am J Pathol. 2008;173:1657–1668. doi: 10.2353/ajpath.2008.080337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YY, Myhre AE, Pettersen SJ, Dahle MK, Foster SJ, Thiemermann C, Bjornland K, Aasen AO, Wang JE. Peptidoglycan of Staphylococcus aureus induces enhanced levels of matrix metalloproteinase-9 in human blood originating from neutrophils. Shock. 2005;24:214–218. doi: 10.1097/01.shk.0000174935.13786.6c. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Jadhav V, Obenaus A, Colohan A, Zhang JH. Matrix metalloproteinase inhibition attenuates brain edema in an in vivo model of surgically-induced brain injury. Neurosurgery. 2007;61:1067–1075. doi: 10.1227/01.neu.0000303203.07866.18. discussion 1075–1066. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Jadhav V, Obenaus A, Colohan A, Zhang JH. Matrix metalloproteinase inhibition attenuates brain edema in an in vivo model of surgically-induced brain injury. Neurosurgery. 2008 doi: 10.1227/01.neu.0000303203.07866.18. [DOI] [PubMed] [Google Scholar]