

Abstract
Many Gram-negative bacteria that cause major diseases and mortality worldwide require the type III secretion system (T3SS) to inject virulence proteins into their hosts and cause infections. A structural component of the T3SS is the needle apparatus, which consists of a base, an external needle, and a tip complex. In Salmonella typhimurium, the external needle is assembled by the polymerization of the needle protein PrgI. On top of this needle sits a tip complex, which is partly formed by the tip protein SipD. How SipD interacts with PrgI during the assembly of the T3SS needle apparatus remains unknown. The central region of PrgI forms an α-helical hairpin, whereas SipD has a long central coiled-coil, which is a defining structural feature of other T3SS tip proteins as well. Using NMR paramagnetic relaxation enhancement, we have identified a specific region on the SipD coiled-coil that interacts directly with PrgI. We present a model of how SipD might dock at the tip of the needle based on our paramagnetic relaxation enhancement results, thus offering new insight about the mechanism of assembly of the T3SS needle apparatus.
Keywords: NMR, Protein Assembly, Protein Secretion, Protein Structure, Protein-Protein Interactions, PrgI, Salmonella, SipD, T3SS, Paramagnetic Relaxation Enhancement
Introduction
The T3SS2 needle apparatus is critical in the pathogenesis of many bacterial pathogens including Salmonella, a leading cause of food-borne outbreaks and illnesses in the United States (1). The Salmonella pathogenicity island 1 (SPI-1) encodes a T3SS, which is essential for bacterial invasion of target host cells (2). The external portion of the T3SS needle apparatus (3) consists of a needle, and on top of the needle sits a tip complex (4–7). In Salmonella, the needle is assembled from the polymerization of ∼120 copies of the PrgI needle protein (8), and part of the tip complex is formed by the SipD tip protein (5). The atomic structures of PrgI (80 residues) (9, 10) and the Shigella MxiH (11) and Burkholderia BsaL (12) needle monomers have been determined. PrgI (9, 10), MxiH (11), and BsaL (12) are small proteins that form well structured α-helical hairpins flanked by flexible N- and C-terminal tails. The T3SS tip proteins that have been structurally characterized are the Yersinia LcrV (13), the Burkholderia BipD (14, 15), and the Shigella IpaD (15) tip proteins. We have also determined the crystal structure of SipD at 1.9 Å resolution (Protein Data Bank (PDB) ID 3NZZ) (25) (Fig. 1). The T3SS tip proteins are relatively large proteins (SipD has 343 residues), and the most prominent structural feature of the T3SS tip proteins is a long central coiled-coil (13–15), which gives these proteins an overall oblong shape.
FIGURE 1.

Crystal structure of SipD (PDB ID 2NZZ) shown with the positions of the 14 spin labels (indicated as spheres) throughout the length of the central coiled-coil (helix α4 and α8). The spheres are colored based on the strength of the PRE effect with PrgI as yellow (weak), pink (moderate), and red (strong). The SipD N-terminal region (helix α1-α3) is colored blue, the central coiled-coil is colored gray, and the rest of SipD is colored cyan. A 16-residue loop in the N-terminal region (residues 118–133, colored green) lacked electron density and was modeled using CHARMM (33) and energy-minimized to remove steric clash. The SipD crystal starts at Ser-47 and ends in Gln-342, and residues 92–94 also lacked electron density. SipD in A is rotated by 180° in B.
The interaction between the needle protein and the tip protein can be characterized by NMR (16). Chemical shift mapping has identified residues of the Shigella MxiH needle protein that are in contact with the IpaD tip protein (16). However, there have been no further experimental data about other T3SS needle-tip interactions. Further, which regions of the tip protein are in contact with the needle protein remains unknown. On the basis of structural analysis, it has been proposed that the coiled-coil motif of the tip protein might be the binding site for the needle protein (11, 17). We have recently assigned the NMR backbone resonances of SipD (18). However, SipD shows no chemical shift perturbation upon titration with PrgI. Here we use NMR paramagnetic relaxation enhancement (PRE) to identify which specific region in the SipD coiled coil interacts with PrgI.
EXPERIMENTAL PROCEDURES
Expression and Purification of PrgI
The expression and purification of the monomeric form of PrgI, where the C-terminal five residues were deleted and replaced with a His tag with the sequence LEH6, had been described previously (9). Briefly, 15N-labeled PrgI was expressed by growing Escherichia coli BL21(DE3) harboring the plasmid pET22b-PrgI in 1 liter of M9 minimal medium supplemented with 1 g/liter 15[N] ammonium chloride and purified by Ni2+ affinity chromatography as described (9).
Cysteine Mutagenesis of SipD
The plasmid used in the E. coli expression and purification of SipD (residues 39–343) has been described previously (18). The expression plasmid expressed a fusion protein arranged as follows: His tag-GB1 domain-tobacco etch virus protease site-SipD (18). To ensure that the spin label was attached to only one cysteine residue in SipD, the single native Cys-244 residue was mutated to serine. Therefore, all the SipD constructs used in the PRE experiments reported herein contained the background C244S mutation. For PRE measurements, a total of 14 cysteine mutants in residues Asp-136, Ala-144, Asp-147, Asn-155, Asp-169, Lys-173, Ala-285, Ala-296, Thr-303, Leu-318, Lys-324, Ser-328, Ser-331, and Glu-335 were introduced by PCR into the C244S background. All the mutagenesis reported herein was confirmed by DNA sequencing.
Expression and Purification of SipD
SipD (residues 39–343) was overexpressed in E. coli in 1 liter of LB medium as a GB1 fusion protein, purified by Ni2+ affinity chromatography, and digested with tobacco etch virus protease as described previously (18). Purified SipD was dialyzed in buffer (20 mm sodium phosphate, pH 7, and 20 mm NaCl) with 4 mm DTT to prevent the dimerization of the SipD cysteine mutants. Protein concentration was estimated by UV absorbance at A280.
Spin Labeling
The S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL) spin label (Toronto Research Chemicals) was attached to the SipD cysteine mutants as follows. Just prior to labeling with MTSL, the DTT in the protein sample was removed by passage through a NAP-5 column (GE Healthcare) that was previously pre-equilibrated with buffer (20 mm sodium phosphate, pH 7.0, and 20 mm NaCl). The MTSL solution was freshly made prior to spin labeling by dissolving in acetone. Typically, 28 mg of MTSL was dissolved in 70 μl of acetone (to make a 1.5 m solution), and this solution was added into 1000 μl of 0.3 mm SipD in buffer (20 mm sodium phosphate, pH 7.0, and 20 mm NaCl) and incubated at room temperature for over 9 h. To remove the excess MTSL, the mixture was passed through a NAP-5 column that was pre-equilibrated with buffer (20 mm sodium phosphate, pH 7.0, and 20 mm NaCl). The extent of spin labeling was confirmed by electrospray ionization mass spectrometry (supplemental Fig. S1). Of the 14 SipD cysteine mutants, 13 were 100% spin-labeled, whereas D169C was only 95% spin-labeled.
Circular Dichroism Spectroscopy
Samples for circular dichroism (CD) spectroscopy contained 5–10 μm protein in buffer (20 mm sodium phosphate, pH 7.0, and 20 mm NaCl). CD spectra were acquired in triplicate per sample using a JASCO J-815 spectropolarimeter. Wavelength scans were collected at 25 °C at a scan rate of 50 nm/min (supplemental Fig. S2).
NMR Spectroscopy
Typical NMR samples contained 0.5 mm 15N-labeled PrgI mixed with 0.5 mm paramagnetic (or MTSL spin-labeled) SipD in 500 μl of buffer (20 mm sodium phosphate, pH 7.0, 20 mm NaCl, 10% D2O). Likewise, control samples contained 0.5 mm 15N-labeled PrgI mixed with 0.5 mm diamagnetic (or without MTSL) SipD. NMR data were acquired at 25 °C on a Bruker Avance 800-MHz spectrometer equipped with a cryoprobe with a Z-axis gradient, processed with NMRPipe (19), and analyzed using NMRView (20). Initially, for all 14 SipD cysteine mutants, PRE was determined by the single time point method (21) by acquiring two two-dimensional 1H-15N HSQC spectra and calculating the peak intensity ratio (Ipar/Idia) for each non-overlapped PrgI peak using our previous NMR assignments for PrgI (9). The peak intensities of residues that are close to the spin label will be reduced (with Ipar/Idia ratio <1), whereas the peak intensities of residues that are far away from the spin label will be unaffected (with Ipar/Idia ∼1). The first HSQC was acquired using a sample containing paramagnetic SipD with 15N-labeled PrgI, whereas the second HSQC was acquired using diamagnetic SipD with 15N-labeled PrgI. Typical HSQC spectra for the single time point method are shown in Fig. 2. In both NMR samples, the concentrations of PrgI, buffer conditions, and HSQC acquisition and processing parameters were kept identical to ensure that the changes in the chemical shifts were only from the effect of the spin label on SipD. The two-dimensional 1H-15N HSQC pulse sequence (22) with water suppression by water flip back, WATERGATE, and pulse-field gradients were used. Typical data acquisition parameters were: 18 scans, 18 ppm 15N sweep width centered at 118 ppm, 2048 1H complex points, and 190 15N complex points.
FIGURE 2.

Representative two-dimensional 1H-15N HSQC spectra used in the PRE determination by the single time point method. Shown are the two-dimensional 1H-15N HSQC spectra of 15N-labeled PrgI mixed with unlabeled SipD A144C/C244S double mutant in a 1:1 molar ratio. The SipD used was without the MTSL spin label (A) and with the MTSL spin label (B). The assignments of PrgI were published previously (9). Some peaks that showed reduced intensities from the paramagnetic effect are indicated by arrows. The two spectra are shown at identical contour levels, and the peak for Leu-31 in B (marked by an asterisk) appeared at a lower contour level.
PREs were also acquired by the two time point method (23) for the SipD cysteine mutants (D136C, A144C, N155C, T303C, L318C, K324C, S328C, and E335C). The PRE 1H transverse relaxation rate (Γ2) from two time points was acquired using the modified two-dimensional 1H-15N HSQC pulse sequence as described (23), which acquires two two-dimensional 1H-15N HSQC spectra in an interleaved manner. The first two-dimensional 1H-15N HSQC spectrum has the normal INEPT delay (of 1/4J), whereas the second spectrum has a longer INEPT delay, T/2, which consists of the usual 1/4J INEPT delay plus an additional time delay for PRE relaxation measurement. For the SipD-PrgI complex, the optimal T value (23) that gave the strongest NMR signal was found to be 10 ms. Typically, for a 0.5 mm SipD-PrgI complex sample at 1:1 molar ratio, the two time point PRE data were acquired with 96 scans and 18 ppm 15N sweep width centered at 118 ppm. Each two-dimensional 1H-15N HSQC spectra had 2048 1H complex points and 300 15N complex points (supplemental Fig. S3). The total acquisition time was 28 h to complete the two interleaved HSQC spectra. The Γ2 was calculated from peak intensities using Equation 1 (23), where Ta and Tb are the two time points and Idia and Ipar are the peak intensities corresponding to diamagnetic and paramagnetic species.
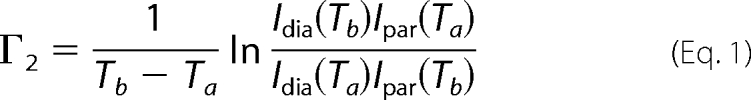 |
Structure Calculation
PRE restraints were derived from Equation 1 (23) and used in structure calculations to model the SipD-PrgI complex using Xplor-NIH (24). Two time point PREs from SipD spin labels Ala-144, Lys-324, Ser-328, and Glu-335 were used in the structure calculations. The structure calculation was performed based on rigid body docking of the lowest energy NMR structure of PrgI (9) that was further refined by residual dipolar coupling (supplemental Fig. S4 and supplemental Fig. S5) and the 1.9 Å resolution crystal structure of SipD (PDB ID 2NZZ) (25). During the structure calculation, the MTSL moiety was covalently attached to the Cβ atoms of A144C, K324C, S328C, and E335C. To account for the conformational flexibility of the MTSL spin label, 100 SipD conformers, each with three different MTSL starting conformations, were generated (supplemental Fig. S6). The orientation of PrgI with respect to SipD was randomized, and the initial structures contained three PrgI molecules distributed in random positions around SipD. A total of 400 structures were calculated for each of the spin labels (A144C, K324C, S328C, and E335C). After the calculations, structures with overall Q-factor (23) below of 0.35 were retained for further analysis.
Salmonella Invasion Assay
The Salmonella invasion assay was used to test the effect of SipD mutations on the ability of Salmonella to invade a cultured human epithelial cell line (Henle 407 cells, ATCC CCL6) following the method described previously (25). Point mutations (M137E, S143R, G151R, S327R, and S332R) were introduced by PCR in the plasmid pRK2-SipD (25), which harbors full-length SipD. Wild-type (SL1344) and sipD− knock-out strains of Salmonella typhimurium used in the invasion assay have been described elsewhere (25).
RESULTS
How the T3SS tip protein interacts with the needle protein during the assembly needle apparatus remains unknown. Thus, the purpose of this study is to identify how the T3SS tip protein interacts with the needle protein. The approach taken was that of NMR PRE method, and the proteins used were the Salmonella SipD tip protein and the PrgI needle protein. We have previously characterized SipD (residues 39–343) (18) and PrgI (residues 1–75) (9) by NMR, and these proteins are well behaved in solution. In this study, we tested the hypothesis that the SipD coiled coil motif (Fig. 1) is the binding site for PrgI.
Engineering of SipD Cysteine Mutants for Spin Labeling
PRE requires the engineering of cysteine residues in specific locations on a protein, and to avoid complications during the spin labeling when multiple cysteine residues are present, the lone native SipD cysteine residue (Cys-244) was mutated into a serine residue. Thus, all the SipD constructs used in this study have the C244S background mutation. The C244S background mutation did not affect the structure of SipD as the crystal structures of wild-type and C244S SipD were virtually identical with Cα r.m.s.d. of 1.4 Å (25). Additionally, the SipD C244S point mutation did not affect the invasiveness of Salmonella toward cultured human epithelial cells (25). We introduced 14 cysteine point mutations positioned throughout the entire length of the SipD central coiled-coil (on helix α4 and α8, Fig. 1) for the attachment of the MTSL spin label.
CD Spectroscopy of SipD Cysteine Mutants
CD spectroscopy was used to assess the proper folding of the 14 SipD cysteine mutants used in this study (supplemental Fig. S2 and supplemental Table S1). All the SipD cysteine mutants used in this study showed CD spectral scans (from 190 to 260 nm), indicative of a well folded and highly α-helical protein with molar ellipticity ([θ]) minima at 222 and 208 nm (supplemental Fig. S2). The CD spectrum for wild-type SipD showed a similar curve to that of the C244S point mutant (supplemental Fig. S2). The CD spectra for the rest of the cysteine mutants used in the spin labeling showed a similar pattern as that of wild-type SipD. Additionally, the ratio of the molar ellipticity at 222 and 208 nm ([θ]222/[θ]208) for all the SipD cysteine mutants showed values from 0.91 to 1.0 (supplemental Table S1), which indicate the presence of α-helices with extensive interhelical contacts (26–29). (In contrast, a [θ]222/[θ]208 ratio of less than 0.8 indicates extended α-helices with few interhelical contacts (26–29).)
Results of Single Time Point PRE
We used the single time point PRE method to qualitatively identify which region within the SipD coiled-coil interacts with PrgI (Fig. 3). The MTSL spin label was attached to 14 SipD cysteine point mutants (in a C244S background) located at various positions throughout the entire length of helix α4 and α8 of the coiled-coil (Fig. 1). (The SipD coiled-coil as depicted in Fig. 1 orients an arbitrary “top/bottom” or “upper/lower” used herein to describe the various parts of the coiled-coil.) For the spin label attached on the upper half region of the coiled-coil on helix α4 (Asn-155, Lys-173, and Asp-169, Fig. 3A) and on helix α8 (Ala-285, Ala-296, and Thr-303, Fig. 3B), the average (Ipar/Idia) intensity ratio was close to 1.0 (Table 1), which indicates that the spin labels in these positions had essentially no paramagnetic relaxation effect on the HSQC peaks of PrgI. Spin labels in Asp-136 (helix α4, Fig. 3C) and Leu-318 (helix α8, Fig. 3D) showed average Ipar/Idia ratios of 0.8 (Table 1) and showed a moderate paramagnetic relaxation effect. Spin labels attached at the bottom of the coiled-coil, in helix α4 (residues Asp-147 and Ala-144, Fig. 3C) and helix α8 (Lys-324, Ser-328, Ser-331, and Glu-335, Fig. 3, D and E), showed the strongest paramagnetic relaxation effect. Spin labels at residues Ser-331 and Glu-335 (Fig. 3E), which are located toward the C-terminal region of SipD helix α8, showed the strongest paramagnetic relaxation effect on the HSQC peaks of PrgI with average Ipar/Idia ratios of 0.3 (Table 1). The observed intermolecular PRE data are qualitatively consistent with each other, which indicates negligible perturbation of the binding caused by cysteine mutation and MTSL conjugation. Qualitatively, the results of single time point PRE indicate that the bottom half of the SipD coiled-coil is the binding site for PrgI.
FIGURE 3.

Results of PRE by single time point method using spin-labeled SipD in complex with 15N-labeled PrgI. The SipD spin labels are at the following positions: Asn-155, Asp-169 and Lys-173 (A); Ala-285, Ala-296, and Thr-303 (B); Asp-136, Ala-144, and Asp-147 (C); Leu-318 and Lys-324 (D), and Ser-328, Ser-331, and Glu-335 (E). The gray line indicates the (Ipar/Idia) intensity ratio at 1. The positions of the spin labels (spheres) in the coiled-coil (helix α4 or α8) of SipD are color-coded with the PRE plots.
TABLE 1.
Average intensity Ipar/Idia ratio from the single time point PRE method
| Spin labeled residue | Average Ipar/Idia |
|---|---|
| Asn-155 | 1.1 ± 0.11 |
| Lys-173 | 1.0 ± 0.14 |
| Ala-285 | 0.99 ± 0.11 |
| Ala-296 | 0.97 ± 0.10 |
| Asp-169 | 0.94 ± 0.11 |
| Thr-303 | 0.85 ± 0.11 |
| Asp-136 | 0.79 ± 0.14 |
| Leu-318 | 0.77 ± 0.11 |
| Asp-147 | 0.69 ± 0.10 |
| Ser-328 | 0.67 ± 0.16 |
| Ala-144 | 0.64 ± 0.19 |
| Lys-324 | 0.48 ± 0.18 |
| Glu-335 | 0.33 ± 0.14 |
| Ser-331 | 0.28 ± 0.17 |
Results of Two Time Point PRE
We also determined the Γ2 values using the two time point method (23), which provides a more accurate measure of the Γ2 over the single time point method (23). As data acquisition for the two time point PRE method took longer than a single time point PRE data, we did not acquire two time point PREs for all the 14 mutants but selected a subset that represented weak, moderate, and strong PRE effects based on Fig. 3. Consistent with the results of single time point PRE, the spin labels at Asn-155 (Fig. 4A) and Thr-303 (Fig. 4B) showed the least PRE effect on PrgI as determined by the two time point method, with average Γ2 values of 1.3 and 4.8 s−1, respectively (Table 2). The spin label at SipD Asn-155 also served as a negative control for a spin-labeled protein that does not interact with PrgI. The spin labels on Asp-136 (Fig. 4C) and Leu-318 (Fig. 4D), which in the single time point data showed a moderate PRE effect with average Ipar/Idia ratios near 0.8 (Table 1), showed average Γ2 values of 18.6 and 20.1 s−1 (Table 2), respectively. Finally, the spin labels that gave a strong PRE effect from the single time point data gave average Γ2 values from 26.8 s−1 for the spin label at Ser-328 to 58.7 s−1 for the spin label at Glu-335 (Table 2). PREs obtained using the two time point method confirmed that the spin labels at the bottom of the SipD coiled-coil had the strongest PRE effect on PrgI (Fig. 4, E–H). Thus, results of the two time point PRE confirmed that the lower half of the SipD coiled-coil is the binding site for PrgI.
FIGURE 4.

Γ2 values acquired using the two time point method (23) for the SipD spin label at the following positions: Asn-155 (A), Thr-303 (B), Asp-136 (C), Leu-318 (D), Ala-144 (E), Lys-324 (F), Ser-328 (G), and Glu-335 (H). The PRE data for Asn-155 (A) and Thr-303 (B) were acquired at 32 scans and 9-h acquisition time, whereas the rest were acquired at 92 scans and 28-h acquisition time.
TABLE 2.
Average PRE Γ2 (s−1) values
| Spin labeled residue | Average Γ2 |
|---|---|
| s−1 | |
| Asn-155 | 1.3 ± 1.8 |
| Thr-303 | 4.8 ± 4.2 |
| Asp-136 | 18.6 ± 8.9 |
| Leu-318 | 20.1 ± 15.0 |
| Ser-328 | 26.8 ± 13.8 |
| Ala-144 | 26.9 ± 20.81 |
| Lys-324 | 42.3 ± 25.7 |
| Glu-335 | 58.7 ± 24.9 |
PRE Structure Calculations
PRE restraints for structure calculations were derived from the two time point PREs from the SipD spin labels at positions Ala-144, Lys-324, Ser-328, and Glu-335 (Fig. 4), which showed a strong PRE effect with PrgI. PrgI forms an α-helical hairpin with helix αA and αB that is flanked by flexible regions in partial helical conformation (Fig. 5) (9). To some extent, almost all parts of PrgI were affected in varying degrees by the SipD spin labels on Ala-144, Lys-324, Ser-328, and Glu-335 (Fig. 5). Regarding SipD, results of structure calculations indicate that PrgI interacted mainly at the lower half of SipD (Fig. 6). Results of PRE structure calculations showed that at the lower half of the SipD coiled-coil, there were two major populated sites for PrgI: one near helix α8 where the majority of PrgI molecules were populated and another near helix α4 with a comparatively smaller population of PrgI. The overall Q-factor (23), which is indicative of the agreement between the observed and calculated PRE, varied between 0.28 and 0.35 in the family of SipD-PrgI structures. Among the four spin labels, PRE restraints from positions Glu-335 and Lys-324 showed a Q-factor of 0.28–0.35, whereas PRE restraints from positions Ala-144 and Ser-328 showed Q-factors of 0.32 and 0.35, respectively. These Q-factors indicated good agreement between observed and calculated PRE values.
FIGURE 5.

Two time point PREs mapped on the surface of PrgI for the SipD spin labels on positions Ala-144 (A), Lys-324 (B), Ser-328 (C), and Glu-335 (D). The PREs are colored according to the strength of the Γ2 values (green being the lowest to red being the highest). The lowest energy structure of the NMR structure of PrgI that was refined by residual dipolar coupling was used.
FIGURE 6.

Results of PRE structure calculation of the SipD-PrgI complex. The possible structures of PrgI bound to SipD are shown as density maps of PrgI around SipD and colored according to the PRE restraints obtained from SipD spin labels at position Ala-144 (blue), Lys-324 (green), Ser-328 (black), and Glu-335 (red). This figure was made using VMD-XPLOR (34); A and B are orthogonal views.
Invasion Assay
Results of Salmonella invasion assay as a function of SipD mutagenesis showed that substitution of bulky side chains at the SipD-PrgI binding interface significantly affected the ability of Salmonella to invade (see Fig. 8). For instance, the point mutations S327R and M137E (which are adjacent to the Ser-328 and Asp-136 spin labels that yielded strong PRE effect) resulted in a significant decrease in invasiveness by 50 and 65%, respectively. The S143R mutation (adjacent to spin label A144C) decreased invasiveness as well, but not as not as significantly as M137E and S327R. On the other hand, G151R has similar invasiveness as the wild type (100% invasiveness), and S332R has slightly increased invasiveness than the wild type (125% invasiveness, adjacent to the spin label S331C).
FIGURE 8.

Results of Salmonella invasion assay as a function of SipD point mutations (M137E, S143R, G151R, S327R, and S332R). Results are shown relative to sipD+ or when exogenous sipD was introduced into a sipD knock-out strain of S. typhimurium strain (sipD−).
DISCUSSION
Currently, there are no studies that probe the SipD-PrgI interaction or any T3SS tip protein-needle protein combination on a per residue basis. Our goal is to identify which region of the SipD coiled-coil is the primary binding site for PrgI. The central coiled-coil (formed by helix α4/α8 in SipD and colored gray in Fig. 1) is a structural characteristic of all T3SS tip proteins that have been determined to date (13–15). We hypothesize that the SipD coiled-coil is the primary binding site for the needle protein based on the structural similarity between the SipD α-helical hairpin (helix α1/α2, Fig. 1) and the PrgI α-helical hairpin (helix αA/αB, Fig. 5). The SipD α-helical hairpin can be superimposed on the PrgI α-helical hairpin with a Cα r.m.s.d. of 3.2 Å. Although a 3.2 Å r.m.s.d. conveys weak structural similarity, the needle monomers of Salmonella, Shigella, and Burkholderia, which share 44–63% sequence identities and are structurally homologous to each other, also share Cα r.m.s.d. of 3.2 Å as well. Thus, there appears to be a size and shape complementarity, albeit weakly, between the tip protein α-helical hairpin and that of its cognate needle protein. For this reason, others have proposed that the N-terminal α-helical hairpins of BipD and IpaD serve as self-chaperones for BipD and IpaD (15).
We have recently reported the NMR assignments of SipD (18) and have successfully used SipD for NMR studies of molecular interactions with bile salts (18). However, our attempts at using NMR chemical shift mapping of 15N-labeled SipD titrated with unlabeled PrgI failed to identify unambiguously which SipD residues were involved in the SipD-PrgI interaction because of the disappearance of SipD residues upon the addition of PrgI and the more than 300 SipD residues that made the analysis complicated (supplemental Fig. S7). We reasoned that the intermediate exchange behavior of the SipD-PrgI interaction was due to the weak binding affinity of SipD to PrgI in solution. Thus, we employed a more sensitive NMR method based on PRE (30, 31) to study the SipD-PrgI interaction. We used the method of paramagnetic relaxation enhancement, wherein a paramagnetic spin label is attached to a cysteine residue and used in titration with 15N-labeled PrgI. We engineered 14 cysteine mutants across the length of the SipD coiled-coil (helix α4/α8) and used CD spectroscopy to ensure that the cysteine mutants did not alter the global fold of SipD (supplemental Fig. S2). The PREs were determined initially by the single time point (Fig. 3) and by the two time point (Fig. 4) methods. Both PRE methods indicate that the lower half of the SipD coiled-coil (adjacent to the SipD helix α1/α2) was the primary binding site for PrgI.
Results of PRE-based structure calculations confirm that PrgI bind at the bottom of SipD, and more important, reveal that PrgI interacts with SipD at two major sites as indicated by the clustering of two populations of PrgI molecules near the SipD helix α4 and α8 (Fig. 6). Galkin et al. (32) recently reported the 18 Å resolution structure of the assembled Salmonella needle based on cryoelectron microscopy (Fig. 7). In the assembled needle, the PrgI α-helical hairpin is oriented toward the top of the needle. Our PRE results together with the cryoEM structure of the PrgI needle (32) constrain the possible orientation of SipD with respect to the needle. Because the lower half of the SipD coiled-coil is the main site for interaction with PrgI, the top of SipD would have to be pointed away from the needle to avoid steric clash with the rest of the PrgI needle (Fig. 7). Based on structural analysis, others have proposed a model of tip-needle interaction where five tip protein molecules are inserted on top of the MxiH needle (11, 17), which is made of 5.6 MxiH monomers per turn (11). The tip protein is thus flanked by two needle monomers in the assembled needle. The interaction of a tip protein molecule with multiple needle monomers is also seen in solution. Results of our PRE structure calculations indicate there are two major sites in SipD that interact with PrgI. PRE results are consistent with a SipD molecule inserting between two PrgI monomers in the assembled needle (Fig. 7).
FIGURE 7.

Possible orientation of SipD in the assembled needle-tip complex as constrained by PRE results and the structure of the PrgI needle (32). Because the lower half of the SipD coiled-coil (marked by red spheres) is the primary binding site for PrgI, SipD would have be oriented in the assembled needle-tip complex with the mixed α/β domain pointed away from the needle. The reverse orientation, where the α/β domain is pointed toward the needle, will result in steric clash. The cryoEM-based model of the PrgI needle was made using coordinates obtained from Drs. Vitold E. Galkin and Edward H. Egelman (32); A and B are orthogonal views. PrgI molecules (numbered 1–6) in the topmost layer of the needle are colored cyan.
A Salmonella invasion assay was used to confirm our PRE findings. Point mutations M137E and S327R that were at the bottom of the SipD coiled-coil, which would be expected to be near the SipD-PrgI binding interface, showed significant reduction in invasiveness (Fig. 8). Unlike the results for M137E and S327R, the mutations S143R, G151R, and S332R essentially had no effect on bacterial invasiveness. This could be attributed to the fact that these residues are not at the binding interface.
In summary, our PRE results demonstrated that the lower half of the SipD coiled-coil (toward the C terminus of helix α8) is the main binding site for PrgI. Further, PRE structure calculations demonstrated that there are two major PrgI binding sites at the bottom of the SipD coiled-coil. Together with the recent cryoEM structure of the PrgI needle (32), our PRE results constrain the possible orientations of SipD in the needle-tip assembly.
Supplementary Material
Acknowledgments
We are grateful to Dr. Dalian Zhong for the SipD expression plasmid, Dr. Yu Wang for Fig. S7, Dr. Vitold E. Galkin and Dr. Edward H. Egelman (University of Virginia) for providing the coordinates of the PrgI needle, and Dr. Wonpil Im (University of Kansas) for the use of the computing cluster facility at the University of Kansas Center for Bioinformatics.
This work was supported by NIH Grants RR017708 and R01AI074856 (to R. N. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S7 and Table S1.
- T3SS
- type III secretion system
- HSQC
- heteronuclear single quantum coherence
- INEPT
- insensitive nuclei enhanced by polarization transfer
- MTSL
- S-(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate
- PRE
- paramagnetic relaxation enhancement
- Γ2
- PRE 1H transverse relaxation rate
- r.m.s.d
- root mean square deviation.
REFERENCES
- 1. Mead P. S., Slutsker L., Dietz V., McCaig L. F., Bresee J. S., Shapiro C., Griffin P. M., Tauxe R. V. (1999) Emerg. Infect. Dis. 5, 607–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galán J. E. (2001) Annu. Rev. Cell Dev. Biol. 17, 53–86 [DOI] [PubMed] [Google Scholar]
- 3. Cornelis G. R. (2006) Nat. Rev. Microbiol. 4, 811–825 [DOI] [PubMed] [Google Scholar]
- 4. Mueller C. A., Broz P., Müller S. A., Ringler P., Erne-Brand F., Sorg I., Kuhn M., Engel A., Cornelis G. R. (2005) Science 310, 674–676 [DOI] [PubMed] [Google Scholar]
- 5. Espina M., Olive A. J., Kenjale R., Moore D. S., Ausar S. F., Kaminski R. W., Oaks E. V., Middaugh C. R., Picking W. D., Picking W. L. (2006) Infect. Immun. 74, 4391–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mota L. J. (2006) Trends. Microbiol. 14, 197–200 [DOI] [PubMed] [Google Scholar]
- 7. Sani M., Botteaux A., Parsot C., Sansonetti P., Boekema E. J., Allaoui A. (2007) Biochim. Biophys. Acta 1770, 307–311 [DOI] [PubMed] [Google Scholar]
- 8. Marlovits T. C., Kubori T., Sukhan A., Thomas D. R., Galán J. E., Unger V. M. (2004) Science 306, 1040–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang Y., Ouellette A. N., Egan C. W., Rathinavelan T., Im W., De Guzman R. N. (2007) J. Mol. Biol. 371, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poyraz O., Schmidt H., Seidel K., Delissen F., Ader C., Tenenboim H., Goosmann C., Laube B., Thünemann A. F., Zychlinsky A., Baldus M., Lange A., Griesinger C., Kolbe M. (2010) Nat. Struct. Mol. Biol. 17, 788–792 [DOI] [PubMed] [Google Scholar]
- 11. Deane J. E., Roversi P., Cordes F. S., Johnson S., Kenjale R., Daniell S., Booy F., Picking W. D., Picking W. L., Blocker A. J., Lea S. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 12529–12533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang L., Wang Y., Picking W. L., Picking W. D., De Guzman R. N. (2006) J. Mol. Biol. 359, 322–330 [DOI] [PubMed] [Google Scholar]
- 13. Derewenda U., Mateja A., Devedjiev Y., Routzahn K. M., Evdokimov A. G., Derewenda Z. S., Waugh D. S. (2004) Structure 12, 301–306 [DOI] [PubMed] [Google Scholar]
- 14. Erskine P. T., Knight M. J., Ruaux A., Mikolajek H., Wong Fat Sang N., Withers J., Gill R., Wood S. P., Wood M., Fox G. C., Cooper J. B. (2006) J. Mol. Biol. 363, 125–136 [DOI] [PubMed] [Google Scholar]
- 15. Johnson S., Roversi P., Espina M., Olive A., Deane J. E., Birket S., Field T., Picking W. D., Blocker A. J., Galyov E. E., Picking W. L., Lea S. M. (2007) J. Biol. Chem. 282, 4035–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang L., Wang Y., Olive A. J., Smith N. D., Picking W. D., De Guzman R. N., Picking W. L. (2007) J. Biol. Chem. 282, 32144–32151 [DOI] [PubMed] [Google Scholar]
- 17. Blocker A. J., Deane J. E., Veenendaal A. K., Roversi P., Hodgkinson J. L., Johnson S., Lea S. M. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 6507–6513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y., Nordhues B. A., Zhong D., De Guzman R. N. (2010) Biochemistry 49, 4220–4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 20. Johnson B. A. (2004) Methods Mol. Biol. 278, 313–352 [DOI] [PubMed] [Google Scholar]
- 21. Gillespie J. R., Shortle D. (1997) J. Mol. Biol. 268, 158–169 [DOI] [PubMed] [Google Scholar]
- 22. Grzesiek S., Bax A. (1993) J. Am. Chem. Soc. 115, 12593–12594 [Google Scholar]
- 23. Iwahara J., Tang C., Marius Clore G. (2007) J. Magn. Reson. 184, 185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwieters C. D., Kuszewski J. J., Clore G. M. (2006) Prog. NMR Spectroscopy 48, 47–62 [Google Scholar]
- 25. Chatterjee S., Zhong D., Nordhues B. A., Battaile K. P., Lovell S., De Guzman R. N. (2011) Protein Sci. 20, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou N. E., Zhu B. Y., Kay C. M., Hodges R. S. (1992) Biopolymers 32, 419–426 [DOI] [PubMed] [Google Scholar]
- 27. Kiss R. S., Kay C. M., Ryan R. O. (1999) Biochemistry 38, 4327–4334 [DOI] [PubMed] [Google Scholar]
- 28. Choy N., Raussens V., Narayanaswami V. (2003) J. Mol. Biol. 334, 527–539 [DOI] [PubMed] [Google Scholar]
- 29. Wang Y., Boudreaux D. M., Estrada D. F., Egan C. W., St. Jeor S. C., De Guzman R. N. (2008) J. Biol. Chem. 283, 28297–28304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang C., Louis J. M., Aniana A., Suh J. Y., Clore G. M. (2008) Nature 455, 693–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tang C., Ghirlando R., Clore G. M. (2008) J. Am. Chem. Soc. 130, 4048–4056 [DOI] [PubMed] [Google Scholar]
- 32. Galkin V. E., Schmied W. H., Schraidt O., Marlovits T. C., Egelman E. H. (2010) J. Mol. Biol. 396, 1392–1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., Karplus M. (1983) J. Comp. Chem. 4, 187–217 [Google Scholar]
- 34. Schwieters C. D., Clore G. M. (2001) J. Magn. Reson. 149, 239–244 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.