

Abstract
The C-type lectin-like receptor CLEC-2 signals via phosphorylation of a single cytoplasmic YXXL sequence known as a hem-immunoreceptor tyrosine-based activation motif (hemITAM). In this study, we show that phosphorylation of CLEC-2 by the snake toxin rhodocytin is abolished in the absence of the tyrosine kinase Syk but is not altered in the absence of the major platelet Src family kinases, Fyn, Lyn, and Src, or the tyrosine phosphatase CD148, which regulates the basal activity of Src family kinases. Further, phosphorylation of CLEC-2 by rhodocytin is not altered in the presence of the Src family kinase inhibitor PP2, even though PLCγ2 phosphorylation and platelet activation are abolished. A similar dependence of phosphorylation of CLEC-2 on Syk is also seen in response to stimulation by an IgG mAb to CLEC-2, although interestingly CLEC-2 phosphorylation is also reduced in the absence of Lyn. These results provide the first definitive evidence that Syk mediates phosphorylation of the CLEC-2 hemITAM receptor with Src family kinases playing a critical role further downstream through the regulation of Syk and other effector proteins, providing a new paradigm in signaling by YXXL-containing receptors.
Keywords: Cell Surface Receptor, Hemostasis, Mouse, Platelet, Receptor-Tyrosine Kinase, Signal Transduction, Src, Thrombosis, Tyrosine-Protein Kinase (Tyrosine Kinase), Tyrosine-Protein Phosphatase (Tyrosine Phosphatase)
Introduction
The C-type lectin-like receptor CLEC-2 is a type II transmembrane protein that is highly expressed on the surface of platelets and megakaryocytes (1, 2) and at a lower level on several other hematopoietic lineages, including monocytes and dendritic cells (3–5). The first ligand to be identified for CLEC-2 was the powerful snake venom toxin, rhodocytin, purified from the Malayan pit viper, Calloselasma rhodostoma (2). F(ab)2 antibody fragments of CLEC-2 antibodies were subsequently shown to mediate activation of human platelets (2, 6). The transmembrane protein podoplanin was identified as an endogenous ligand, inducing powerful activation of platelets and CLEC-2-expressing cell lines (2, 6–8). Podoplanin is expressed on the surface of a wide variety of cells such as kidney podocytes, lung type I alveolar cells, and lymphatic endothelial cells but is absent from vascular endothelial cells and platelets. The activation of CLEC-2 by podoplanin is essential for the separation of the lymphatic and blood vasculatures (9–12). In addition, the expression of podoplanin on the surface of certain tumors is implicated in the process of tumor metastasis through activation of CLEC-2 (8, 13). CLEC-2 has recently been reported to play a role in supporting platelet activation at arteriolar rates of flow on collagen in some (6, 11) but not all studies (14).
CLEC-2 is expressed on resting platelets as a noncovalent homodimer (15, 16) and signals through a single YXXL sequence in its cytoplasmic tail known as a hem-immunoreceptor tyrosine-based activation motif (hemITAM)8 (2, 15–17). The minimal signaling unit for activation of Syk by CLEC-2 has been proposed to be the cross-linking of two CLEC-2 dimers (15, 16), activating a similar signaling cascade to that of the platelet collagen receptor GPVI-FcRγ-chain complex, with critical roles for LAT (linker for activation of T cells), SLP-76 (Src homology 2 domain-containing leukocyte protein of 76 kDa), phosphoinositide 3-kinases, and phospholipase C (PLC) γ2 (2, 17). CLEC-2 signaling occurs in lipid rafts and is dependent on actin polymerization, release of the secondary mediators ADP and thromboxane A2, and activation of the small G protein Rac (18).
Phosphorylation of the GPVI-FcRγ-chain ITAM is mediated by Src family kinases leading to subsequent binding and activation of Syk. Thus, ITAM phosphorylation is preserved in the absence of Syk or the Syk inhibitor R406 (19, 20). The Src family kinases Lyn and Fyn mediate phosphorylation of the GPVI-FcRγ-chain ITAM (21–23). Fyn and Lyn bind to a proline-rich region in the cytosolic tail of GPVI via their Src homology 3 domains, allowing for rapid initiation of platelet activation (21, 24). In addition, they also support activation by a second pathway of activation that is independent of the GPVI proline-rich motif (25). Recently, it has been proposed that GPVI-associated Lyn is in an active conformation, suggesting that Lyn initiates GPVI-induced platelet activation, with Fyn supporting sustained GPVI signaling (23).
Src family kinases also play a role in CLEC-2-mediated signaling, because structurally distinct broad range Src family kinase inhibitors such as PP2 and PD 173956 inhibit human platelet activation (2, 17). However, it has recently been reported that phosphorylation of CLEC-2 in human platelets stimulated by rhodocytin is blocked by selective inhibitors of Src family or Syk tyrosine kinases, implicating these kinases in hemITAM phosphorylation (20). One potential explanation for these observations is that Syk mediates CLEC-2 phosphorylation and that Src family kinases regulate Syk. Thus, the proximal events in signaling by hemITAM receptors appear to be distinct from those used by ITAM receptors.
In the present study, we have investigated the molecular events underlying CLEC-2 phosphorylation in mouse platelets using mutant mice deficient in the major platelet Src family kinases (Fyn, Lyn, and Src), the tyrosine kinase Syk, and the global regulator of Src family kinases in platelets, CD148. The results provide the first demonstration that Syk and not Src family kinases mediate hemITAM phosphorylation and identify Src family kinases regulating platelet activation downstream of Syk.
EXPERIMENTAL PROCEDURES
Reagents
Rhodocytin was purified from C. rhodostoma venom as described previously (2). Rat anti-mouse CLEC-2 IgG mAb and rabbit anti-Syk (BR15) polyclonal antibody were from previously described sources (4, 26). CLEC-2 IgM antibody was a gift from Dr. Caetano Reis e Sousa. Hamster anti-mouse CD148 antibody (8A-1) was generated as described (27). Mouse anti-phosphotyrosine antibody 4G10 was purchased from Upstate Biotechnology Inc. pY1217 PLCγ2 antibody was from Cell Signaling Technology. HRP-conjugated secondary anti-rabbit IgG and γ-bind protein G-Sepharose were from GE Healthcare. Anti-rat Fc antibody was from Abcam. All other reagents were purchased from Sigma-Aldrich.
Animals
fyn−/−, lyn−/−, and src−/− breeding pairs were from Jackson Laboratories (Bar Harbor, ME). fyn−/− lyn−/− double knock-out mice were raised from mixed breeding of fyn−/− and lyn−/− mice. CD148−/− mice were generated as described previously (28). Experimental controls were wild-type littermates. No differences were observed in platelet function between the different wild-type controls. The mice were genotyped by PCR amplification of mouse tail genomic DNA. Lack of protein expression was confirmed by Western blotting platelet lysates. The mice were housed under specific pathogen-free conditions in accordance with institutional guidelines approved by United Kingdom Home Office and were used between 8 and 12 weeks of age.
Generation of Chimeric Mice
Radiation chimeras were generated as described previously (19). Briefly, 8-week-old C57BL/6 mice received two doses of irradiation each of 500 Gy 3 h apart. The mice were then reconstituted with an intravenous injection of 1–2 × 106 fetal liver cells obtained from embryonic days 14–16 lyn+/+ src+/+, lyn−/− src−/−, fyn+/+ src+/+, fyn−/− src−/−, syk+/+, syk−/−, and clec-2+/+ or clec-2−/− mouse livers. The genotype of the reconstituting fetal liver cells was confirmed in each case by PCR. Lack of protein expression was confirmed by Western blotting platelet lysates. Reconstituted mice were used for experiments at 8 weeks after irradiation.
Preparation of Mouse Platelets
Whole blood was drawn from the inferior vena cava into acid citrate dextrose ( volume) from CO2-asphyxiated mice following isofluorane anesthesia. Washed mouse platelets were obtained as described previously (29). Briefly, platelets were obtained by centrifugation using prostaglandin I2 to prevent activation during the isolation procedure. Washed platelets were then resuspended in modified HEPES Tyrode buffer (134 mm NaCl, 2.9 mm KCl, 12 mm NaHCO3, 0.34 mm NaH2PO4, 1 mm MgCl2, 5.5 mm glucose, 1 mm MgCl2, 20 mm HEPES, and 5 mm glucose, pH 7.3) at a density of 2 × 108 platelets/ml. Aggregation was monitored by light transmission using a Born lumi-aggregometer (Chronolog, Havertown, PA).
Immunoprecipitation Studies
Washed resting or stimulated platelets (300 μl at 2 × 108 platelets/ml) were lysed with an equal volume of 2× Nonidet P-40 lysis buffer (20 mm Tris-HCl, pH 7.6, 300 mm NaCl, 2 mm EGTA, 2 mm EDTA, 2% Nonidet P-40, 2 mm PMSF, 5 mm Na3VO4, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μg/ml pepstatin). Cell debris was removed by centrifugation at 15,000 × g for 10 min. An aliquot of whole cell lysate was dissolved in reducing Laemmli sample buffer. Syk immunoprecipitation was carried out as standard protocol (30, 31). Platelet lysate was precleared, 2 μl of anti-Syk antibody and a 15-μl bed volume of protein A-Sepharose were added, and each sample was rotated at 4 °C for 2 h. The pellet was washed four times sequentially in lysis buffer before the addition of 20 μl of reducing Laemmli sample buffer. Modifications from standard immunoprecipitation protocols were made for mouse CLEC-2 immunoprecipitations. The amount of CLEC-2 mAb in each sample was normalized to 3 μg. 20 μl of bed volume of γ-bind protein G-Sepharose was added to each sample and allowed to capture the antibody for 1 h with rotation at 4 °C. Following four washes with 1× lysis buffer, 20 μl of nonreducing Laemmli sample buffer was added. 75% of the sample proteins was separated on a SDS-polyacrylamide gel and transferred onto a PVDF membrane. After blocking in 2% BSA, the membranes were incubated with 4G10 antibody overnight, washed, and then incubated with HRP-conjugated secondary antibody. Immunoprecipitated proteins were visualized by chemoluminescence (ECL; Pierce). For CLEC-2 immunoprecipitations, the remaining 25% was treated in the same way but incubated with CLEC-2 mAb overnight.
Platelet Surface Protein Cross-linking
After platelet stimulation, 1.5 mm Sulfo-EGS was added and allowed to incubate at room temperature for 30 min. The reaction was quenched with the addition of Tris-HCl (pH 7.5; 25 mm) and allowed to incubate for a further 20 min at room temperature. The samples were lysed with the addition of an equal volume of 2× ice-cold Nonidet P-40 lysis buffer.
Cell Lines
CHO cells were transfected with pcDNA3 containing full-length mouse podoplanin using a calcium phosphate transfection method. Stable transfectants were obtained using medium containing 1 mg/ml geneticin (G418), and clonal cell populations were obtained following serial dilutions into 96-well plates. Primary human lymphatic endothelial cells were obtained from Promocell GmbH (Heidelberg, Germany) and cultured in endothelial cell growth medium according to the manufacturer instructions. Surface expression of podoplanin was assessed by flow cytometry.
Statistical Analysis
Statistical significance was evaluated using a two-tailed Student's t test. A p value <0.05 was considered statistically significant.
RESULTS
Differential Role of Src Family Kinases in Platelet Activation by a CLEC-2 Monoclonal Antibody and by Rhodocytin
A rat anti-mouse IgG CLEC-2 monoclonal antibody (CLEC-2 mAb) induces concentration-dependent CLEC-2 phosphorylation and platelet aggregation, which is abolished in CLEC-2-deficient mouse platelets (Fig. 1). The time to onset of aggregation decreases with increasing concentrations of antibody (Fig. 1B). Rhodocytin also stimulated concentration-dependent phosphorylation with the delay in onset also decreasing with increasing concentrations (Fig. 1, B and C). Concentrations of CLEC-2 mAb (10 μg/ml) and rhodocytin (30 nm) that were just sufficient to induce maximal CLEC-2 phosphorylation and aggregation were chosen for further experiments, unless stated.
FIGURE 1.

Dose response curves in response to CLEC-2 mAb and rhodocytin. A, CLEC-2-deficient and litter-matched wild-type washed platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb. B, murine washed platelets (2 × 108 platelets/ml) were stimulated with increasing concentrations of CLEC-2 mAb or rhodocytin. Platelet aggregation was measured as a change in light transmission, using a lumi-aggregometer. Representative aggregation traces are shown. The addition of the agonist is indicated by an arrowhead. The data represent the means and standard error of at least eight independent experiments at 3 min of stimulation. C, CLEC-2 was immunoprecipitated (IP), and immunoprecipitates were immunoblotted with an anti-phosphotyrosine antibody and anti-CLEC-2 mAb as described under “Experimental Procedures.” The percentage of tyrosine phosphorylation was measured at 3 min of stimulation and is represented as the means and standard error of four independent experiments. WB, Western blotting.
We have previously reported that CLEC-2-mediated activation of human platelets is abolished in the presence of Src family kinase inhibitors (2, 17). Consistent with this, we show that the Src family kinase inhibitor PP2 also blocks aggregation of mouse platelets stimulated by CLEC-2 mAb and by rhodocytin (Fig. 2A), confirming a critical role for Src family kinases in CLEC-2-induced platelet activation. Tyrosine phosphorylation of both CLEC-2 and the downstream kinase Syk is completely blocked by PP2 in response to CLEC-2 mAb. Strikingly, however, phosphorylation of CLEC-2 by rhodocytin is not altered in the presence of PP2 (Fig. 2B). The inhibition of aggregation to the snake toxin by PP2 (Fig. 2A) is explained by the partial reduction in Syk phosphorylation and inhibition of tyrosine phosphorylation of residue Tyr-1217 of the downstream protein PLCγ2 (Fig. 2B), a residue shown to be correlated with its activity (32). These results demonstrate that Src family kinases do not mediate phosphorylation of CLEC-2 in mouse platelets stimulated by rhodocytin but that they play a role in the hemITAM phosphorylation induced by a CLEC-2 mAb.
FIGURE 2.

Effect of Src family kinase inhibition on murine platelet in response to CLEC-2 mAb and rhodocytin. A, murine washed platelets (2 × 108 platelets/ml) were incubated for 10 min with or without 20 μm PP2 and then stimulated with 10 μg/ml CLEC-2 mAb or 30 nm rhodocytin. Platelet aggregation was measured as a change in light transmission using a lumi-aggregometer. The addition of the agonist is indicated by an arrowhead. Representative aggregation traces from three to six independent experiments are shown. B, CLEC-2 and Syk were immunoprecipitated (IP), and immunoprecipitates were immunoblotted with an anti-phosphotyrosine antibody. CLEC-2 immunoprecipitates were also immunoblotted with anti-CLEC-2 mAb as described under “Experimental Procedures.” Whole cell lysates (WCL) were probed with pY1217 PLCγ2 antibody and reprobed with PLCγ2 antibodies. The percentage of tyrosine phosphorylation was measured at 3 min of stimulation and is represented as the means and standard error of three to six independent experiments. *, p < 0.05; **, p < 0.005 (significant difference according to two-tailed Student's t test). WB, Western blotting.
Distinct Roles for Fyn, Lyn, and Src in Platelet Activation by CLEC-2
The major Src family kinases known to be expressed in mouse platelets are Fyn, Lyn, and Src, although there is evidence for the presence of other isoforms (33, 34). To dissect the role of individual Src family kinases in CLEC-2-mediated signaling, we used mice deficient in the three major platelet Src family kinases, Fyn, Lyn, or Src, after first establishing that the absence of one Src family kinase did not alter expression of the others (supplemental Fig. S1). CLEC-2 and αIIbβ3 surface expression in platelets deficient in each of the Src family kinases was similar to that of their litter-matched controls (not shown).
Aggregation of platelets deficient in Src or Fyn induced by CLEC-2 mAb (3 and 10 μg/ml) or by rhodocytin (10 and 30 nm) were similar to those of litter-matched controls (Fig. 3, A–C and not shown). In contrast, there was a marked delay in the onset of aggregation to 10 μg/ml CLEC-2 mAb in Lyn-deficient platelets, with 50% aggregation being reached at 250 ± 18 s compared with 140 ± 6 s for control platelets (Fig. 3, A and C). There was, however, no significant change in the time of onset or magnitude of aggregation induced by rhodocytin (10 and 30 nm) in Lyn-deficient platelets (Fig. 3, B and C, and not shown).
FIGURE 3.

Lyn plays a critical role in CLEC-2-mediated signaling by antibody ligation but is not involved in response to rhodocytin. Fyn-, Lyn-, or Src-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb (A) or 30 nm rhodocytin (B). Platelet aggregation was measured as a change in light transmission, using a lumi-aggregometer. Representative aggregation traces are shown. The addition of the agonist is indicated by an arrowhead. C, data represent the means of the time to get 50% of aggregation and standard error of three to six independent experiments. **, p < 0.005 (significant difference versus wild type, according to two-tailed Student's t test). D, Fyn- or Lyn-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb or 30 nm rhodocytin for 3 min. CLEC-2 and Syk were immunoprecipitated (IP), and immunoprecipitates were immunoblotted with an anti-phosphotyrosine antibody. CLEC-2 immunoprecipitates were also immunoblotted with anti-CLEC-2 antibody as described under “Experimental Procedures.” The percentage of tyrosine phosphorylation was measured at 3 min of stimulation and represented as the means and standard error of three to six independent experiments. **, p < 0.005 (significant difference versus wild type, according to two-tailed Student's t test). WB, Western blotting.
Phosphorylation of CLEC-2 and Syk in response to the CLEC-2 mAb was abolished at 180 s in the absence of Lyn in contrast to the robust response observed in control platelets. This is consistent with the delay in platelet activation observed in Lyn-deficient platelets. Phosphorylation of CLEC-2 can, however, be seen following full aggregation of Lyn-deficient platelets (not shown). In contrast, a small reduction in CLEC-2 and Syk tyrosine phosphorylation was observed in response to the CLEC-2 mAb in Fyn-deficient platelets in some but not all experiments, although this did not reach statistical significance (Fig. 3D). There was no change in phosphorylation of CLEC-2 following CLEC-2 mAb stimulation in the absence of Src (not shown). Consistent with the results obtained with PP2 (Fig. 2B), phosphorylation of CLEC-2 induced by rhodocytin was not altered in the absence of Src, Fyn, or Lyn (Fig. 3D and not shown). These data demonstrate that Lyn is the major kinase involved in CLEC-2 platelet activation following CLEC-2 mAb ligation.
To investigate whether platelet activation is further delayed in the absence of more than one Src family kinases, we generated mice doubly deficient in Fyn/Lyn, Lyn/Src, and Fyn/Src. Mice doubly deficient in Fyn/Lyn were generated by breeding of heterozygotes, whereas the other two sets of double knock-outs were generated as radiation chimeras as described under “Experimental Procedures.” As shown in Fig. 4A, the loss of two of the major platelet Src family kinases did not alter expression of the remaining major Src family kinases. Interestingly, a further delay in the onset of aggregation in platelets from a mouse doubly deficient in Fyn/Lyn or Lyn/Src stimulated with CLEC-2 mAb was observed (Fig. 4, B and D) when compared with Lyn-deficient platelets (Fig. 3, A and C). In contrast, platelets doubly deficient in Fyn/Src exhibited only a very minor delay in onset of platelet aggregation (Fig. 4, B and D). CLEC-2 phosphorylation was abolished in platelets deficient in Fyn/Lyn and Lyn/Src at 180 s (not shown), as was the case for Lyn-deficient platelets (Fig. 3D), but was unchanged in platelets deficient in Fyn/Src (Fig. 4E and not shown). These data demonstrate that Fyn and Src contribute to platelet activation by the CLEC-2 mAb in the absence of Lyn but that neither plays a significant role in the presence of Lyn. In contrast, the response to rhodocytin was not altered in mouse platelets doubly deficient in Fyn/Lyn, Lyn/Src, or Fyn/Src. Given that platelet activation by both CLEC-2 mAb and by rhodocytin is abolished in the presence of the broad spectrum inhibitor PP2, these results demonstrate that there are other Src family kinases that contribute to platelet activation downstream of CLEC-2 and that there may be considerable redundancy between Src family kinases in mediating this effect.
FIGURE 4.

A, whole cell lysates prepared from deficient platelets and their litter-matched wild-type platelets were immunoblotted with anti-Lyn, anti-Fyn, anti-Src, and anti-tubulin antibodies. B and C, washed platelets deficient in Fyn/Lyn, Fyn/Src, and Lyn/Src (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb (B) or 30 nm rhodocytin (C). Platelet aggregation was measured as a change in light transmission, using a lumi-aggregometer. Representative aggregation traces from three independent experiments are shown. The addition of the agonist is indicated by an arrowhead. D, data represent the means of the time to get 50% of aggregation and standard error of three independent experiments. **, p < 0.005 (significant difference versus wild type, according to two-tailed Student's t test). E, Fyn/Src-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb for 3 min. CLEC-2 was immunoprecipitated (IP), and immunoprecipitates were immunoblotted with an anti-phosphotyrosine antibody. CLEC-2 immunoprecipitates were also immunoblotted with anti-CLEC-2 antibody as described under “Experimental Procedures.” Representative data from two independent experiments are shown. WB, Western blotting.
CD148 Is Dispensable for Platelet Activation by CLEC-2 Ligation
Src family kinases are regulated by phosphorylation on excitatory and inhibitory tyrosines. Recently, we have demonstrated a reciprocal increase in phosphorylation of the inhibitory site and a decrease in phosphorylation of the activatory site of all Src family kinases in platelets deficient in the receptor-like protein tyrosine phosphatase CD148. This has a net effect of greatly reducing the activity of Src family kinases, leading to a marked inhibition of platelet activation by the ITAM receptor GPVI and by integrin αIIbβ3 (35). In contrast, activation of mouse platelets by CLEC-2 mAb (3 and 10 μg/ml) or by rhodocytin (10 and 30 nm) is not significantly altered in the absence of CD148 (Fig. 5 and not shown), demonstrating that Src family kinase activity is not rate-limiting for platelet activation by either of the CLEC-2 receptor ligands. This therefore provides further evidence for a fundamental difference in the proximal events underlying platelet activation by CLEC-2 from those of the platelet ITAM receptor GPVI.
FIGURE 5.

CD148 deficiency does not impair CLEC-2 signaling. A, CD148-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb or 30 nm rhodocytin. Platelet aggregation was measured as a change in light transmission, using a lumi-aggregometer. Representative aggregation traces of four independent experiments are shown. The addition of the agonist is indicated by an arrowhead. B, CD148-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb or 30 nm rhodocytin for 3 min. CLEC-2 and Syk were immunoprecipitated (IP), and immunoprecipitates were immunoblotted with an anti-phosphotyrosine antibody. CLEC-2 immunoprecipitates were also immunoblotted with anti-CLEC-2 mAb as described under “Experimental Procedures.” The percentage of tyrosine phosphorylation was measured at 3 min of stimulation and represented as means and standard error of four independent experiments. WB, Western blotting.
The Tyrosine Kinase Syk Underlies Phosphorylation of CLEC-2
The role of the tyrosine kinase Syk in mediating CLEC-2 phosphorylation and platelet aggregation was investigated using Syk-deficient chimeric mice (Fig. 6A). Phosphorylation of CLEC-2 and aggregation of platelets induced by the CLEC-2 mAb or by rhodocytin were completely blocked in Syk-deficient platelets (Fig. 6, B and C), demonstrating that Syk is essential for phosphorylation of the CLEC-2 hemITAM. Thus, the role of Lyn in mediating phosphorylation of CLEC-2 in response to the CLEC-2 mAb may be mediated via phosphorylation of Syk (Fig. 3D), which is regulated by both autophosphorylation and phosphorylation by Src family kinases (36).
FIGURE 6.
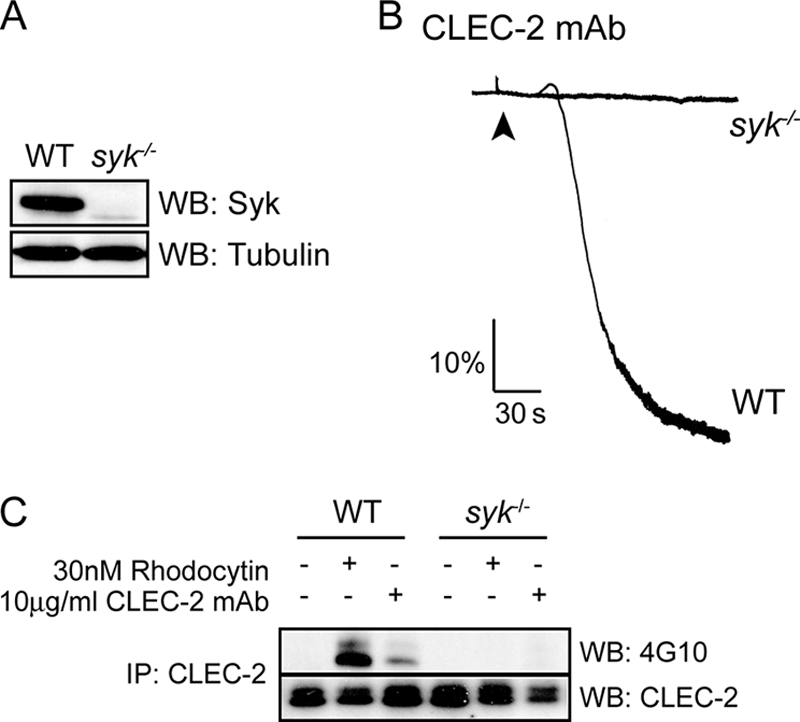
Syk is a key regulator of CLEC-2 signaling. A, whole cell lysates prepared from wild-type and Syk-deficient platelets were immunoblotted with anti-Syk and anti-tubulin antibodies. B, Syk-deficient washed platelets and their litter-matched control platelets (2 × 108 platelets/ml) were stimulated with 10 μg/ml CLEC-2 mAb. Platelet aggregation was measured as a change in light transmission, using a lumi-aggregometer. Representative aggregation traces of three independent experiments are shown. The addition of the agonist is indicated by an arrowhead. C, after 3 min of stimulation with 10 μg/ml CLEC-2 mAb, CLEC-2 was immunoprecipitated (IP) from Syk-deficient platelets and their litter-matched control platelets (2 × 108 platelets/ml) lysates and immunoblotted with an anti-phosphotyrosine antibody and anti-CLEC-2 mAb as described under “Experimental Procedures.” A representative blot of three independent experiments is shown. WB, Western blotting.
Role of Src Family Kinases in CLEC-2 Signaling Induced by Podoplanin-expressing Cell Lines and Other Multivalent Ligands
The differential role of Src family kinases in mediating phosphorylation of CLEC-2 by CLEC-2 mAb and by rhodocytin could reflect the degree of cross-linking of CLEC-2 (37, 38). Thus, the tetrameric rhodocytin (37) is able to cluster CLEC-2 to a much greater degree than the dimeric IgG mAb. This is illustrated in platelets treated with the chemical cross-linker SulfoEGS (Fig. 7A). Rhodocytin induced a much greater reduction in the monomeric form of CLEC-2 relative to the CLEC-2 mAb, whereas neither ligand induced clustering of the membrane tyrosine phosphatase CD148 (not shown), thereby demonstrating that this effect was not due to nonspecific cross-linking.
FIGURE 7.

Role of Src family kinases in CLEC-2-mediated aggregation following multivalent ligands and podoplanin ligation. A, washed platelets (2 × 108/ml) under basal, CLEC-2 mAb (10 μg/ml) or rhodocytin-stimulated (30 nm) conditions had their surface proteins cross-linked with the addition of 1.5 mm Sulfo-EGS cross-linking reagent, with a linker length of 1.6 nm (16 Å) as described under “Experimental Procedures.” Monomeric CLEC-2 was quantified, and the data represent the means and standard error of three independent experiments. **, p < 0.005 (significant difference versus wild type, according to two-tailed Student's t test). mAb, CLEC-2 mAb; Rh, rhodocytin. B, Lyn-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) were stimulated with 2 μg/ml CLEC-2 mAb for 2 min and then with 10 μg/ml anti-rat Fc antibody for 5 min. Representative aggregation traces of three independent experiments are shown. The addition of the agonists is indicated by an arrowhead. C, Lyn-deficient washed platelets and their litter-matched wild-type platelets (2 × 108 platelets/ml) incubated with or without 20 μm PP2 were stimulated with IgM CLEC-2 antibody. The data represent the means of the time to get 50% of aggregation and standard error of three to six independent experiments. *, p < 0.05 (significant difference versus wild type, according to two-tailed Student's t test). D, Lyn-deficient platelets and their litter-matched control platelets (2 × 108 platelets/ml) incubated with or without 20 μm PP2 were stimulated in plasma with 1.5 × 105 lymphatic endothelial cells (hLEC) or mouse podoplanin expressing CHO cells (mPodoCHO). Platelet aggregation was measured as a change in light transmission, using a lumi-aggregometer. Representative aggregation traces of three independent experiments are shown. The addition of the agonist is indicated by an arrowhead.
In light of this result, we asked whether cross-linking of the CLEC-2 mAb using an anti-rat Fc antibody would induce a more powerful degree of platelet activation and whether this would reduce the dependence on Lyn. The addition of a cross-linking anti-rat Fc antibody to a concentration of CLEC-2 mAb that induced shape change converted the response to a rapid aggregation. Interestingly, a delay in aggregation was still observed in the absence of Lyn following cross-linking by an anti-rat Fc antibody (Fig. 7B) and also in response to a novel pentavalent IgM anti-CLEC-2 antibody (Fig. 7C), although in both cases this was less than in response to the IgG antibody (Fig. 3A). Aggregation to the IgM antibody was abolished in the presence of the Src family kinase inhibitor, PP2 (Fig. 7C).
To investigate the dependence of platelet aggregation to podoplanin on Lyn, we used two podoplanin-expressing cell lines, namely mouse podoplanin-transfected CHO cells and human lymphatic endothelial cells. Neither cell line induced activation of platelets deficient in CLEC-2, confirming that activation was via CLEC-2 (not shown). The two cell lines induce rapid mouse platelet activation, which was blocked in the presence of PP2, confirming an essential role for Src family kinases in mediating activation. However, the response to either of the cell lines was not altered in the absence of Lyn (Fig. 7D). These data show that the dependence on Lyn is agonist-dependent, with the dependence being reduced or abolished for ligands that induce oligomerization of CLEC-2.
DISCUSSION
The present study provides an unequivocal demonstration that the tyrosine kinase Syk mediates CLEC-2 phosphorylation following stimulation by rhodocytin and that this is independent of Src family kinase activation. Phosphorylation of CLEC-2 in response to the snake toxin is unaltered in platelets deficient or doubly deficient in the major platelet Src family kinases, namely Fyn, Lyn, and Src, or in the global regulator of Src kinases, the membrane tyrosine phosphatase CD148. The ability of broad spectrum Src family kinase inhibitors such as PP2 to block platelet aggregation by rhodocytin is explained by partial or complete inhibition of phosphorylation of Syk and downstream proteins such as PLCγ2, respectively. The fact that platelet aggregation by rhodocytin is not altered in mice deficient or doubly deficient in the above Src family kinases demonstrates that one or more other Src family kinases must play a role in mediating aggregation or that there is considerable redundancy between the Src family kinases in mediating activation. These results reveal a new pathway of platelet activation by the hemITAM receptor CLEC-2, which is distinct from that mediated by the ITAM receptor, GPVI. CLEC-2 signals through the sequential action of Syk and Src family kinases, whereas GPVI induces sequential activation of Src family and Syk kinases, with Src family kinases playing a further role downstream of Syk.
The present study also shows that the role of individual members of Src family kinases in CLEC-2 signaling is ligand-dependent. Thus, phosphorylation of CLEC-2 in response to the IgG CLEC-2 antibody is markedly delayed in the absence of Lyn and further delayed in the combined absence of Fyn/Lyn or Lyn/Src. On the other hand, phosphorylation is not altered in mice doubly deficient in Fyn/Src, demonstrating that neither of these Src family kinases plays a critical role in the presence of Lyn. The delay in activation seen in the absence of Lyn is likely to be mediated by a loss of phosphorylation of Syk, which is regulated by Src family kinases and regulates hemITAM phosphorylation. In contrast, activation of platelets by rhodocytin is not altered in mice that are either single or doubly deficient in Fyn, Lyn, or Src, whereas the response is abolished in the absence of Syk. Platelet activation by two podoplanin-expressing cell lines resembles the mechanism underlying activation by rhodocytin in that activation is also independent of Lyn but blocked by the Src kinase inhibitor, PP2.
The differential role of Lyn and other Src family kinases in mediating platelet activation by rhodocytin, podoplanin-expressed cell lines, and the CLEC-2 mAb may be related to the degree of clustering of CLEC-2. The antibody to CLEC-2 is an IgG and thus induces dimerization of the receptor, generating a weak intracellular signal. The further clustering of CLEC-2 with a secondary antibody induces rapid and powerful activation. In contrast, the tetrameric rhodocytin and polymeric podoplanin-expressing cells induce a much greater degree of receptor clustering. The differential dependence on Lyn may also be due to spatial aspects of the ligand-CLEC-2 interaction, because this may influence the degree of Syk recruitment.
We have recently reported that phosphorylation of CLEC-2 in human platelets in response to rhodocytin is abolished in the presence of inhibitors of Src or Syk kinases (20). It was unclear, however, from this study which of these kinases mediated receptor phosphorylation. The demonstration that phosphorylation of CLEC-2 in mouse platelets by rhodocytin is abolished in the absence of Syk but not in the presence of the Src kinase inhibitor PP2 shows that the hemITAM phosphorylation is mediated by Syk. The explanation for the differing dependence of CLEC-2 phosphorylation on Src family kinases in the two species could reflect, for example, a difference in the expression levels of CLEC-2 or of Src family and Syk kinases.
An important question that emerges from this work is how receptor clustering of CLEC-2 brings about activation of Syk. One explanation is that it is brought about as a result of mass action as modeled by Cooper and Qian (39). The key feature of this model is that a constitutively active kinase is associated with the receptor in the absence of agonist stimulation. Consistent with this, we have previously reported constitutive activation of Syk in nonstimulated platelets and transfected cell lines (40). Thus, although there may be other components that regulate Src family and Syk kinases upon ligand engagement, this model predicts that clustering alone is sufficient to mediate receptor phosphorylation. The reason why phosphorylation of CLEC-2 is mediated by Syk rather than by one or more other Src family kinases, as is the case for GPVI-FcRγ-chain ITAM, may be related to their juxtaposition in the cell. For example, we have recently shown that Syk is localized to plasma membrane in platelets by binding to filamin A and that a loss of filamin A markedly impairs signaling by the tyrosine kinase (41).
In summary, this study demonstrates for the first time that Syk mediates phosphorylation of the CLEC-2 hemITAM and that Src family kinases play a critical role further downstream through regulation of Syk and other effector proteins including PLCγ2. It is now of considerable interest to establish whether this pathway is unique to CLEC-2 or whether it is also used by other hemITAM receptors.
Supplementary Material
Acknowledgments
We thank Reis e Sousa for the CLEC-2 antibodies. We thank Drs Craig E. Hughes and Steve G. Thomas who provided cells for the chimeric mice, Ian Ricketts and Jenny Ullah in the Biomedical Service Unit for generating the radiation chimeric mice and maintaining the mouse colonies, Dr. Brenda L. Finney for help with the platelet studies, and Dr. Beata Grygielska for technical support.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
- ITAM
- immunoreceptor tyrosine-based activation motif
- GP
- glycoprotein
- PLC
- phospholipase C.
REFERENCES
- 1. Senis Y. A., Tomlinson M. G., García A., Dumon S., Heath V. L., Herbert J., Cobbold S. P., Spalton J. C., Ayman S., Antrobus R., Zitzmann N., Bicknell R., Frampton J., Authi K. S., Martin A., Wakelam M. J., Watson S. P. (2007) Mol. Cell Proteomics 6, 548–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Suzuki-Inoue K., Fuller G. L., García A., Eble J. A., Pöhlmann S., Inoue O., Gartner T. K., Hughan S. C., Pearce A. C., Laing G. D., Theakston R. D., Schweighoffer E., Zitzmann N., Morita T., Tybulewicz V. L., Ozaki Y., Watson S. P. (2006) Blood 107, 542–549 [DOI] [PubMed] [Google Scholar]
- 3. Colonna M., Samaridis J., Angman L. (2000) Eur. J. Immunol. 30, 697–704 [DOI] [PubMed] [Google Scholar]
- 4. Kerrigan A. M., Dennehy K. M., Mourão-Sá D., Faro-Trindade I., Willment J. A., Taylor P. R., Eble J. A., Reis e Sousa C., Brown G. D. (2009) J. Immunol. 182, 4150–4157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sobanov Y., Bernreiter A., Derdak S., Mechtcheriakova D., Schweighofer B., Düchler M., Kalthoff F., Hofer E. (2001) Eur. J. Immunol. 31, 3493–3503 [DOI] [PubMed] [Google Scholar]
- 6. May F., Hagedorn I., Pleines I., Bender M., Vögtle T., Eble J., Elvers M., Nieswandt B. (2009) Blood 114, 3464–3472 [DOI] [PubMed] [Google Scholar]
- 7. Christou C. M., Pearce A. C., Watson A. A., Mistry A. R., Pollitt A. Y., Fenton-May A. E., Johnson L. A., Jackson D. G., Watson S. P., O'Callaghan C. A. (2008) Biochem. J. 411, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Suzuki-Inoue K., Kato Y., Inoue O., Kaneko M. K., Mishima K., Yatomi Y., Yamazaki Y., Narimatsu H., Ozaki Y. (2007) J. Biol. Chem. 282, 25993–26001 [DOI] [PubMed] [Google Scholar]
- 9. Bertozzi C. C., Schmaier A. A., Mericko P., Hess P. R., Zou Z., Chen M., Chen C. Y., Xu B., Lu M. M., Zhou D., Sebzda E., Santore M. T., Merianos D. J., Stadtfeld M., Flake A. W., Graf T., Skoda R., Maltzman J. S., Koretzky G. A., Kahn M. L. (2010) Blood 116, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schacht V., Ramirez M. I., Hong Y. K., Hirakawa S., Feng D., Harvey N., Williams M., Dvorak A. M., Dvorak H. F., Oliver G., Detmar M. (2003) EMBO J. 22, 3546–3556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Suzuki-Inoue K., Inoue O., Ding G., Nishimura S., Hokamura K., Eto K., Kashiwagi H., Tomiyama Y., Yatomi Y., Umemura K., Shin Y., Hirashima M., Ozaki Y. (2010) J. Biol. Chem. 285, 24494–24507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uhrin P., Zaujec J., Breuss J. M., Olcaydu D., Chrenek P., Stockinger H., Fuertbauer E., Moser M., Haiko P., Fässler R., Alitalo K., Binder B. R., Kerjaschki D. (2010) Blood 115, 3997–4005 [DOI] [PubMed] [Google Scholar]
- 13. Kunita A., Kashima T. G., Morishita Y., Fukayama M., Kato Y., Tsuruo T., Fujita N. (2007) Am. J. Pathol. 170, 1337–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hughes C. E., Navarro-Nunez L., Finney B. A., Mourao-Sa D., Pollitt A. Y., Watson S. P. (2010) J. Thromb. Haemostasis 8, 2328–2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hughes C. E., Pollitt A. Y., Mori J., Eble J. A., Tomlinson M. G., Hartwig J. H., O'Callaghan C. A., Fütterer K., Watson S. P. (2010) Blood 115, 2947–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watson A. A., Christou C. M., James J. R., Fenton-May A. E., Moncayo G. E., Mistry A. R., Davis S. J., Gilbert R. J., Chakera A., O'Callaghan C. A. (2009) Biochemistry 48, 10988–10996 [DOI] [PubMed] [Google Scholar]
- 17. Fuller G. L., Williams J. A., Tomlinson M. G., Eble J. A., Hanna S. L., Pöhlmann S., Suzuki-Inoue K., Ozaki Y., Watson S. P., Pearce A. C. (2007) J. Biol. Chem. 282, 12397–12409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pollitt A. Y., Grygielska B., Leblond B., Désiré L., Eble J. A., Watson S. P. (2010) Blood 115, 2938–2946 [DOI] [PubMed] [Google Scholar]
- 19. Poole A., Gibbins J. M., Turner M., van Vugt M. J., van de Winkel J. G., Saito T., Tybulewicz V. L., Watson S. P. (1997) EMBO J. 16, 2333–2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spalton J. C., Mori J., Pollitt A. Y., Hughes C. E., Eble J. A., Watson S. P. (2009) J. Thromb. Haemost. 7, 1192–1199 [DOI] [PubMed] [Google Scholar]
- 21. Ezumi Y., Shindoh K., Tsuji M., Takayama H. (1998) J. Exp. Med. 188, 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quek L. S., Pasquet J. M., Hers I., Cornall R., Knight G., Barnes M., Hibbs M. L., Dunn A. R., Lowell C. A., Watson S. P. (2000) Blood 96, 4246–4253 [PubMed] [Google Scholar]
- 23. Schmaier A. A., Zou Z., Kazlauskas A., Emert-Sedlak L., Fong K. P., Neeves K. B., Maloney S. F., Diamond S. L., Kunapuli S. P., Ware J., Brass L. F., Smithgall T. E., Saksela K., Kahn M. L. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 21167–21172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki-Inoue K., Tulasne D., Shen Y., Bori-Sanz T., Inoue O., Jung S. M., Moroi M., Andrews R. K., Berndt M. C., Watson S. P. (2002) J. Biol. Chem. 277, 21561–21566 [DOI] [PubMed] [Google Scholar]
- 25. Bori-Sanz T., Inoue K. S., Berndt M. C., Watson S. P., Tulasne D. (2003) J. Biol. Chem. 278, 35914–35922 [DOI] [PubMed] [Google Scholar]
- 26. Clark E. A., Shattil S. J., Ginsberg M. H., Bolen J., Brugge J. S. (1994) J. Biol. Chem. 269, 28859–28864 [PubMed] [Google Scholar]
- 27. Lin J., Zhu J. W., Baker J. E., Weiss A. (2004) J. Immunol. 173, 2324–2330 [DOI] [PubMed] [Google Scholar]
- 28. Zhu J. W., Brdicka T., Katsumoto T. R., Lin J., Weiss A. (2008) Immunity 28, 183–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Suzuki-Inoue K., Inoue O., Frampton J., Watson S. P. (2003) Blood 102, 1367–1373 [DOI] [PubMed] [Google Scholar]
- 30. McCarty O. J., Larson M. K., Auger J. M., Kalia N., Atkinson B. T., Pearce A. C., Ruf S., Henderson R. B., Tybulewicz V. L., Machesky L. M., Watson S. P. (2005) J. Biol. Chem. 280, 39474–39484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pearce A. C., Senis Y. A., Billadeau D. D., Turner M., Watson S. P., Vigorito E. (2004) J. Biol. Chem. 279, 53955–53962 [DOI] [PubMed] [Google Scholar]
- 32. Watanabe D., Hashimoto S., Ishiai M., Matsushita M., Baba Y., Kishimoto T., Kurosaki T., Tsukada S. (2001) J. Biol. Chem. 276, 38595–38601 [DOI] [PubMed] [Google Scholar]
- 33. Pestina T. I., Stenberg P. E., Druker B. J., Steward S. A., Hutson N. K., Barrie R. J., Jackson C. W. (1997) Arterioscler. Thromb. Vasc. Biol. 17, 3278–3285 [DOI] [PubMed] [Google Scholar]
- 34. Lannutti B. J., Shim M. H., Blake N., Reems J. A., Drachman J. G. (2003) Exp. Hematol. 31, 1268–1274 [DOI] [PubMed] [Google Scholar]
- 35. Senis Y. A., Tomlinson M. G., Ellison S., Mazharian A., Lim J., Zhao Y., Kornerup K. N., Auger J. M., Thomas S. G., Dhanjal T., Kalia N., Zhu J. W., Weiss A., Watson S. P. (2009) Blood 113, 4942–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mócsai A., Ruland J., Tybulewicz V. L. (2010) Nat. Rev. Immunol. 10, 387–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hooley E., Papagrigoriou E., Navdaev A., Pandey A. V., Clemetson J. M., Clemetson K. J., Emsley J. (2008) Biochemistry 47, 7831–7837 [DOI] [PubMed] [Google Scholar]
- 38. Watson A. A., Eble J. A., O'Callaghan C. A. (2008) Protein Sci. 17, 1611–1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cooper J. A., Qian H. (2008) Biochemistry 47, 5681–5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mori J., Pearce A. C., Spalton J. C., Grygielska B., Eble J. A., Tomlinson M. G., Senis Y. A., Watson S. P. (2008) J. Biol. Chem. 283, 35419–35427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Falet H., Pollitt A. Y., Begonja A. J., Weber S. E., Duerschmied D., Wagner D. D., Watson S. P., Hartwig J. H. (2010) J. Exp. Med. 207, 1967–1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.