


Abstract
Some topoisomerase inhibitors trap covalent topoisomerase–DNA complexes as topoisomerase–drug–DNA ternary complexes. Ternary complex formation results in inhibition of DNA replication and generation of permanent double-strand breaks. Recent demonstrations of the stimulation of covalent topoisomerase–DNA complex formation by DNA lesions suggest that DNA damage may act as an endogenous topoisomerase poison. We have investigated the effects of abasic (AP) sites on topoisomerase IV (Topo IV). AP sites can stimulate the formation of covalent Topo IV–DNA complexes when they are located either within the 4 base overhang generated by DNA scission or immediately 5′ to the point of scission (the –1 position). Thus, the AP site acts as a position-specific, endogenous topoisomerase poison. Both EDTA and salt can reverse covalent Topo IV–DNA complexes induced by AP sites located within the 4 base overhang. Interestingly, an AP site at the –1 position inhibits EDTA-mediated reversal of formation of the covalent Topo IV–DNA complex. Furthermore, we find that, unlike quinolone-induced covalent Topo IV–DNA complexes, AP site-induced covalent Topo IV–DNA complexes do not inhibit the helicase activities of the DnaB and T7 Gene 4 proteins. These results suggest that the AP site-induced poisoning of Topo IV does not arrest replication fork progression.
INTRODUCTION
Topoisomerases are responsible for altering the linking number of DNA (1). These essential enzymes break and rejoin DNA strands by forming a covalent linkage between the enzyme and the DNA at the site of DNA scission. This covalent topoisomerase–DNA complex is normally a fleeting catalytic intermediate. The steady-state level of the covalent topoisomerase–DNA complex depends on the cleavage–religation equilibrium. If the equilibrium is shifted to either stimulate strand cleavage or inhibit religation, the covalent topoisomerase–DNA complex can persist, as if the topoisomerase were trapped on the DNA.
DNA gyrase was recognized, shortly after its discovery (2), as the cellular target of quinolone antibacterial drugs (3,4). Kreuzer and Cozzarelli (5) have demonstrated that quinolones block DNA replication not by depriving the cell of DNA gyrase but by converting DNA gyrase into a poison. These topoisomerase inhibitors trap a covalent topoisomerase–DNA complex as a topoisomerase–drug–DNA ternary complex, which leads to inhibition of DNA replication, generation of permanent double-strand breaks and subsequent cell death (6–9). Anticancer drugs that target human topoisomerases also convert their targets into poisons in a similar manner (10). Based on their unique mode of action, these topoisomerase inhibitors are often referred to as ‘topoisomerase poisons’ (6–8).
Because these topoisomerase poisons act at the enzyme–DNA interface to affect the local structure of the DNA and the catalytic activity of the topoisomerase, it seems possible that DNA damage may affect the topoisomerase–DNA interaction. As a result, the level of covalent topoisomerase–DNA complex increases. In fact, recent studies in eukaryotic systems have demonstrated that several commonly formed DNA lesions stimulate topoisomerase-catalyzed cleavage of DNA. For instance, topoisomerase I (Topo I) is shown to be responsible for the generation of single-strand breaks and the formation of covalent protein–DNA complexes after UV radiation (11,12). Abasic (AP), apurinic and apyrimidinic, sites stimulate eukaryotic Topo I- and topoisomerase II (Topo II)-catalyzed cleavage (13–16). AP sites induce local structural alterations in the duplex DNA, which contribute to their effect on the catalytic activity of Topo II (17). Thus, it is proposed that some DNA lesions may act as position-specific endogenous topoisomerase poisons.
Here we have examined the effect of AP sites on topoisomerase IV (Topo IV), a prokaryotic type II topoisomerase. We found that, as is the case with eukaryotic Topo II, AP sites could stimulate the formation of covalent Topo IV–DNA complexes when these lesions were located within the 4 base overhang generated by DNA scission (+1, +2, +3 and +4 positions). Interestingly, an AP site located immediately 5′ to the point of scission (the –1 position) also stimulated covalent Topo IV–DNA complex formation. In addition, EDTA-mediated reversal of formation of the covalent Topo IV–DNA complex was inhibited when the AP site was located at the –1 position. Thus, the AP site at the –1 position seemed to affect prokaryotic and eukaryotic type II topoisomerases in a distinct manner. In contrast, AP site-induced covalent Topo IV–DNA complexes were reversed by either EDTA or salt when AP sites were located within the 4 base overhang generated by DNA scission. Furthermore, we found that AP site-induced covalent Topo IV–DNA complexes could not inhibit the functional activities of DnaB and T7 Gene 4 helicases. These results suggest that AP site-induced covalent Topo IV–DNA complexes could not block the progression of replication forks.
MATERIALS AND METHODS
DNAs
The construction of a recombinant M13 containing a defined Topo IV-binding site (M13-T440) and the preparation of the single-stranded circular DNA of M13-T440 were as described previously (18).
Partial duplex DNAs were prepared according to Shea and Hiasa (18). Briefly, a 63 nt oligo T4C (5′-CCGGCTCGTATCTAGACTCCTAAAAATCCGGGGTATACCCCGGATTTTTAGGAGTGTGTCGCG-3′) was synthesized (IDT, Coralville, IA) and hybridized to the M13-T440 single-stranded DNA to prepare a primer–template. The oligo in the primer–template was then 3′-end-labeled by incorporation of one residue of [32P]dAMP (Amersham Pharmacia Biotech, Piscataway, NJ) with Klenow enzyme (Roche Molecular Biochemicals, Indianapolis, IN). A set of oligos with the same DNA sequence as T4C was also synthesized (IDT). These oligos contained an AP site at various positions (underlined). AP sites were introduced into these oligos by replacing each nucleotide between or immediately adjacent to the two points of scission (underlined) with a dSpacer (Glen Research, Sterling, VA). The dSpacer is 5′-O-dimethoxytrityl-1′,2′-dideoxyribose-3′-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramide and the incorporation of a dSpacer into an oligo results in a stable AP site in the oligo. These oligos were used to prepare the partial duplex DNAs containing an AP site at various positions.
Proteins
Purified ParC and ParE proteins were mixed on ice to form active Topo IV (19). Purified DnaB and T7 Gene 4 proteins were generous gifts of Kenneth Marians (Memorial Sloan-Kettering Cancer Center) and Smita Patel (Robert Wood Johnson Medical School), respectively.
Topo IV–catalyzed cleavage assay
Reaction mixtures (12.5 µl) containing 40 mM HEPES–KOH (pH 7.6), 10 mM magnesium acetate, 10 mM dithiothreitol, 50 µg/ml bovine serum albumin, 2 mM ATP, 10 fmol (as molecule) T440 DNA, 300 fmol (as tetramer) Topo IV and 100 µM norfloxacin, when indicated, were incubated at 37°C for 10 min. SDS was added to a final concentration of 1% and the reaction mixtures were incubated at 37°C for 5 min. EDTA and proteinase K were then added to 25 mM and 100 µg/ml, respectively, and incubation was continued for an additional 15 min. The DNA products were purified by extraction of the reaction mixtures with phenol/chloroform (1:1 v/v) and ethanol precipitation, heat denatured at 95°C for 5 min and then analyzed by electrophoresis through 12% polyacrylamide (19:1 acrylamide:bisacrylamide) gels (140 × 160 × 1.2 mm) at 14 V/cm for 2 h using 50 mM Tris–borate (pH 8.3) and 1 mM EDTA as the electrophoresis buffer (TBE buffer). Gels were dried under vacuum onto DE81 paper (Whatman, Clifton, NJ) and autoradiographed with Hyperfilm MP film (Amersham Pharmacia Biotech). Quantification was performed using a Molecular Dynamics STORM 840 PhosphorImager (Amersham Pharmacia Biotech).
EDTA-mediated reversal assay
Reaction mixtures (12.5 µl) were assembled as described in the previous section. Topo IV was bound to the partial duplex DNA containing a single AP site during the first stage of incubation at 37°C for 10 min. Reactions were terminated by adding either EDTA or SDS to 25 mM and 1%, respectively, and incubating at 37°C for 5 min. Then, either SDS or EDTA, together with proteinase K, was added to each reaction mixture to 1%, 25 mM and 100 µg/ml, respectively. Incubation was further continued at 37°C for an additional 15 min. The DNA products were purified and analyzed as described in the previous section.
Kinetics of EDTA-mediated reversal of the AP site-induced covalent Topo IV–DNA complex were measured as follows. Standard reactions containing Topo IV and T440 (–1) DNA were increased in size 6-fold (75 µl). After 10 min incubation at 37°C, EDTA was added to the reaction mixtures to a final concentration of 25 mM and incubation was continued at 37°C. Portions (12.5 µl each) of the reaction mixtures were withdrawn at the indicated times and SDS was added to 1% to terminate the reactions. The reaction mixtures were incubated at 37°C for an additional 5 min, proteinase K was added to a final concentration of 100 µg/ml and incubation was further continued for 15 min. The DNA products were purified and analyzed as described in the previous section.
Salt-mediated reversal assay
Reaction mixtures (12.5 µl) were assembled as described in the previous section and Topo IV was bound to the partial duplex DNA containing an AP site during the first stage of incubation at 37°C for 10 min. Then the reaction mixtures were incubated either in the absence or presence of 0.5 M NaCl at 37°C for 5 min. Reactions were terminated by adding SDS to a final concentration of 1% and incubating at 37°C for 5 min. Then EDTA and proteinase K were added to final concentrations of 25 mM and 100 µg/ml, respectively, and incubation was further continued at 37°C for an additional 15 min. The DNA products were purified and then analyzed as described in the previous section.
Helicase assays
The strand displacement assay for the DnaB and T7 Gene 4 proteins was performed as previously described (18). Briefly, another set of oligos containing an AP site at various positions and a 22 nt 3′-non-hybridized tail was synthesized (IDT). These oligos were 5′-end-labeled and then hybridized to the M13-T440 single-stranded DNA to prepare the partial duplex DNAs containing an AP site and a 3′-non-hybridized tail.
Standard reaction mixtures (12.5 µl) contained 40 mM HEPES–KOH (pH 7.6 at 23°C), 10 mM magnesium acetate, 3.5 mM ATP, 10 mM dithiothreitol, 50 µg/ml bovine serum albumin and 10 fmol (as molecule) DNA substrate. Three hundred femtomoles (as tetramer) of Topo IV was first bound to the DNA in a first stage incubation of 5 min at 37°C. Either DnaB (250 fmol as hexamer) or T7 Gene 4 protein (250 fmol as hexamer) was then added and the reaction mixtures were incubated during the second stage for 10 min at 37°C. Reactions were terminated by adding EDTA to 25 mM, followed by the addition of a quarter volume of a dye mixture containing 15% glycerol, 2% sarkosyl, 0.05% xylene cyanol, 0.05% bromophenol blue and 50 mM EDTA. Aliquots were analyzed by electrophoresis through either vertical 1% agarose gels (140 × 120 × 3 mm) at 5 V/cm for 2 h in a running buffer of 50 mM Tris–HCl (pH 7.9 at 23°C), 40 mM sodium acetate, and 1 mM EDTA (TAE buffer) or 9% polyacrylamide (19:1 acrylamide:bisacrylamide) gels (140 × 160 × 1.2 mm) at 14 V/cm for 2 h using TBE buffer. Gels were dried under vacuum onto DE81 paper (Whatman) and autoradiographed with Hyperfilm MP film (Amersham Pharmacia Biotech). Strand displacement was quantified by scanning images with a Molecular Dynamics STORM 840 PhosphorImager (Amersham Pharmacia Biotech).
RESULTS
DNA substrate
The defined Topo IV-binding site is designed as perfect palindromes with two points of DNA scission offset 2 bp from the center (19). The partial duplex DNAs used in this study contained a 40 bp defined Topo IV-binding site [5′-ACTCCTAAAAATCCGGGG↓TATA(↑)CCCCGGATTTTTAGGAGT-3′ (19)] in the middle of the 64 bp duplex region (Fig. 1). These partial duplex DNAs were prepared by annealing oligos containing a single AP site at various positions. Thus, AP sites were present on only one (top) strand (Fig. 1) of the duplex. However, because the DNA sequence of the defined Topo IV-binding site is palindromic (19), AP sites on the other (bottom) strand (Fig. 1) would affect Topo IV in the same manner as those at equivalent positions on the opposite (top) strand (Fig. 1). Note that the DNA sequence of the defined Topo IV-binding site is palindromic, but not symmetrical. For instance, the G residue at the –1 position on the top strand is not equivalent to the C residue at the +5 position, but is equivalent to the G residue complementary to the C residue at the +5 position. As shown in Figure 1, positions where AP sites were introduced on the annealed (top) strand were numbered, from –2 to +6, relative to the point of scission. Based on their positions, AP sites were referred to as AP(–2), AP(–1), AP(+1), AP(+2), AP(+3), AP(+4), AP(+5) and AP(+6). Accordingly, partial duplex DNAs containing an AP site were also referred to as T440(–2), T440(–1), T440(+1), T440(+2) T440(+3), T440(+5) and T440(+6). The partial duplex DNA without an AP site is referred to as T440(0) in this study.
Figure 1.

The defined Topo IV-binding site. Schematic presentation of the defined Topo IV-binding site (19) in the T440 partial duplex DNA. The DNA sequence around two points of scission (arrows) is shown. Nucleotides (large letters) replaced with an AP are numbered by their position relative to the point of scission. The closed circle represents the 32P-labeled nucleotide at the 3′-end of the annealed oligo. Details are described in the text.
AP sites stimulate covalent Topo IV–DNA complex formation in a position-specific manner
Osheroff and colleagues (14–16) have recently demonstrated that AP sites can stimulate DNA cleavage catalyzed by human and Drosophilia melanogaster Topo II proteins. Stimulation of eukaryotic Topo II-catalyzed cleavage by an AP site is observed only when AP sites are located within the 4 base overhang generated by DNA scission. Thus, the AP site acts as a position-specific endogenous topoisomerase poison of eukaryotic Topo II. It has been suggested that the structural alterations induced by the AP site affect the Topo II–DNA interaction and trap the Topo II as a covalent Topo II–DNA complex (17). Here we have examined whether the AP site could act as an endogenous topoisomerase poison of Topo IV, a prokaryotic type II topoisomerase.
The standard DNA cleavage assay was performed to assess the effect of AP sites on Topo IV. The partial duplex DNAs containing a single AP site at various positions were incubated with Topo IV at 37°C for 10 min and reactions were terminated by addition of SDS (Fig. 2). As is the case with eukaryotic Topo II proteins, AP sites located within the 4 base overhang generated by DNA scission stimulated covalent Topo IV–DNA complex formation (Fig. 2A). Interestingly, AP(–1), the AP site located immediately 5′ to the point of scission also stimulated Topo IV-catalyzed cleavage (Fig. 2B). An AP site at the equivalent position does not stimulate Topo II-catalyzed cleavage of DNA (14–16). AP(–2), AP(+5) and AP(+6) did not stimulate Topo IV-catalyzed cleavage. These results demonstrate that AP sites located between positions –1 and +4 affected the catalytic activity of Topo IV and stimulated formation of covalent Topo IV–DNA complexes. Thus, the AP site could act as a position-specific endogenous topoisomerase poison of Topo IV. A cleavage assay using a short duplex DNA as substrate confirmed that AP site-stimulated, Topo IV-catalyzed cleavage created a double-strand break, not a single-strand break (data not shown). In addition, no DNA cleavage was observed when a catalytically inactive Topo IV (20,21) was incubated with any of the partial duplex DNAs (data not show). Thus, observed AP site-stimulated DNA cleavage was catalyzed by Topo IV.
Figure 2.
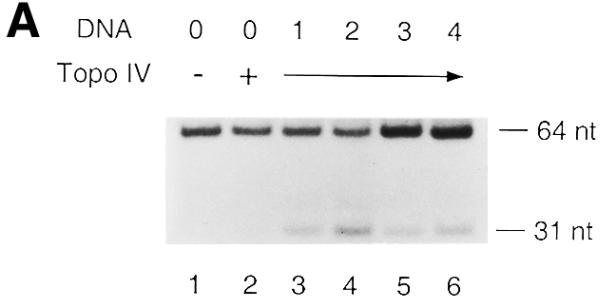
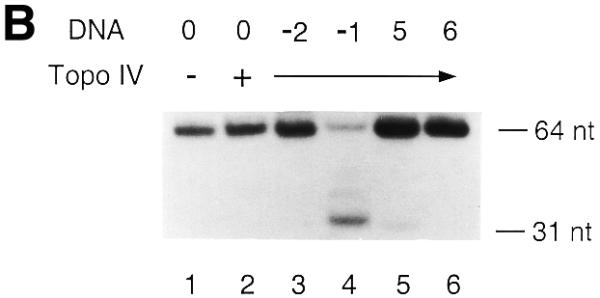
AP sites can stimulate the formation of covalent Topo IV–DNA complexes. The DNA cleavage assay using Topo IV (24 nM) and the T440 DNA containing an AP site as DNA substrate (0.8 nM) was performed and the DNA products were analyzed as described in Materials and Methods. AP sites were located within the 4 base overhang generated by DNA scission (A) and immediately outside the two points of scission (B). Experiments were repeated three times and representative results are shown. The results of the quantification are shown in Table 1. Lane 0, T440(0); lane –2, T440(–2); lane –1, T440(–1); lane 1, T440(+1); lane 2, T440(+2); lane 3, T440(+3); lane 4, T440(+4); lane 5, T440(+5); lane 6, T440(+6). Size markers were 5′-end-labeled 64 and 31 nt oligos.
The extent of AP site-stimulated Topo IV-catalyzed cleavage varied depending on the position of the AP site (Fig. 2). Both the distance from the point of scission and the type of AP site, either apurinic or apyrimidinic, seemed to determine the extent of stimulation (Table 1). However, the AP site could affect not only the catalytic activity of Topo IV but also the affinity of Topo IV for the defined Topo IV-binding site. Thus, the extent of AP site-stimulated DNA cleavage could be affected by the different affinities of Topo IV for the partial duplex DNA containing an AP site. We examined this possibility by measuring norfloxacin-stimulated Topo IV-catalyzed cleavage of T440 DNA (Table 1). All T440 DNAs were cleaved to a similar extent when norfloxacin was present, except that a slight reduction in cleavage was observed when either T440(+1) or T440(+3) DNA was used as the substrate. The differences in AP site-stimulated Topo IV-catalyzed cleavage were much greater than those in norfloxacin-stimulated Topo IV-catalyzed cleavage (Table 1). These results show that AP sites do not affect norfloxacin-stimulated covalent Topo IV–DNA complex formation at the defined Topo IV-binding site. Thus, it seemed reasonable to assume that AP sites did not greatly affect the affinity of Topo IV for the defined Topo IV-binding site. It was likely that the various extents of Topo IV-catalyzed cleavage stimulated by different AP sites were due to distinct efficiencies of topoisomerase poisoning by each AP site.
Table 1. Stimulation of Topo IV–catalyzed cleavage of T440 partial duplex DNAs containing an AP site by either the AP site or norfloxacin.
| DNA |
–Norfloxacin
(%)a |
+Norfloxacin
(%)b |
–Norfloxacin/+norfloxacin
(%)c |
| T440(0) | 6.5 | 78.3 | 8.3 |
| T440(–2) | 4.8 | 70.6 | 6.8 |
| T440(–1) | 56.2 | 74.5 | 75.4 |
| T440(+1) | 26.9 | 64.1 | 42.0 |
| T440(+2) | 40.2 | 86.3 | 46.6 |
| T440(+3) | 18.4 | 59.2 | 31.1 |
| T440(+4) | 19.6 | 77.2 | 25.3 |
| T440(+5) | 7.4 | 76.5 | 9.7 |
| T440(+6) | 3.0 | 82.4 | 3.6 |
Experiments were repeated three times.
aError range was ±2.8%.
bError range was ±5.9 %.
cAverage values were used for the calculation.
AP(–1) inhibits EDTA-mediated disassembly of the covalent Topo IV–DNA complex
A unique characteristic of topoisomerase–drug–DNA ternary complexes is that these complexes are completely reversible (5). For instance, addition of EDTA results in religation of the DNA strands and reversal of formation of topoisomerase–drug–DNA ternary complexes. The exact mechanism of EDTA-mediated reversal of ternary complex formation is not fully understood but it has been proposed that chelating Mg2+ leads to religation of the DNA strands and disassembly of the ternary complex (5). We examined the reversibility of formation of AP site-induced covalent Topo IV–DNA complexes.
Topo IV was incubated with T440 DNA substrate containing an AP site and incubation was terminated by adding either EDTA or SDS. After an additional 5 min incubation, Topo IV was denatured and removed and the DNA products were analyzed (Fig. 3). The Topo IV–norfloxacin–T440(0) ternary complex was used as a control. As expected, formation of the Topo IV–norfloxacin–T440(0) ternary complex was completely reversed under these conditions. The 5 min incubation in the presence of EDTA also reversed formation of Topo IV–T440(+1), Topo IV–T440(+2) (Fig. 3), Topo IV–T440(+3) and Topo IV–T440(+4) (data not shown) complexes. However, the majority of Topo IV–T440(–1) complexes remained as the covalent Topo IV–DNA complex (Fig. 3). These results demonstrated that formation of all the AP site-induced covalent Topo IV–DNA complexes, except that induced by AP(–1), were reversible.
Figure 3.

Reversibility of formation of the AP site-induced covalent Topo IV–DNA complexes. An EDTA-mediated reversal assay was performed as described in Materials and Methods to assess reversibility of AP(–1)-, AP(+1)- and AP(+2)-induced covalent Topo IV–DNA complex formation. The Topo IV–norfloxacin–T440(0) ternary complex was used as a control. Reactions were terminated by addition of either SDS (open bars) or EDTA (closed bars). Experiments were repeated twice. The average values are shown and the error range was 4%. Abbreviations are as in the legend to Figure 2.
We measured the time course of EDTA-mediated reversal of formation of the Topo IV–T440(–1) complex. As shown in Figure 4, disassembly of Topo IV–T440(–1) complexes was very slow and it took 15 min to disassemble half of the complexes. In contrast, disassembly of the Topo IV–norfloxacin–T440(0), Topo IV–T440(+1) and Topo IV–T440(+2) complexes was complete within 2 min (Fig. 4B). Thus, AP(–1) inhibited EDTA-mediated disassembly of the covalent Topo IV–DNA complex. One possible explanation is that AP(–1), but not AP sites within the 4 base overhang, could stabilize Mg2+ in the covalent Topo IV–DNA complex and prevent Mg2+ from being chelated by EDTA.
Figure 4.
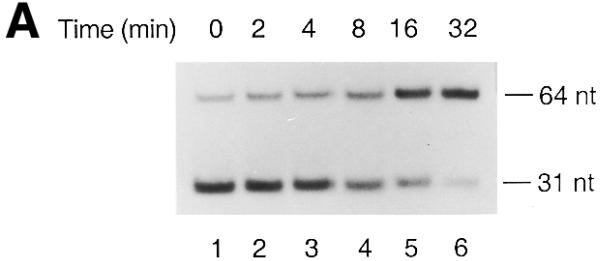

AP(–1) inhibits EDTA-mediated reversal of formation of the covalent Topo IV–DNA complex. The time course of EDTA-mediated reversal of Topo IV–T440(–1) complex formation was measured as described in Materials and Methods. Size markers are as in the legend to Figure 2. (A) The DNA products were analyzed by PAGE. (B) The relative amounts of cleaved DNA (normalized to the amounts of cleaved DNA at 0 min) were measured at the times indicated. Open circle, Topo IV–T440(–1); square, Topo IV–T440(+1); triangle, Topo IV–T440(+2); closed circle, Topo IV–norfloxacin–T440(0).
AP site-induced covalent Topo IV–DNA complexes are sensitive to salt
Next, we assessed salt sensitivity of AP site-induced covalent Topo IV–DNA complexes. After the binding reaction, the Topo IV–T440(–1), Topo IV–T440(+1), Topo IV–T440(+2) and Topo IV–norfloxacin–T440(0) complexes were incubated either in the absence or presence of 0.5 M NaCl at 37°C for 5 min. All the binary and ternary complexes were sensitive to salt and disassembled under these conditions (Fig. 5). The Topo IV–T440(–1) complex was more resistant to salt-mediated disassembly than were the other complexes. However, salt-mediated disassembly of Topo IV–T440(–1) complexes (Fig. 5) was more effective than EDTA-mediated disassembly (Figs 3 and 4).
Figure 5.

AP site-induced covalent Topo IV–DNA complexes are sensitive to salt. Salt sensitivity of AP site-induced covalent Topo IV–DNA complexes was assessed as described in Materials and Methods. The relative amounts of cleaved DNA after a 5 min incubation in the absence (open bars) or presence (closed bars) of 0.5 M NaCl were measured. T440(–1), T440(+1) and T440(+2) were used as substrate and the Topo IV–norfloxacin–T440(0) ternary complex was used as a control. Experiments were repeated twice and the average values are shown. Error range was 6%. Abbreviations are as in the legend to Figure 2.
AP site-induced Topo IV–DNA complexes cannot inhibit the activities of replicative helicases
The poisoning of topoisomerases by topoisomerase inhibitors results in inhibition of DNA replication (21). Recent studies (12–15), as well as our observations described in previous sections, suggested that an AP site could act as an endogenous topoisomerase poison. If both exogenous and endogenous topoisomerase poisons have the same biological consequences, DNA damage-induced covalent topoisomerase–DNA complexes would also lead to inhibition of DNA replication. We have demonstrated that a Topo IV–quinolone–DNA ternary complex inhibits replicative helicase activity, which leads to the arrest of replication forks (18). To examine if an AP site-induced covalent Topo IV–DNA complex could arrest replication fork progression, we assessed the effect of AP site-induced covalent Topo IV–DNA complexes on DnaB and T7 Gene 4 helicases.
A strand displacement assay for DnaB was performed (Fig. 6A and B). T440(0), T440(–1) or T440(+2) was incubated with DnaB in either the absence or presence of Topo IV. In the absence of Topo IV DnaB could displace 84.0, 90.5 and 89.7% of the annealed oligo from T440(0), T440(–1) and T440(+2), respectively (Fig. 6A). In the presence of Topo IV DnaB-catalyzed displacement of T440(0), T440(–1) and T440(+2) was reduced to 74.0, 76.4 and 76.9%, respectively (Fig. 6A). The displaced DNA strands were the intact oligo and no strand breaks were observed (Fig. 6B). These results show that the presence of Topo IV, either in the absence or presence of an AP site, partially inhibited DnaB-catalyzed strand displacement activity. This partial inhibition of DnaB helicase by Topo IV alone has been noted in previous studies (18).
Figure 6.
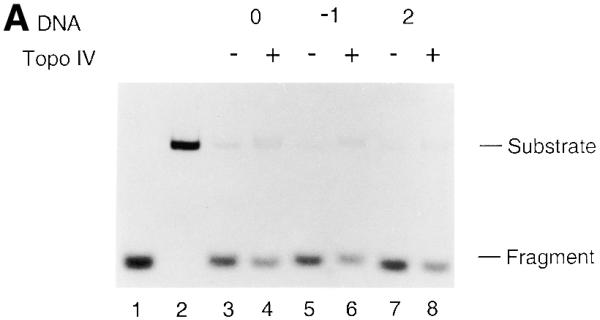
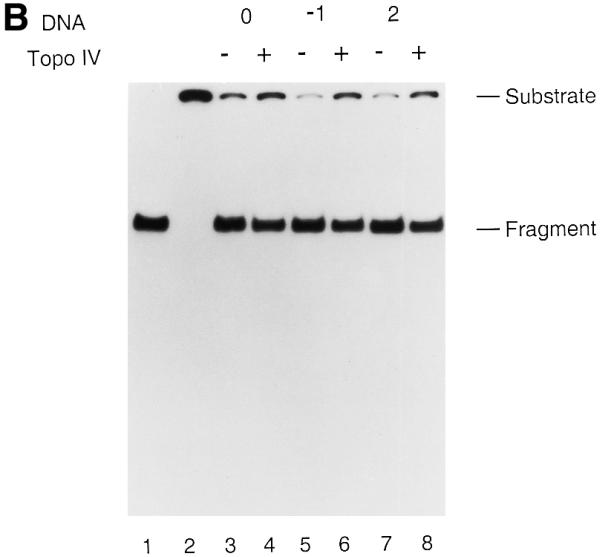
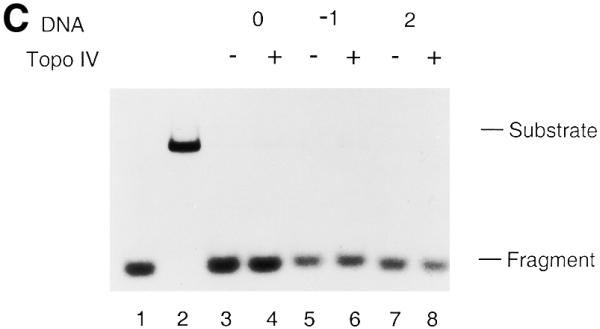
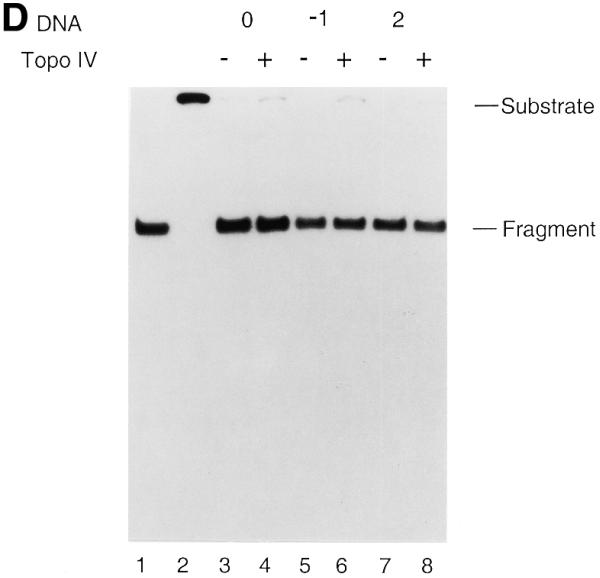
AP site-induced covalent Topo IV–DNA complexes cannot inhibit the functional activities of replicative helicases. Strand displacement assays for the DnaB (A and B) and T7 Gene 4 proteins (C and D) were performed as described in Materials and Methods. Reactions were incubated in the absence or presence of Topo IV. No norfloxacin was present. The DNA products were analyzed by electrophoresis through either 1% native agarose gels (A and C) or 9% native polyacrylamide gels (B and D). Relative amounts of strand displacement were as follows. For DnaB (A): lane 3, 84.0%; lane 4, 74.0%; lane 5, 90.5%; lane 6, 76.4%; lane 7, 89.7%; lane 8, 76.9%. For T7 Gene 4 (C): lane 3, 90.9%; lane 4, 90.7%; lane 5, 91.5%; lane 6, 88.4%; lane 7, 93.7%; lane 8, 91.4%. Lanes 1 and 2 in all panels were heat-denatured and native T440(0), respectively. Experiments were repeated twice and representative results are shown. Error range for DnaB and T7 Gene 4 helicase assays were 5 and 4%, respectively. Abbreviations are as in the legend to Figure 2.
To avoid partial inhibition of strand displacement by Topo IV alone, we performed the strand displacement assay using T7 Gene 4 protein (Fig. 6C and D). In the absence of Topo IV the T7 Gene 4 protein displaced 90.9, 91.5 and 93.7% of the annealed oligo, respectively, when T440(0), T440(–1) and T440(+2) were used as substrate (Fig. 6C). Even in the presence of Topo IV, Gene 4 helicase could displace 90.7, 88.4 and 91.4% of the oligo from T440(0), T440(–1) and T440(+2), respectively (Fig. 6C). Once again, the displaced DNA strands were the intact oligo and no strand breaks were observed (Fig. 6D).
These results demonstrated that neither the presence of AP sites nor AP site-induced covalent Topo IV–DNA complexes inhibited the functional activities of replicative helicases. In contrast, >80% of the activities of the DnaB and T7 Gene 4 helicases are inhibited by Topo IV–quinolone–DNA ternary complexes (18). Thus, in contrast to quinolone-induced covalent Topo IV–DNA complexes, AP site-induced covalent Topo IV–DNA complexes could not inhibit translocation of DNA helicases. It was likely that poisoning of Topo IV by an AP site would not cause inhibition of DNA replication.
DISCUSSION
Topoisomerase poisons are thought to act at the interface between the DNA and the topoisomerase in the ternary complex (6–8). The quinolone drug has been shown to cause a local structural perturbation in the DNA, which stimulates the Topo IV-catalyzed strand breakage reaction (20). Subtle structural alterations in the DNA at the topoisomerase-binding site can affect the catalytic activity of the enzyme. Thus, it seems reasonable that DNA lesions might also affect the catalytic activity of the topoisomerase. In fact, recent studies have demonstrated that commonly formed DNA lesions can stimulate eukaryotic Topo I- and Topo II-catalyzed cleavage of DNA (13–16). It is likely that DNA damage causes local structural alterations (17), which either stimulate the strand breakage reaction or inhibit religation. Thus, DNA damage may act as a position-specific, endogenous topoisomerase poison.
We have shown here that an AP site can stimulate Topo IV-catalyzed cleavage of DNA. AP sites stimulate eukaryotic Topo II-catalyzed DNA cleavage only when these lesions are located within the 4 base overhang generated by DNA scission (14–16). However, we found that AP sites could stimulate Topo IV-catalyzed cleavage when they were located not only within the 4 base overhang (between positions +1 and +4) (Fig. 2A) but also immediately 5′ to the point of scission (position –1) (Fig. 2B). Thus, AP(–1) was capable of trapping Topo IV, but not eukaryotic Topo II, as a covalent enzyme–DNA complex.
Formation of topoisomerase–drug–DNA ternary complexes can be reversed by EDTA, salt and heat treatment (5). The mechanisms of disassembly of ternary complexes are not fully understood. However, it has been proposed that EDTA chelates Mg2+, which forces the topoisomerase in the ternary complex to religate the DNA strands and disassociate from the DNA (5). On the other hand, salt and heat treatment are likely to reduce the affinity of the topoisomerase–DNA interaction, which would result in disassociation of the ternary complexes.
The reversal assay revealed another unique effect of AP(–1) on Topo IV. Normally, EDTA can disassemble topoisomerase–drug–DNA ternary complexes in a short period of time. In fact, Topo IV–norfloxacin–T440(0) ternary complexes and Topo IV–T440 binary complexes formed with T440(+1), T440(+2), T440(+3) and T440(+4) were completely disassembled within 2 min when incubated in the presence of EDTA (Fig. 4). However, EDTA-mediated disassembly was not effective with the covalent Topo IV–T440(–1) complex (Figs 3 and 4). Thus, AP(–1) seems to inhibit the religation reaction. These results suggest that an AP site at the –1 position can stabilize Mg2+ and make it resistant to EDTA-mediated chelation. It is interesting to speculate that Mg2+ in the Topo IV–DNA complex may be stabilized by a hydrogen bond formed with a water molecule trapped at AP(–1), as is the case with AP nuclease (22). It seems reasonable to assume that the Mg2+ in the Topo IV–DNA complex is located at or close to the base at the –1 position. Note that AP site-induced formation of covalent Topo I–DNA complexes was irreversible (13).
Topoisomerase poisoning causes inhibition of DNA replication, which triggers cytotoxic events (6–8). We have shown that a Topo IV–norfloxacin–DNA ternary complex inhibits the replicative helicase and causes replication fork arrest (18). We examined if AP site-induced covalent Topo IV–DNA complexes could inhibit replicative helicases. Interestingly, AP site-induced covalent Topo IV–DNA complexes had no effect on DnaB- and T7 Gene 4-catalyzed strand displacement (Fig. 6). AP site-induced covalent Topo IV–DNA complexes may not be stable enough to block replication fork progression. Our recent studies on DNA gyrase have demonstrated that formation of quinolone-induced covalent DNA gyrase–DNA complexes is necessary but not sufficient to arrest replication fork progression and that the stability of the DNA gyrase–quinolone–DNA ternary complex is critical for replication fork arrest (23).
What might happen in the cell when topoisomerase is trapped at an AP site? AP sites, as well as other types of DNA damage, need to be repaired before DNA replication takes place. DNA lesions remaining on the template strand cause errors during DNA replication and generate mutations in the genome. The AP site-induced covalent Topo IV–DNA complex is likely to inhibit the action of AP nuclease and other repair proteins. When a replication fork collides with Topo IV trapped at an AP site, the AP site-induced covalent Topo IV–DNA complex would be disassembled and the AP site on the template strand would be exposed. As a result of DNA replication, a mutation could be generated at the AP site. Thus, AP site-induced topoisomerase poisoning might potentiate mutations.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Kenneth Marians for his gifts of purified proteins and critical comments on this manuscript. We also thank Dr Smita Patel for her gift of T7 Gene 4 protein. This study was supported by NIH Grant GM59465.
References
- 1.Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- 2.Gellert M., Mizuuchi,K., O’Dea,M.H. and Nash,H.A. (1976) DNA gyrase: an enzyme that introduces superhelical turns into DNA. Proc. Natl Acad. Sci. USA, 73, 3872–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugino A., Peebles,C.L., Kreuzer,K.N. and Cozzarelli,N.R. (1977) Mechanism of action of nalidixic acid: purification of Escherichia coli nalA gene product and its relationship to DNA gyrase and a novel nicking-closing enzyme. Proc. Natl Acad. Sci. USA, 74, 4767–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gellert M., Mizuuchi,K., O’Dea,M.H., Itoh,T. and Tomizawa,J. (1977) Nalidixic acid resistance: a second genetic character involved in DNA gyrase activity. Proc. Natl Acad. Sci. USA, 74, 4772–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kreuzer K.N. and Cozzarelli,N.R. (1979) Escherichia coli mutants thermosensitive for deoxyribonucleic acid gyrase subunit A: effects on deoxyribonucleic acid replication, transcription and bacteriophage growth. J. Bacteriol ., 140, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maxwell A. (1992) The molecular basis of quinolone action. J. Antimicrob. Chemother., 30, 409–416. [DOI] [PubMed] [Google Scholar]
- 7.Chen A.Y. and Liu,L.F. (1994) DNA topoisomerases: essential enzymes and lethal targets. Annu. Rev. Pharmacol. Toxicol., 34, 191–218. [DOI] [PubMed] [Google Scholar]
- 8.Froelich-Ammon S.J. and Osheroff,N. (1995) Topoisomerase poisons: harnessing the dark side of enzyme mechanism. J. Biol. Chem., 270, 21429–21432. [DOI] [PubMed] [Google Scholar]
- 9.Chen C.-R., Malik,M., Snyder,M. and Drlica,K. (1996) DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J. Mol. Biol., 258, 627–637. [DOI] [PubMed] [Google Scholar]
- 10.Tewey K.M., Rowe,T.C., Yang,L., Halligan,B.D. and Liu,L.F. (1984) Adriamycin-induced DNA damage is mediated by mammalian DNA topoisomerase II. Science, 266, 466–468. [DOI] [PubMed] [Google Scholar]
- 11.Rosenstein B.S., Subramanian,M.T. and Muller,M.T. (1997) The involvement of topoisomerase I in the induction of DNA-protein crosslinks and DNA single-strand breaks in cells of ultraviolet-irradiated human and frog cell lines. Radiat. Res., 148, 575–579. [PubMed] [Google Scholar]
- 12.Lanza A., Tornaletti,S., Rodolfo,C., Scanavini,M.C. and Pedrini,A.M. (1996) Human DNA topoisomerase I-mediated cleavages stimulated by ultraviolet light-induced DNA damage. J. Biol. Chem., 271, 6978–6986. [DOI] [PubMed] [Google Scholar]
- 13.Pourquier P., Ueng,L.-M., Kohlhagen,G., Mazumder,A., Gupta,M., Kohn,K.W. and Pommier,Y. (1997) Effects of uracil incorporation, DNA mismaches and abasic sites on cleavage and religation activities of mammalian topoisomerase I. J. Biol. Chem., 272, 7792–7796. [DOI] [PubMed] [Google Scholar]
- 14.Kingma P.S., Corbett,A.H., Burcham,P.C., Marnett,L.J. and Osheroff,N. (1995) Abasic sites stimulate double-stranded DNA cleavage mediated by topoisomerase II. J. Biol. Chem., 270, 21441–21444. [DOI] [PubMed] [Google Scholar]
- 15.Kingma P.S. and Osheroff,N. (1997) Apurinic sites are position specific topoisomerase II poisons. J. Biol. Chem., 272, 1148–1155. [DOI] [PubMed] [Google Scholar]
- 16.Kingma P.S. and Osheroff,N. (1997) Spontaneous DNA damage stimulates topoisomerase II-mediated DNA cleavage. J. Biol. Chem., 272, 7488–7493. [DOI] [PubMed] [Google Scholar]
- 17.Cline S.D., Jones,W.R., Stone,M.P. and Osheroff,N. (1999) DNA abasic lesions in a different light: solution structure of an endogenous topoisomerase II poison. Biochemistry, 38, 15500–15507. [DOI] [PubMed] [Google Scholar]
- 18.Shea M.E. and Hiasa,H. (1999) Interactions between DNA helicases and frozen topoisomerase IV-quinolone-DNA ternary complexes. J. Biol. Chem., 274, 22747–22754. [DOI] [PubMed] [Google Scholar]
- 19.Peng H. and Marians,K.J. (1993) Escherichia coli topoisomerase IV: purification, characterization, subunit structure and subunit interactions. J. Biol. Chem., 268, 24481–24490. [PubMed] [Google Scholar]
- 20.Marians K.J. and Hiasa,H. (1997) Mechanism of quinolone action: a drug-induced structural perturbation of the DNA precedes strand cleavage by topoisomerase IV. J. Biol. Chem., 272, 9401–9409. [DOI] [PubMed] [Google Scholar]
- 21.Hiasa H., Yousef,D.O. and Marians,K.J. (1996) DNA strand cleavage is required for replication fork arrest by a frozen topoisomerase-quinolone-DNA ternary complex. J. Biol. Chem., 271, 26424–26429. [DOI] [PubMed] [Google Scholar]
- 22.Mol C.D., Kuo,C.-F., Thayer,M.M., Cunningham,R.P. and Tainer,J.A. (1995) Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature, 374, 381–386. [DOI] [PubMed] [Google Scholar]
- 23.Hiasa H. and Shea,M.E. (2000) DNA gyrase-mediated wrapping of the DNA strand is required for the replication fork arrest by the DNA gyrase-quinolone-DNA ternary complex. J. Biol. Chem., 275, 34780–34786. [DOI] [PubMed] [Google Scholar]