

Abstract
Expansion of a GAA·TTC repeat in the first intron of the frataxin (FXN) gene causes an mRNA deficit that results in Friedreich ataxia (FRDA). The region flanking the repeat on FRDA alleles is associated with more extensive DNA methylation than is seen on normal alleles and histone modifications typical of repressed genes. However, whether these changes are responsible for the mRNA deficit is controversial. Using chromatin immunoprecipitation and cell lines from affected and unaffected individuals, we show that certain marks of active chromatin are also reduced in the promoter region of the FXN gene in patient cells. Thus, the promoter chromatin may be less permissive for transcription initiation than it is on normal alleles. Furthermore, we show that the initiating form of RNA polymerase II and histone H3 trimethylated on lysine 4, a chromatin mark tightly linked to transcription initiation, are both present at lower levels on FRDA alleles. In addition, a mark of transcription elongation, trimethylated H3K36, shows a reduced rate of accumulation downstream of the repeat. Our data thus suggest that repeat expansion reduces both transcription initiation and elongation in FRDA cells. Our findings may have implications for understanding the mechanism responsible for FRDA as well as for therapeutic approaches to reverse the transcription deficit.
Keywords: Ataxia, Chromatin Histone Modification, Chromatin Remodeling, DNA Transcription, Epigenetics, Gene Silencing, Genetic Diseases, Friedreich Ataxia, Trinucleotide Repeat
Introduction
Friedreich ataxia (FRDA),3 an autosomal recessive disorder with early onset, is the most common inherited ataxia (1). In addition to ataxia, diabetes and hypertrophic cardiomyopathy are also commonly seen, with congestive heart failure being a frequent cause of early mortality. The disorder results from mutations in the frataxin (FXN) gene. Most affected individuals have large expansions of a GAA·TTC repeat in intron 1 of both FXN alleles (1). Expanded alleles produce lower than normal amounts of FXN mRNA, resulting in a deficiency of frataxin, a protein essential for mitochondrial iron metabolism (1). In general, the repeat number is directly related to the age of onset and the severity of symptoms (2).
Brain, peripheral blood leukocytes, and lymphoblastoid cells from individuals with FRDA all show a similar FXN mRNA deficit, and the region flanking the repeat in these cells is aberrantly methylated and enriched for histone modifications characteristic of genes that are transcriptionally repressed (3–7). Because the repeat is relatively close to the promoter, the repeat-mediated chromatin changes could affect transcription initiation if they were to spread to the promoter. This could produce a promoter chromatin configuration that was less permissive for transcription initiation (8). Because intragenic DNA methylation can reduce transcription elongation (8), repeat expansion may also affect the ability of RNA polymerase II (RNAPII) to transcribe through the intron.
An alternative explanation for the FXN mRNA deficit in FRDA has been suggested based on the observation that long GAA·TTC repeats block transcription elongation on unchromatinized templates in vitro or when propagated in bacteria (9–12). This phenomenon has been attributed to the ability of these repeats to form triple-stranded DNA structures during transcription that trap RNA polymerase on the template. Should this mechanism operate at the FXN locus in FRDA cells, the major effect would presumably be downstream of transcription initiation.
To better understand how repeat expansion affects transcription, we have extended our previous chromatin immunoprecipitation studies on affected and unaffected alleles to include a variety of additional marks of active chromatin and of the initiating form of RNAPII. Our data suggest that problems with both transcription initiation and elongation contribute significantly to the FXN mRNA deficit responsible for FRDA pathology. Our data provide clues to the molecular basis of repeat-mediated disease pathology in FRDA as well as a clear rationale for the use of chromatin-modifying agents in the treatment of this disorder.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
Lymphoblasts from individuals without FRDA (GM06895, GM06865, and GM06906) and those with FRDA (GM04079, GM15850, GM16207, GM16209, and GM16243) were obtained from the Coriell Cell Repository (Camden, NJ). The repeat numbers in these patient cell lines are 340/420, 650/1030, 280/830, 800/800, and 670/1170, respectively. Lymphoblasts were grown in RPMI 1640 medium supplemented with 10% fetal calf serum and 1× antibiotic-antimycotic under standard conditions (Invitrogen). The following antibodies (catalogue numbers given in parentheses) were purchased: H4K5Ac (07-327), H3K4Me2 (07-030), H3K9Ac (07-352), H3K9Me2 (07-441), H3K4Me3 (04-745), and normal rabbit IgG (12-370) from Millipore (Temecula, CA), antibody to RPB1 subunit of yeast RNA polymerase II (8WG16 clone) (W0011) from NeoClone Biotechnology, and anti-H4K16Ac (39167) from Active Motif (Carlsbad, CA). Antibodies to the RNAPII C-terminal domain, repeat YSPTSPS (phospho-Ser-5) (ab5131) and YSPTSPS (phospho-Ser-2) (ab5095), and H3K36Me3 (ab9050) were from Abcam (Cambridge, MA). Rabbit preimmune serum, used as a control for chromatin immunoprecipitation with the H4K16Ac antibody, was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Primers were purchased from Integrated DNA Technologies (Coralville, IA). The sequences of the primers used in this study are given in Table 1.
TABLE 1.
Primers used in this study
F, forward; R, reverse; LK, linker.
| 5′ to 3′ | |
|---|---|
| Primers for ChIP PCRs | |
| Promoter F | CCCCACATACCCAACTGCTG |
| Promoter R | GCCCGCCGCTTCTAAAATTC |
| Exon 1 3′ end F | AGCAGCATGTGGACTCTC |
| Exon 1 R2 | TGGGCTGGGCTGGGTGACGCCAGG |
| Up GAA F | GAAACCCAAAGAATGGCTGTG |
| Up GAA R | TTCCCTCCTCGTGAAACACC |
| Down GAA F | CTGGAAAAATAGGCAAGTGTGG |
| Down GAA R | CAGGGGTGGAAGCCCAATAC |
| Exon 2 F | GCCTCAACCAGATTTGGAAT |
| Exon 2 R | GCCCAAAGTTCCAGATTT |
| GAPDH exon 1 F | TCGACAGTCAGCCGCATCT |
| GAPDH intron 1 R | CTAGCCTCCCGGGTTTCTCT |
| MYO-D exon 1 F | CCGCCTGAGCAAAGTAAATGA |
| MYO-D exon 1 R | GGCAACCGCTGGTTTGG |
| Primers for reverse transcription | |
| LK exon 1 3′ end | actggagcacgaggacactCAGGTCGCATCGATGTC |
| LK exon 2 | actggagcacgaggacactGCCCAAAGTTCCAGATTT |
| Exon 1 R2 | TGGGCTGGGCTGGGTGACGCCAGG |
| Primers for SYBR Green PCR | |
| LK primer | ACTGGAGCACGAGGACACT |
| Exon 3 F | CAGAGGAAACGCTGGACTCT |
| Exon 4 R | AGCCAGATTTGCTTGTTTGG |
| GUS F | CTCATTTGGAATTTTGCCGATT |
| GUS R | CCGAGTGAAGATCCCCTTTTTA |
| Exon 2 F | GCCTCAACCAGATTTGGAAT |
| Exon 1 3′ end F | AGCAGCATGTGGACTCTC |
Real-time PCR Analysis of FXN Transcription
Total RNA was purified from cells using the TRIzol® reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription with either random hexamers or strand-specific primers was done on DNase I-treated RNA using SuperScript III first strand synthesis kit from Invitrogen as per the manufacturer's recommendation. Strand-specific primers were used that contained a linker sequence at the exon 1 and exon 2 as described previously (13). Real-time PCR was then done with Power SYBR® Green PCR master mix (Applied Biosystems, Foster City, CA) using a primer homologous to the linker sequence and exon-specific primers. PCR with RNA that had not been reverse-transcribed produced no product, demonstrating that the PCR product obtained using this strategy was not due to contaminating DNA. Primer sequences are listed in Table 1.
Chromatin Immunoprecipitation Assays
The ChIP assay kit from Millipore was used for histone ChIPs according to the manufacturer's instructions with slight modifications as described previously (14). For RNAPII ChIP assays, modified wash steps were used: 1× with low salt wash buffer (Millipore) for 5 min, 1× with wash buffer II (50 mm HEPES-KOH, pH 7.5, 500 mm NaCl, 1 mm EDTA pH 8.0, 1% Triton X-100, 0.1% sodium deoxycholate) for 3 min, 1× with wash buffer III (10 mm Tris, pH 8.0, 250 mm LiCl, 1 mm EDTA, pH 8.0, 0.5% IGEPAL-CA630, 0.5% sodium deoxycholate) for 3 min followed by two washes with Tris-EDTA, pH 8.0, for 5 min each. The amount of FXN DNA immunoprecipitated was determined using quantitative real-time PCR, a Power SYBR Green PCR master mix, and primers specific for different regions of the gene (Table 1). The immunoprecipitated material was normalized to 5% of input and expressed relative to GAPDH. For RNAPII ChIPs, the background at MyoD was subtracted from the final values. The ChIP experiments were performed at least in triplicate, and each PCR reaction was done in triplicate. Student's t test was used to calculate the p values (GraphPad Software, Inc., La Jolla, CA).
RESULTS
Repeat Expansion Results in Reduced Levels of a Subset of Marks of Active Chromatin on the Promoter of FRDA Alleles
To study the extent of the repeat-mediated chromatin changes on FRDA alleles, we examined a number of marks typical of active chromatin in three regions of the FXN gene in cells from unaffected individuals and those with FRDA. These regions comprise a promoter segment located immediately upstream of a previously identified transcription start site (TSS), referred to as TSS1 (15), and the previously described Up GAA and Down GAA regions that flank the FRDA repeat (3), as illustrated in Fig. 1A. We found that in addition to reduced levels of marks of active chromatin in the region immediately flanking the GAA·TTC repeats, some of these marks were also reduced in the promoter region of FRDA alleles. Specifically, in four patient cell lines, all five marks of active chromatin examined, H4K5Ac, H4K16Ac, H3K9Ac, H3K14Ac, and H3K4Me2, were significantly lower in the Up GAA and Down GAA regions than in cells from unaffected individuals (Fig. 1). Furthermore, although no differences were seen in the levels of H3K9Ac and H3K14Ac in the promoter region, levels of H4K16Ac, H4K5Ac, and H3K4Me2 were significantly lower on patient alleles. Notably, although the difference between the levels of these chromatin marks in unaffected and affected cells was much less than in the regions flanking the repeat (2–3-fold as compared with 8–10-fold), the differences were still significant. Because our finding of altered chromatin in the promoter region in lymphoblastoid cells is different from previously published reports (3, 16), we tested two additional cell lines from individuals without FRDA, GM06895 and GM06906, to assess whether these differences could be attributed to variation in the levels of these marks on unaffected alleles. Although the absolute level of each histone modification did differ in each of these cell lines, the same trend was found for all the chromatin marks tested (data not shown).
FIGURE 1.

Association of active histone marks with the promoter, Up GAA, and Down GAA region of the frataxin gene in unaffected (GM06865) and FRDA cells (GM15850, GM16243, GM16209, and GM04079). A, schematic representation of the FXN gene showing the positions of the regions analyzed by real-time PCR in ChIP assays. B, chromatin immunoprecipitated with indicated antibodies was normalized to 5% of input and expressed relative to GAPDH. Error bars indicate S.D.
The reduced levels of marks of active chromatin seen on the promoter would be consistent with the recent finding that the FXN promoter in the brains of two individuals with FRDA was enriched for two repressive chromatin marks, H3K9Me2 and H3K9Me3 (5), and that fibroblasts from two different FRDA individuals also show enrichment for H3K9Me2 and another repressive chromatin mark, H3K27Me3, in a region slightly closer to the frataxin open reading frame (ORF) than the region we examined (17). Taken together, these observations suggest that the repeat-mediated chromatin changes seen in intron 1 do extend into the promoter. Thus, in principle, repeat expansion could contribute to the FXN mRNA deficit in FRDA cells via an effect on transcription initiation.
The Major FXN Transcription Start Site Is Located 62 bp Upstream of the ORF in Lymphoblastoid Cells
In addition to TSS1, which was defined on the basis of a clone isolated from a 5′ rapid amplification of cDNA ends experiment (15), a variety of other FXN cDNA clones with different 5′ ends have also been described (18). Given that some chromatin marks have a restricted distribution with respect to the TSS, we carried out primer extension on RNA isolated from normal and patient cells using a primer within the open reading frame to define the major TSS in lymphoblastoid cells. Very long exposures of the gel-resolved primer extension products showed no detectable bands in the region of TSS1. Instead, a single product terminating 159 bases downstream of TSS1 was the major product seen. We refer to this putative TSS, which is 62 bp upstream of the ORF, as TSS2 (Fig. 2, left panel). Thus, the most prevalent transcript from the FXN gene originates closer to the ORF than TSS1, at least in lymphoblastoid cells. The same putative TSS was seen in both unaffected and affected individuals. In contrast to what is seen in most mammalian promoters, TSS2 was not associated with either a TATAA box (19, 20) or an Initiator (Inr) element (21).
FIGURE 2.

FXN transcription start site in lymphoblastoid cells. Left panel, sequencing gel showing the product of primer extension with end-labeled Exon 1 R2 primer and RNA from unaffected (UN) and FRDA cells using SuperScript III reverse transcriptase according to standard procedures. G, A, T, and C lanes represent the sequencing reactions from the corresponding region of DNA in plasmid FXN81658 (4) using the same primer. MW, pBR322 DNA digested with MspI. Right panel, DNA sequence of the 5′ end of the FXN locus showing the position of previously reported transcription start site, TSS1 (15), and the transcription start site TSS2 identified in this study. The numbers alongside the sequence indicate the position of bases with respect to the first base of the open reading frame, which is shown in the open box. The sequence between the two brackets is the sequence −221 to −121 bases upstream of the frataxin open reading frame, which we showed previously to be important for FXN promoter activity in muscle cells (32). The region highlighted in gray corresponds to the remnants of an ancient L2 repeated DNA element.
Further support for the idea that transcription initiates closer to the ORF can be seen in the distribution of histone H3 trimethylated at lysine 4 (H3K4Me3) on normal alleles. Although H3K4Me2 has a broad distribution within active genes, H3K4Me3 levels are highest immediately downstream of transcription initiation (22–24). As can be seen in Fig. 3A, when a primer pair from the TSS1 region was used to interrogate chromatin immunoprecipitated with an antibody specific to H3K4Me3, the levels of this mark were much lower than what is seen when a primer pair within the open reading frame, close to TSS2, is used. This supports the idea that TSS2 is closer to the start of transcription than TSS1. Because H3K4Me3 deposition is directly related to the activity of the initiating form of RNAPII, its levels are thought to more clearly reflect the extent of transcription initiation than some other chromatin modifications (25–28). The levels of H3K4Me3 on patient alleles were ∼50% of that seen on the unaffected controls. Given that these patient cells have FXN mRNA levels that are 20–30% of that of unaffected individuals (Fig. 3B), the reduced level of H3K4Me3 suggests that a transcription initiation problem may account for a large proportion of the FXN mRNA deficit in FRDA.
FIGURE 3.

Transcription initiation in FRDA cells. A, the abundance of H3K4Me3 on different regions of the FXN gene in unaffected and FRDA cells relative to its abundance on GAPDH. The differences in the levels of H3K4Me3 in unaffected and FRDA cells were only significant for the exon 1 region (p < 0.05). B, FXN mRNA level in unaffected and FRDA cells relative to GUS. Real-time PCR for FXN mRNA was done with primers exon 3 F and exon 4 R as described previously (3). C, relative abundance of DNA immunoprecipitated with an antibody specific to RNAPII phosphorylated at Ser-5 in unaffected and FRDA cells. The asterisks indicate those regions of patient alleles that are significantly different from the corresponding regions of both unaffected alleles tested at p = 0.05 or better. D, relative abundance of DNA immunoprecipitated by the 8WG16 antibody to RNAPII in unaffected and FRDA cells. The asterisks indicate those regions of patient alleles that are significantly different from the corresponding regions of both unaffected alleles tested at p = 0.05 or better. Error bars in all panels indicate S.D.
Evidence of Reduced Transcription Initiation Is Seen on FRDA Alleles
To confirm that transcription initiation is indeed impaired on FRDA alleles, we determined the enrichment of TSS1 and TSS2 on the affected and unaffected alleles with two different antibodies to RNAPII. The first antibody we tested is specific to RNAPII phosphorylated on serine 5 (Ser(P)-5), the initiating form of RNAPII (29). As can be seen from Fig. 3C, levels of this form of RNAPII are higher on the promoter and exon 1 regions tested than the Up GAA region. This was true of both affected and unaffected cells. However, FRDA cells showed ∼50% less enrichment for the form of RNAPII detected by this antibody, consistent with the H3K4Me3 data and the idea that initiation occurs less frequently on FRDA alleles.
Further support for an effect on transcription initiation was obtained with 8WG16, an antibody to a subunit of yeast RNAPII (30). This antibody recognizes a more complex array of human RNAPII isoforms than the first antibody, including those phosphorylated at Ser-5. As with the first RNAPII antibody, we saw strong enrichment for the epitopes recognized by the 8WG16 antibody in exon 1 on unaffected alleles with ∼50% less enrichment being seen on FRDA alleles (Fig. 3D). The distribution of 8WG16 epitopes was narrower than those detected with the Ser(P)-5 antibody with significantly more enrichment being seen in the TSS2 region than the TSS1 region.
A Transcription Elongation Problem May Also Contribute to Reduced FXN mRNA
To assess the contribution of a block to transcription elongation to the mRNA deficit in FRDA, we first looked for evidence of a transcript that was truncated in the vicinity of the repeat by comparing the levels of exon 1 and exon 2 in both affected and unaffected individuals. To avoid any potential contribution from the antisense transcript produced from the FXN locus (17), we used strand-specific RT-PCR to examine the levels of FXN mRNA specific to exon 1 and exon 2 in unaffected and FRDA cells. When a primer pair located downstream of TSS2 was used to compare the levels of exon 1 using quantitative PCR, a 4–5-fold higher product yield was seen in unaffected individuals as compared with individuals with FRDA (Fig. 4, upper panel). This was comparable with the difference in total FXN mRNA seen with primers from exons 3 and 4 (Fig. 3B). The levels of exon 2 were also 4–5 times higher in unaffected cells (Fig. 4, lower panel). Thus, no patient-specific decline in exon 2 levels relative to those of exon 1 was detected. However, it was apparent that the absolute yield of PCR product from exon 2 was 3–4-fold lower than that of exon 1 in both affected and unaffected cells despite the very similar efficiency of amplification seen when DNA was used as a template. Thus, it may be that transcription termination occurs frequently on both normal and patient alleles in the region downstream of the exon 1 primer pair.
FIGURE 4.
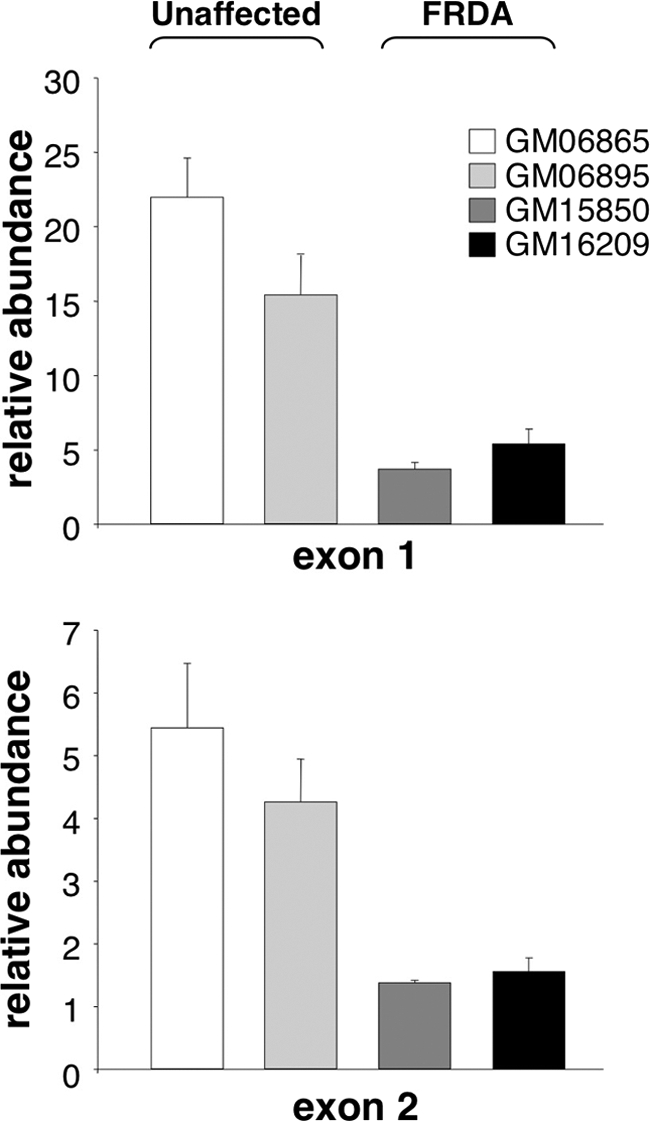
FXN mRNA levels in unaffected and FRDA cells. The amount of exon 1 and exon 2 was determined using strand-specific PCR as described under “Experimental Procedures.” The levels of FXN mRNA containing exon 1 (upper panel) and exon 2 (lower panel) are shown relative to GUS. Error bars indicate S.D.
Because a truncated transcript might be unstable, we used a second approach to assess an effect of the repeats on transcription elongation. We first tested antibodies to RNAPII phosphorylated at Ser-2, the elongating form of RNAPII. Unfortunately, although enrichment was seen on a highly transcribed gene (GAPDH), we did not get a good enrichment on the FXN locus even on normal alleles due, perhaps in part, to the low transcription rate of the gene. However, trimethylation of H3K36 occurs co-transcriptionally in a manner dependent on the level of elongating RNAPII (31). Thus, this histone modification is considered a reasonable proxy measure of the amount of elongation. We found that H3K36Me3 levels rose more slowly in FRDA cells than they did in cells from unaffected individuals, with no significant accumulation of H3K36Me3 on FRDA alleles (Fig. 5). In particular, there were much lower levels of H3K36Me3 on the Down GAA region relative to the Up GAA region that could be consistent with a repeat-mediated effect on transcription elongation.
FIGURE 5.

The abundance of H3K36Me3 on different regions of the FXN gene in unaffected and FRDA cells. The amount of DNA immunoprecipitated with an antibody to H3K36Me3 was determined as described under “Experimental Procedures” and is shown relative to GAPDH. Error bars indicate S.D.
DISCUSSION
The data in this study provide information relevant to both the normal functioning of the FXN gene and the aberrant expression from FRDA alleles. We have shown, using a combination of primer extension to map the 5′ end of the transcript and chromatin immunoprecipitation with antibodies to proteins that are enriched close to the start of transcription, that the major TSS (TSS2) on the normal FXN gene is located closer to the frataxin ORF than previously appreciated, at least in lymphoblastoid cells. This finding is consistent with our previous work using reporter assays in muscle cells (32), in which we showed that there was significant promoter activity located in the sequences downstream of TSS1. This work showed that the most important regions for the positive regulation of the FXN promoter in muscle cells are located between −221 and −121 bases upstream of the start of the frataxin ORF (Fig. 2, right panel, bracketed sequence). This is the only significant region of homology between primates and rodents and represents the ancient remnant of an L2-like element, a family of long interspersed repeated DNA elements (32). Taken together, these data suggest that the major TSS in both lymphoblastoid and muscle cells lies closer to the frataxin ORF than previously appreciated. Examination of the region in the vicinity of TSS2 suggests that, unlike TSS1 that colocalizes with an Initiator (Inr)-like element, TSS2 is not associated with TATA-like or Initiator elements, motifs typical of most mammalian promoters. Our identification of this TSS should help facilitate a better understanding of the factors important for the optimal expression from this unusual promoter.
In addition, our observation of the higher yield of exon 1 relative to exon 2 in both normal and patient cells (Fig. 4) suggests that transcription frequently terminates downstream of exon 1 even in normal cells. Because normal cells contain a GAA·TTC repeat tract that is far too short to cause triplex DNA or Sticky DNA formation, a repeat-mediated basis for this transcription termination seems unlikely. Because intragenic methylation can affect transcription elongation (8), the elongation problem seen in cells from unaffected individuals may be related to the DNA methylation present in the region upstream of the FRDA repeat even on normal alleles (4). An alternative explanation for this termination is suggested by the broader distribution of epitopes that are recognized by antibodies to RNAPII phosphorylated at Ser-5 as compared with the 8WG16 antibody (Fig. 3). This phenomenon has been attributed to an isoform of RNAPII that carries, in addition to the Ser-5 modification, other modifications that prevent recognition by 8WG16 (33). Such an RNAPII isoform is often seen on inducible genes and would be consistent with the observation that iron is a positive regulator of FXN expression (34). Because such genes often generate transcripts that terminate near the first exon junction (35), the high frequency of premature transcription termination on both normal and FRDA alleles may be related to some conserved property of this class of genes.
The promoters of normal and FRDA alleles show differences in the extent of their enrichment for marks of transcriptionally permissive chromatin. Specifically, FRDA promoters are associated with reduced levels of H4K5Ac and H4K16Ac but not H3K9Ac and H3K14Ac. Because H3 acetylation is thought to occur co-transcriptionally, it is unclear what significance these modifications have upstream of transcription. On the other hand, acetylation of H4 is thought to precede transcription. Thus, the reduced H4 acetylation on the FRDA promoter likely reflects a chromatin configuration that is less permissive for transcription (36). In particular, H4K16 acetylation is thought to play an important role in maintaining an open chromatin configuration (37).
We also found that FRDA alleles were associated with reduced levels of RNAPII that is recognized by an antibody that is specific for the isoform phosphorylated at Ser-5. This result is at odds with data published while this work was in progress in which cells from an affected individual and his unaffected sibling showed no significant difference in the region of TSS1 in the amount of DNA immunoprecipitated with the same antibody (16). A number of explanations for this difference are possible including the fact that maximal enrichment for this form of RNAPII is seen in the vicinity of TSS2, and although we always find a significant difference between affected and unaffected alleles at TSS2, the difference is not always significant at TSS1. The fact that another antibody that recognizes the initiating form of RNAPII, 8WG16, also shows lower levels of initiating RNAPII on FRDA alleles and that levels of H3K4Me3, a histone modification characteristic of the early phases of transcription initiation, are also lower than normal would be consistent with our Ser(P)-5 RNAPII data.
Thus, our data suggest that there is a reduced efficiency of transcription initiation on FRDA alleles for which the repressive chromatin marks on the promoter are the most parsimonious explanation. It seems reasonable to think that the effect on transcription initiation might vary with repeat number and be more marked on those alleles with a larger number of repeats that facilitate a broader spread of repressive chromatin marks from intron 1.
A role for the repeat-mediated chromatin changes in the FRDA transcript deficit is consistent with the fact that histone deacetylase inhibitors increase transcription from FRDA alleles (3, 6, 38, 39). This idea has been challenged by the recent demonstration that an inhibitor of H3K9 methylation does not reactivate FRDA alleles. This has led to the suggestion that the chromatin changes are not responsible for the FXN mRNA deficit and thus that the histone deacetylase inhibitors are acting indirectly to increase FXN transcription (16). However, it may be that H3K9 demethylation is not sufficient to reverse all of the key determinants of repressive chromatin on FRDA alleles. Our demonstration of a repeat-mediated effect on transcription initiation lends support to the idea that the chromatin changes on FRDA alleles have consequences for transcription and thus that histone deacetylase inhibitors may be acting, at least in part, on the underlying cause of FRDA.
The lack of significant accumulation of H3K36Me3 downstream of the GAA·TTC suggests that a problem with transcription elongation may also contribute to the mRNA deficit in FRDA. The relative contribution of the elongation problem and the initiation problem remains to be assessed. The question of whether the elongation problem results from the chromatin changes in the body of the gene or the formation of DNA structures that trap RNAPII on the template also remains to be seen.
In conclusion, our data demonstrate that FRDA alleles show problems with both transcription initiation and elongation and suggest that repeat-induced chromatin changes are responsible for some, if not all, of the resultant FXN mRNA deficit. This lends support to the idea that histone deacetylase inhibitors, which are currently in clinical trials for the treatment of FRDA, act directly to correct the underlying problem caused by the expanded repeats. The contribution of any non-chromatin-based effects of the repeat to the FXN mRNA deficit remains an open question.
Acknowledgments
We thank the members of the Usdin workgroup for careful reading of this manuscript and helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grant DK057810 from the Intramural Program of the NIDDK (to K. U.).
- FRDA
- Friedreich ataxia
- RNAPII
- RNA polymerase II
- TSS
- transcription start site.
REFERENCES
- 1. Pandolfo M. (2002) Adv. Exp. Med. Biol. 516, 99–118 [DOI] [PubMed] [Google Scholar]
- 2. Montermini L., Richter A., Morgan K., Justice C. M., Julien D., Castellotti B., Mercier J., Poirier J., Capozzoli F., Bouchard J. P., Lemieux B., Mathieu J., Vanasse M., Seni M. H., Graham G., Andermann F., Andermann E., Melançon S. B., Keats B. J., Di Donato S., Pandolfo M. (1997) Ann. Neurol. 41, 675–682 [DOI] [PubMed] [Google Scholar]
- 3. Herman D., Jenssen K., Burnett R., Soragni E., Perlman S. L., Gottesfeld J. M. (2006) Nat. Chem. Biol. 2, 551–558 [DOI] [PubMed] [Google Scholar]
- 4. Greene E., Mahishi L., Entezam A., Kumari D., Usdin K. (2007) Nucleic Acids Res. 35, 3383–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al-Mahdawi S., Pinto R. M., Ismail O., Varshney D., Lymperi S., Sandi C., Trabzuni D., Pook M. (2008) Hum. Mol. Genet 17, 735–746 [DOI] [PubMed] [Google Scholar]
- 6. Rai M., Soragni E., Jenssen K., Burnett R., Herman D., Coppola G., Geschwind D. H., Gottesfeld J. M., Pandolfo M. (2008) PLoS ONE 3, e1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soragni E., Herman D., Dent S. Y., Gottesfeld J. M., Wells R. D., Napierala M. (2008) Nucleic Acids Res. 36, 6056–6065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lorincz M. C., Dickerson D. R., Schmitt M., Groudine M. (2004) Nat. Struct. Mol. Biol. 11, 1068–1075 [DOI] [PubMed] [Google Scholar]
- 9. Grabczyk E., Usdin K. (2000) Nucleic Acids Res. 28, 2815–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sakamoto N., Chastain P. D., Parniewski P., Ohshima K., Pandolfo M., Griffith J. D., Wells R. D. (1999) Mol. Cell 3, 465–475 [DOI] [PubMed] [Google Scholar]
- 11. Krasilnikova M. M., Kireeva M. L., Petrovic V., Knijnikova N., Kashlev M., Mirkin S. M. (2007) Nucleic Acids Res. 35, 1075–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bidichandani S. I., Ashizawa T., Patel P. I. (1998) Am. J. Hum. Genet. 62, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Filippova G. N., Thienes C. P., Penn B. H., Cho D. H., Hu Y. J., Moore J. M., Klesert T. R., Lobanenkov V. V., Tapscott S. J. (2001) Nat. Genet. 28, 335–343 [DOI] [PubMed] [Google Scholar]
- 14. Biacsi R., Kumari D., Usdin K. (2008) PLoS Genet 4, e1000017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Campuzano V., Montermini L., Moltò M. D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., Zara F., Cañizares J., Koutnikova H., Bidichandani S. I., Gellera C., Brice A., Trouillas P., De Michele G., Filla A., De Frutos R., Palau F., Patel P. I., Di Donato S., Mandel J. L., Cocozza S., Koenig M., Pandolfo M. (1996) Science 271, 1423–1427 [DOI] [PubMed] [Google Scholar]
- 16. Punga T., Bühler M. (2010) EMBO Mol. Med. 2, 120–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Biase I., Chutake Y. K., Rindler P. M., Bidichandani S. I. (2009) PLoS One 4, e7914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strausberg R. L., Feingold E. A., Grouse L. H., Derge J. G., Klausner R. D., Collins F. S., Wagner L., Shenmen C. M., Schuler G. D., Altschul S. F., Zeeberg B., Buetow K. H., Schaefer C. F., Bhat N. K., Hopkins R. F., Jordan H., Moore T., Max S. I., Wang J., Hsieh F., Diatchenko L., Marusina K., Farmer A. A., Rubin G. M., Hong L., Stapleton M., Soares M. B., Bonaldo M. F., Casavant T. L., Scheetz T. E., Brownstein M. J., Usdin T. B., Toshiyuki S., Carninci P., Prange C., Raha S. S., Loquellano N. A., Peters G. J., Abramson R. D., Mullahy S. J., Bosak S. A., McEwan P. J., McKernan K. J., Malek J. A., Gunaratne P. H., Richards S., Worley K. C., Hale S., Garcia A. M., Gay L. J., Hulyk S. W., Villalon D. K., Muzny D. M., Sodergren E. J., Lu X., Gibbs R. A., Fahey J., Helton E., Ketteman M., Madan A., Rodrigues S., Sanchez A., Whiting M., Madan A., Young A. C., Shevchenko Y., Bouffard G. G., Blakesley R. W., Touchman J. W., Green E. D., Dickson M. C., Rodriguez A. C., Grimwood J., Schmutz J., Myers R. M., Butterfield Y. S., Krzywinski M. I., Skalska U., Smailus D. E., Schnerch A., Schein J. E., Jones S. J., Marra M. A. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16899–16903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mishina M., Kurosaki T., Yamamoto T., Notake M., Masu M., Numa S. (1982) EMBO J. 1, 1533–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sassone-Corsi P., Corden J., Kédinger C., Chambon P. (1981) Nucleic Acids Res. 9, 3941–3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smale S. T., Baltimore D. (1989) Cell 57, 103–113 [DOI] [PubMed] [Google Scholar]
- 22. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 23. Santos-Rosa H., Schneider R., Bannister A. J., Sherriff J., Bernstein B. E., Emre N. C., Schreiber S. L., Mellor J., Kouzarides T. (2002) Nature 419, 407–411 [DOI] [PubMed] [Google Scholar]
- 24. Schneider R., Bannister A. J., Myers F. A., Thorne A. W., Crane-Robinson C., Kouzarides T. (2004) Nat. Cell Biol. 6, 73–77 [DOI] [PubMed] [Google Scholar]
- 25. Lee J. H., Skalnik D. G. (2008) Mol. Cell Biol. 28, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee J. H., Tate C. M., You J. S., Skalnik D. G. (2007) J. Biol. Chem. 282, 13419–13428 [DOI] [PubMed] [Google Scholar]
- 27. Wang P., Lin C., Smith E. R., Guo H., Sanderson B. W., Wu M., Gogol M., Alexander T., Seidel C., Wiedemann L. M., Ge K., Krumlauf R., Shilatifard A. (2009) Mol. Cell Biol. 29, 6074–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu M., Wang P. F., Lee J. S., Martin-Brown S., Florens L., Washburn M., Shilatifard A. (2008) Mol. Cell Biol. 28, 7337–7344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Komarnitsky P., Cho E. J., Buratowski S. (2000) Genes Dev. 14, 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirakata M., Okano Y., Pati U., Suwa A., Medsger T. A., Jr., Hardin J. A., Craft J. (1993) J. Clin. Invest. 91, 2665–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krogan N. J., Kim M., Tong A., Golshani A., Cagney G., Canadien V., Richards D. P., Beattie B. K., Emili A., Boone C., Shilatifard A., Buratowski S., Greenblatt J. (2003) Mol. Cell Biol. 23, 4207–4218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greene E., Entezam A., Kumari D., Usdin K. (2005) Genomics 85, 221–230 [DOI] [PubMed] [Google Scholar]
- 33. Brookes E., Pombo A. (2009) EMBO Rep. 10, 1213–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li K., Besse E. K., Ha D., Kovtunovych G., Rouault T. A. (2008) Hum. Mol. Genet. 17, 2265–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stock J. K., Giadrossi S., Casanova M., Brookes E., Vidal M., Koseki H., Brockdorff N., Fisher A. G., Pombo A. (2007) Nat. Cell Biol. 9, 1428–1435 [DOI] [PubMed] [Google Scholar]
- 36. Rybtsova N., Leimgruber E., Seguin-Estévez Q., Dunand-Sauthier I., Krawczyk M., Reith W. (2007) Nucleic Acids Res. 35, 3431–3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shogren-Knaak M., Ishii H., Sun J. M., Pazin M. J., Davie J. R., Peterson C. L. (2006) Science 311, 844–847 [DOI] [PubMed] [Google Scholar]
- 38. Rai M., Soragni E., Chou C. J., Barnes G., Jones S., Rusche J. R., Gottesfeld J. M., Pandolfo M. (2010) PLoS One 5, e8825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xu C., Soragni E., Chou C. J., Herman D., Plasterer H. L., Rusche J. R., Gottesfeld J. M. (2009) Chem. Biol. 16, 980–989 [DOI] [PMC free article] [PubMed] [Google Scholar]