

Abstract
The proprotein convertase PCSK9 plays a key role in cholesterol homeostasis by binding the LDL receptor and targeting it toward degradation. PCSK9 is strongly expressed in the liver and is found in human and mouse plasma as mature (∼62 kDa) and inactivated (∼55 kDa) forms. Ex vivo data showed that human PCSK9 is inactivated by cleavage at Arg218↓ by the overexpressed convertases furin and PC5/6A. Analysis of the plasma of human heterozygotes for R218S and F216L mutations revealed a ∼50% reduction in the levels of the ∼55-kDa form. To identify the convertase(s) responsible for cleavage at Arg218 in vivo, we inactivated the genes of furin and/or PC5/6 specifically in hepatocytes. The PCSK9-inactivated form was strongly reduced in mice lacking furin in hepatocytes (Fur-hKO) and only slightly reduced in PC5/6-hKO plasma. In agreement with a key role of furin in regulating PCSK9 activity in vivo, we observed an overall 26% drop in the LDL receptor protein levels of Fur-hKO livers, likely due to the compound effects of a 35% increase in PCSK9 mRNA levels and the loss of PCSK9 cleavage, suggesting a higher activity of PCSK9 in these mice. Overexpression of PCSK9 in primary hepatocytes obtained from these mice revealed that only full-length, membrane-bound, but not soluble, furin is the cognate convertase. We conclude that in hepatocytes furin regulates PCSK9 mRNA levels and is the key in vivo-inactivating protease of circulating PCSK9.
Keywords: Cholesterol, Gene Knock-out, Hepatocyte, Lipoprotein Receptor, Mouse Genetics, Protein Processing, Serine Protease, Furin, PCSK9, Proprotein Convertase
Introduction
A large number of secretory proteins are produced as precursors that are cleaved at specific sites to generate mature bioactive products. Most of these specific cleavages occur after basic residues and are achieved by one or more of the seven basic amino acid-specific members of the proprotein convertase (PC)2 family, who share identities to bacterial subtilisins and yeast kexin (genes PCSK1 to PCSK7) (1). Four of them, furin, PC5/6, PACE4, and PC7, are widely or ubiquitously expressed and responsible for most of the processing events occurring in the constitutive secretory pathway or at the cell surface, including receptors, viral glycoproteins, or TGFβ-like growth factors (2). Although these PCs exhibit a functional redundancy ex vivo, their inactivation leads to specific phenotypes revealing that, in vivo, each PC fulfills key processing events. Furin KO in mice resulted in numerous embryonic malformations including the absence of axial rotation and heart looping leading to death around embryonic day (E)11 (3). PC5/6 KO leads to death at birth with an altered antero-posterior pattern, including extra vertebrae, lack of tail, kidney agenesis, hemorrhages, collapsed alveoli, and retarded ossification (4). PACE4 KO led to an altered left-right patterning, including cyclopism and craniofacial and cardiac malformations in some embryos (5). The above phenotypes are largely due to processing defects of various TGFβ-like growth factors. Finally, PC7 KO mice exhibit no obvious phenotypes (6).
The two other members of the PC family, SKI-1 (also known as S1P) and PCSK9 (proprotein convertase of the subtilisin kexin type 9), belong to the pyrolysin and proteinase K subfamilies of subtilases, respectively (7). PCSK9, whose gene is the third locus for dominant familial hypercholesterolemia (8), enhances the degradation of the low density lipoprotein receptor (LDLR) in vivo (9). It binds the first of three EGF-like repeats, EGF-A, present in the extracellular domain of the LDLR (10). The complex is internalized and directed toward endosomes/lysosomes for degradation, thereby preventing recycling of the LDLR to the cell surface (11, 12). Human hypocholesterolemic subjects (13, 14) or mice (15, 16) lacking functional PCSK9 are characterized by low levels of LDL cholesterol due to increased levels of cell surface LDLR, leading to a higher clearance of LDL particles. PCSK9 is now considered one of the most promising new targets in the treatment of familial hypercholesterolemia. PCSK9 is produced as a ∼75-kDa precursor that undergoes autocatalytic cleavage in the endoplasmic reticulum, and the secreted mature form remains associated with the N-terminal inhibitory prosegment (17), whose C terminus is retained in the narrow catalytic pocket of PCSK9 (18). In agreement, PCSK9 has no other substrate known but itself and triggers LDLR degradation independently of its enzymatic activity (19, 20).
Human PCSK9 circulates as a mature form (∼62 kDa) bound to its prosegment, as well as an N-terminally truncated inactive form (∼55 kDa) cleaved at RFHR218↓, R standing for an essential Arg residue for cleavage. This cleavage causes a structural change that results in the detachment of the prosegment (21). Interestingly, three human dominant gain of function mutations found in hypercholesterolemic families occur in this sequence, which partially or totally prevent PCSK9 inactivation: R215H (22), F216L (8), and R218S (23). Ex vivo, furin and soluble PC5/6A, the shortest of the two PC5/6 isoforms, are the only two convertases inactivating PCSK9 at this site (21).
To determine which convertase is responsible for the in vivo inactivation of PCSK9, and because circulating PCSK9 originates from hepatocytes (16), we herein inactivated furin and PC5/6 genes specifically in hepatocytes using a Cre-lox system. Our data validate the relevance of the RFHR218 site for PCSK9 inactivation by showing that the plasma of heterozygote F216L and R218S patients exhibit ∼50% lower levels of inactivated PCSK9. We also show that membrane-bound furin from hepatocytes is the key inactivating PC involved in cleavage at Arg218↓ and that PCSK9 inactivation likely occurs at the hepatocyte cell surface and not in other tissues.
EXPERIMENTAL PROCEDURES
F216L and R218S Hypercholesterolemic Patients
Plasma was obtained from two French patients and kept at −20 °C. The 66-year-old woman carrying the F216L mutation (8) exhibited 317 and 211 mg/dl of total and LDL cholesterol, respectively, before treatment. However, the plasma used in this study was recently obtained under statin treatment, with total cholesterol and LDL cholesterol levels of 190 and 96 mg/liter, respectively. The 56-year-old man, who carries the R218S mutation (23), presented tendinous xanthoma and arcus corneae. He was recruited through the “Réseau National de Recherche sur les Hypercholestérolémies Familiales” at age 45 with 402 and 293 mg/dl of total cholesterol and LDL cholesterol, respectively. The plasma used in this study was recently obtained, after stopping statin treatment for 3 weeks. It contained 350 and 260 mg/dl of total cholesterol and LDL cholesterol, respectively.
Mice and Genotyping
Furinflox/flox and Pcsk5flox/flox mice were generated as described previously (4, 24). Mice were housed in a 12-h light/dark cycle and fed a standard diet (2018 Teklad global 18% protein rodent diet; Harlan Laboratories). All procedures were approved by the bioethics committee for animal care of the Clinical Research Institute of Montreal. Tg(Alb-cre) mice were purchased from The Jackson Laboratory (stock no. 003574). PCSK9 transgenic and KO mice were described previously (16). Mice were genotyped by PCR analysis of tail DNA using specific pairs of primers for Furin and Pcsk5 floxed or deleted alleles (Δ2 and Δ1, respectively), as well as for cre (Table 1).
TABLE 1.
Oligonucleotides used for genotyping and QPCR
| PCR primers | |
|---|---|
| Genotyping | |
| Pcsk5flox | 5′-CAGAATTGCTGTGCTCTGGAversus5′-GTATTGGCATTTCCCTCAGC |
| Pcsk5 D1 | 5′-GGGATCGGCCAGTAGCCAGACTATACGGversus5′-CATAAAATGTATTGGCATTTCCCTCAGC-3′ |
| Furinflox | 5′-ATGCTCAAGGCCAGAAGATCversus5′-AATCTGTTCCCTGCTGAGGA |
| Furin D2 | 5′-GCTGTATTTATTCCGGAGACversus5′-AATCTGTTCCCTGCTGAGGA |
| Cre | 5′-TGCCAGGATCAGGGTTAAAGversus5′-TGCATGATCTCCGGTATTGA |
| QPCR | |
| PC5/6 | 5′-ACTCTTCAGAGGGTGGCTAversus5′-GCTGGAACAGTTCTTGAATC |
| Furin | 5′-CATGACTACTCTGCTGATGGversus5′-GAACGAGAGTGAACTTGGTC |
| LDLR | 5′-GTATGAGGTTCCTGTCCATCversus5′-CCTCTGTGGTCTTCTGGTAG |
| PCSK9 | 5′-CAGGGAGCACATTGCATCCversus5′-TGCAAAATCAAGGAGCATGGG |
| Mouse S16 | 5′-GCTACCAGGGCCTTTGAGATGversus5′-AGGAGCGATTTGCTGGTGTGG |
Generation of Hepatocyte-specific Knock-out (hKO) Mice
The conditional Furin and Pcsk5 mice carry floxed alleles, in which the first coding exon (exon 2 in Furin or exon 1 in Pcsk5) is flanked with loxP sites, which are excisable by Cre recombinase. Herein, we will refer to these control conditional mice as WT mice as their expression levels of furin and PC5/6 mRNA were not different from those of WT mice. To knock-out furin and PC5/6 specifically in hepatocytes, Furinflox/+ or Pcsk5flox/+ mice were crossed to Tg(Alb-cre) mice (25). Heterozygote mice (Furinflox/+ or Pcsk5flox/+) carrying the Cre transgene were then back-crossed to Furinflox/+ or Pcsk5flox/+ to select Furinflox/flox;Tg(Alb-cre)+/0 or Pcsk5flox/flox;Tg(Alb-cre)+/0 homozygotes. By intercrossing these mice, [Fur-PC5/6]hKO mice (Furinflox/flox;Pcsk5flox/flox;Tg(Alb-cre)+/0) that carry only one allele of the transgene were selected.
Quantitative RT-PCR (QPCR) Analysis
RNA from livers were extracted using TRIzol, as recommended by the manufacturer (Invitrogen). cDNA synthesis and QPCR were performed as described previously (26). Primers from neighboring exons were used to measure furin, PC5/6, LDLR, PCSK9, and mouse S16 ribosomal protein expression (Table 1).
Isolation, Culture, and Transfection of Primary Hepatocytes
Hepatocytes were isolated from 8-to-10-week-old male livers using the two-step collagenase perfusion method. After anesthesia of mice by 2% isoflurane inhalation, the peritoneal cavity was opened, and the liver was perfused in situ via the inferior vena cava for 6 min at 37 °C with calcium-free HEPES buffer I (142 mm NaCl, 6.7 mm KCl, 10 mm HEPES, pH 7.6) and for 8 min with calcium-supplemented HEPES buffer II (4.7 mm CaCl2, 66.7 mm NaCl, 6.7 mm KCl, 100 mm HEPES, pH 7.4) containing 0.5 mg/ml collagenase type V (Sigma Aldrich). The perfusion rates were set to 8 and 6 ml/min, respectively. In 3.5-cm Petri dishes coated with fibronectin (0.5 mg/ml, Sigma Aldrich), 5.105 cells were seeded in Williams' medium E supplemented with 10% fetal bovine serum (Invitrogen). After 2 h, the medium was replaced with hepatozyme medium (Invitrogen) for 12 h prior to treatment. All transfections were performed using 6 μg of DNA and Effectene transfection reagent, as recommended by the manufacturer (Qiagen, Mississauga, Ontario, Canada).
Western Blotting and Antibodies
PCSK9 was immunoprecipitated from plasma (50 μl) or hepatocytes culture media (4 ml) using a mouse (12) or human (27) PCSK9 antibody (1:200). The proteins were then separated on 8% gels by tricine SDS-PAGE and transferred to a PVDF membrane (PerkinElmer Life Sciences). PCSK9 was revealed using the same PCSK9 antibody (1:3000) and anti-rabbit IgG (1:2000; TrueBlot, eBioscience, San Diego, CA). Media from primary hepatocytes (30 μl of 1 ml) or liver protein extracts (30 μg) were analyzed by Western blotting as above, using mouse LDLR (1:1000; R&D Systems), actin (1:3000; Sigma) and transferrin receptor (1:1000; BD Transduction Laboratories) antibodies and corresponding secondary antibodies conjugated to horseradish peroxidase (1:10,000; Invitrogen), or HRP-conjugated V5-monoclonal antibody (1:5000; Invitrogen). The antigen-antibody complexes were visualized using the enhanced chemiluminescence kit (ECL; Amersham Biosciences or Pierce). Quantitation was performed using ImageJ software, and normalization to actin.
Histology
Livers were collected and fixed in 4% paraformaldehyde at 4 °C for 24 h, washed in 70% ethanol, and embedded in paraffin. Sections were cut at 4-μm thickness and stained with hematoxylin and eosin (Sigma Aldrich).
RESULTS
Plasma Levels of Human PCSK9 in R218S and F216L Heterozygotes
Patients carrying one R218S (23) or F216L (8) PCSK9 allele exhibit hypercholesterolemia. Because these mutations occur in a typical basic amino acid-specific PC cleavage site RFHR218↓ (1), we hypothesized that the hypercholesterolemic phenotype is due to reduced PCSK9 inactivation at this site (21). To verify this hypothesis, plasma PCSK9 from two patients was immunoprecipitated and analyzed by Western blotting (Fig. 1). The ∼50% reduced levels of the ∼55 kDa cleaved form in both patients suggest that R218S and F216L mutations indeed prevent cleavage of PCSK9.
FIGURE 1.
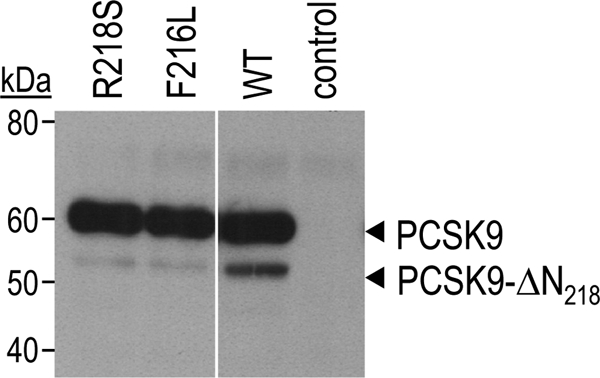
Reduced PCSK9 inactivation in R218S or F216L heterozygote patients. Circulating PCSK9 from subjects carrying the R218S or F216L mutation or no mutation (WT) in the typical PC cleavage site RFHR218↓ was immunoprecipitated and analyzed by Western blotting. No primary antibody was added into the control sample.
Inactivation of Furin and PC5/6 in Hepatocytes
Circulating PCSK9 originates from hepatocytes (16). Thus, to examine the in vivo contribution of the PC-candidates furin and PC5/6 to its inactivation, we generated hepatocyte-specific knock-outs of furin (Fur-hKO) or PC5/6 (PC5/6-hKO) and their combination [Fur-PC5/6]hKO. This was done using a transgene Tg(Alb-cre) expressing Cre under the control of the albumin promoter in mice carrying Furin and/or Pcsk5 conditional floxed alleles. We assessed the efficiency of the Furin and Pcsk5 gene inactivation by measuring hepatic furin and PC5/6 mRNA levels in single or double knock-out mice (Fig. 2). The PC5/6-hKO resulted in a 98% drop in PC5/6 mRNA levels, showing that PC5/6 is mainly expressed in hepatocytes. In contrast, in Fur-hKO livers, only a 42% drop was observed in furin mRNA levels, likely due to furin expression in other cell types than hepatocytes, as Furin floxed alleles were not detectable in purified Fur-hKO primary hepatocytes (data not shown). In double [Fur-PC5/6]hKO livers, furin and PC5/6 mRNA drops were similar to that obtained in single Fur- or PC5/6-hKO livers (Fig. 2).
FIGURE 2.

Efficacy of furin and/or PC5/6 KO in hepatocytes. Furin and PC5/6 transcript levels were determined by QPCR in WT (open bars) and hKO (gray bars) livers from littermates and were normalized to WT levels. Means ± S.E. and p values (bilateral Student's t test) are given and n = 5 to 14 mice per group. **, p ≤ 0.005 and ***, p ≤ 0.0005.
The liver specificity of the Cre expression was assessed by genomic PCR that detected intact (flox) or recombined (Δ1 or Δ2) alleles at the Pcsk5 or Furin loci, respectively. Among all tissues tested, recombined alleles were detected only in liver (supplemental Fig. S1). No difference was observed between body or liver weight of WT and any hKO mice, indicating that lack of furin and/or PC5/6 or both in hepatocytes did not affect their growth (supplemental Fig. S2). No gross liver alterations were observed in paraffin sections stained with hematoxylin and eosin (supplemental Fig. S3). As the albumin promoter is only active at late embryonic stages, reaching a maximum 6 weeks after birth (28), the liver hemorrhages observed in complete PC5/6 knock-out mice (4) are likely due to an early role of PC5/6 in vessel development.
Furin from Hepatocytes Generates Circulating Inactivated Form of PCSK9
We analyzed PCSK9 inactivation in the plasma of mice lacking furin or PC5/6 by immunoprecipitation and Western blotting (Fig. 3) using the same anti-mouse PCSK9 and a Trueblot protocol (see “Experimental Procedures”). Although no signal was detected in PCSK9 KO plasma, WT plasma exhibited two bands corresponding to mature PCSK9 and inactivated form (ΔN221) cleaved at the mouse RFHR221↓ site, equivalent to the human RFHR218↓ site (21). Transgenic mice overexpressing V5-tagged mouse PCSK9 at low levels in the liver (16) exhibited the same two endogenous proteins, plus two equivalent forms that had a slightly higher molecular weight due to the presence of the C-terminal V5 tag (15 extra amino acids). Although PCSK9-ΔN221 levels in PC5/6-hKO plasma were unaffected, those in Fur-hKO (Fig. 3) or [Fur-PC5/6]hKO (data not shown) plasma were markedly reduced, indicating that furin from hepatocytes is the major PC responsible for the in vivo cleavage of PCSK9 in mice.
FIGURE 3.
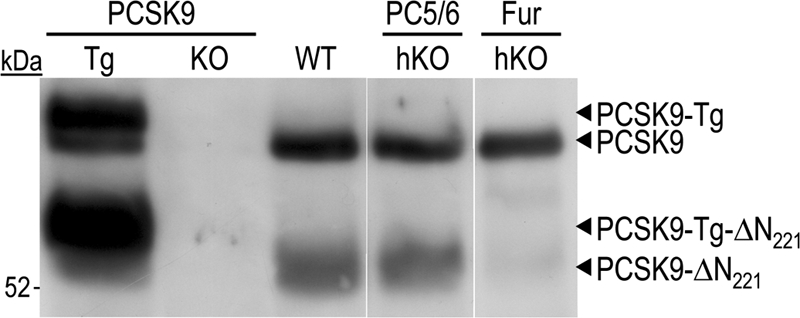
Inactivation of circulating PCSK9 by furin from hepatocytes. Plasma PCSK9 from PCSK9 transgenic and KO mice, as well as WT, Fur-hKO, and PC5/6-hKO mice was analyzed by immunoprecipitation and Western blotting. Endogenous and transgenic mature PCSK9 and inactivated PCSK9-ΔN221 migrations are shown.
LDLR Protein Levels Are Decreased in Liver Lacking Furin in Hepatocytes
Because PCSK9 triggers LDLR degradation (9), we verified whether the absence of furin or PC5/6 from hepatocytes affect LDLR protein levels in mice. Although no significant change in LDLR protein levels were observed in mice lacking PC5/6, those lacking furin in hepatocytes exhibited ∼26% lower LDLR levels (p < 0.005), with no significant change in the control transferrin receptor (Fig. 4). Because QPCR analysis of the same livers demonstrated that LDLR mRNA levels were not affected in Fur-hKO mice (Fig. 5), this indicated that the loss of furin favored LDLR protein degradation, likely due to the absence of PCSK9 cleavage. PCSK9 being a key regulator of the LDLR, we also checked its mRNA levels. Surprisingly, the mRNA levels were increased by 35% (p = 0.03) in Fur-hKO livers (Fig. 5). Thus, lower LDLR levels may also be due to higher PCSK9 mRNA levels. However, the 35% increase in PCSK9 mRNAs did not result in increased levels of circulating PCSK9 (Fig. 3).
FIGURE 4.
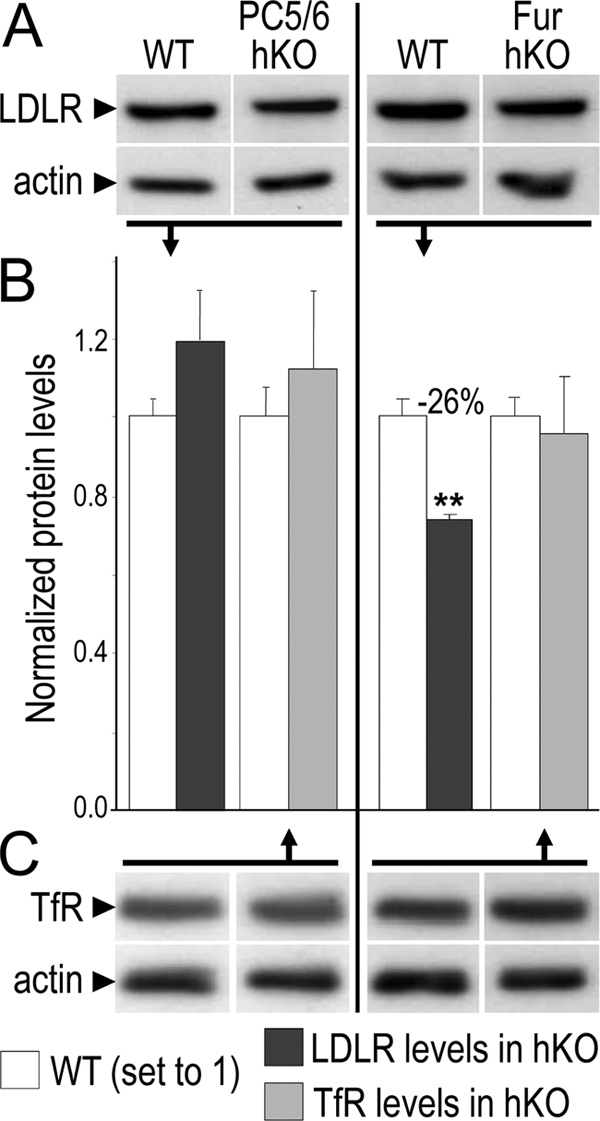
LDLR protein levels in hKO mice. Protein levels of the LDLR (A), transferrin receptor (TfR; C), and actin (A and C) were analyzed by Western blotting in liver extracts of three mice per genotype. Representative Western blots are shown. Normalized values are given as mean ± S.E. (B). **, p ≤ 0.005 values.
FIGURE 5.
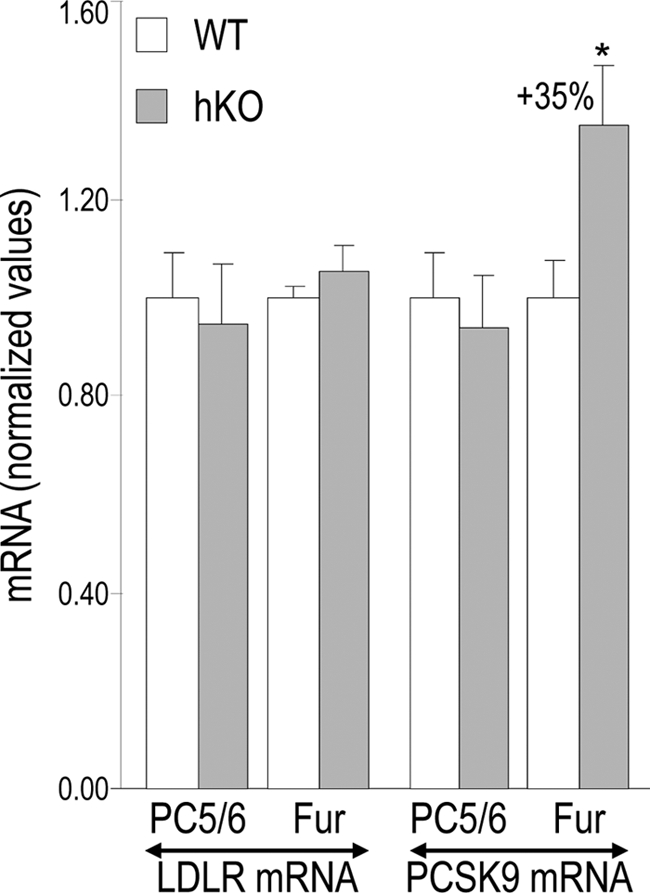
LDLR and PCSK9 mRNA levels in hKO mice. LDLR and PCSK9 mRNA levels were determined by QPCR in WT (open bars) and hKO (gray bars) livers from littermates and were normalized to WT levels. Means ± S.E. and p values (bilateral Student's t test) are given and n = 4 to 15 mice per group. *, p ≤ 0.05.
Inactivation of PCSK9 in Primary Hepatocytes
We next analyzed primary hepatocytes, as they represent the closest cellular model of the in vivo situation. PCSK9 was analyzed in 48-h conditioned media by immunoprecipitation and Western blotting (Fig. 6). Similar to the in vivo observations (Fig. 3), the absence of furin had a major effect. PCSK9-ΔN221 constituted 6% of the PCSK9 species in Fur-hKO hepatocytes, versus 23% in WT hepatocytes, representing a 74% drop in PCSK9-ΔN221. In hepatocytes, the lack of PC5/6 also resulted in a significant 44% drop in PCSK9-ΔN221 (13% versus 23%). When mouse PCSK9 antibody was used for blotting, we observed in the media of hepatocytes the presence of a novel ∼4-kDa lighter form of PCSK9 (∼50 kDa; PCSK9ΔC) (Fig. 6, top panel). The ∼50-kDa form was also detected in primary hepatocytes of transgenic mice expressing mouse PCSK9 C-terminally tagged with V5 (16). However, the latter form was not recognized by mAb V5 (Fig. 6, lower panel), indicating that PCSK9-ΔC lacks the last 30–40 residues, possibly by cleavage at the putative site RADR669↓.
FIGURE 6.
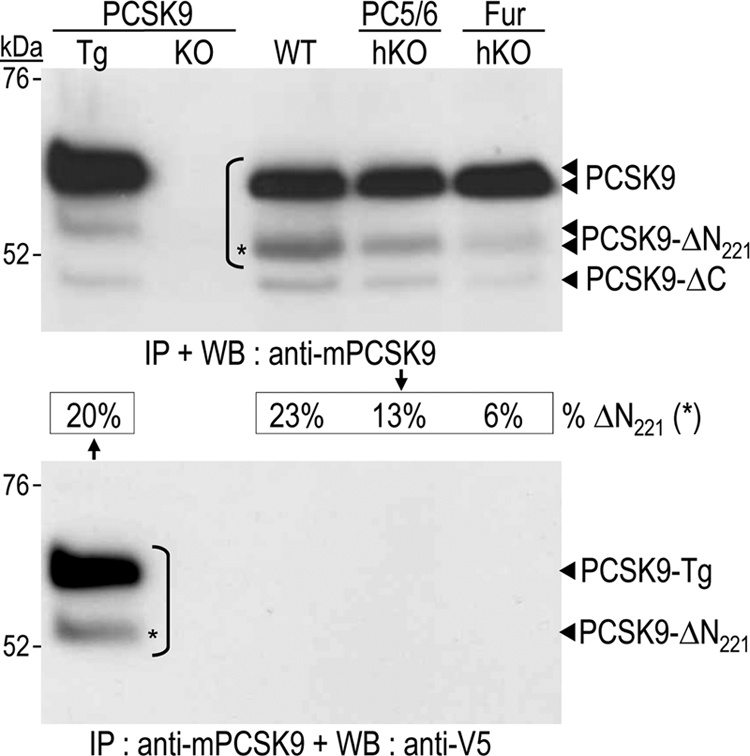
Inactivation of PCSK9 in primary hepatocytes. Hepatocytes were isolated from PCSK9 transgenic and KO mice, as well as WT, PC5/6-hKO, and Fur-hKO mice. Secreted PCSK9 was analyzed in 48-h conditioned media by immunoprecipitation (IP) with a mouse PCSK9 antibody and Western blotting (WB) with either the same mouse PCSK9 antibody (upper panel) or a V5 tag antibody (lower panel). The percentages of endogenous PCSK9 that underwent cleavage after Arg221 (PCSK9-ΔN221) are indicated.
When primary hepatocytes were transfected with a construct expressing a human V5-tagged PCSK9, the level of PCSK9-ΔN218 was reduced by ∼2.6-fold in Fur-hKO medium, and by ∼1.3-fold in PC5/6-hKO medium (Fig. 7). To further assess the role of the typical furin site RFHR218↓ in PCSK9 inactivation, we expressed the human gain of function F216L and R218S PCSK9 mutants in WT hepatocytes. Although the F216L substitution reduced by ∼3-fold the PCSK9 truncation, R218S abolished it (Fig. 7, right lanes), as reported previously in HEK293 cells (21).
FIGURE 7.

Impairment of PCSK9 cleavage by natural mutations of the furin site in primary hepatocytes. Primary hepatocytes from WT, PC5/6-hKO, or Fur-hKO mice were not transfected (NT) or transiently transfected with plasmids coding for V5-tagged human PCSK9 or its natural gain of function mutants F216L and R218S. Conditioned media were subjected to Western blotting 48 h post transfection using a V5 tag antibody.
To enhance the rate of PCSK9 cleavage and better determine the respective contributions of furin and PC5/6A, we co-expressed in double [Fur-PC5/6]hKO hepatocytes PCSK9 and either furin or PC5/6A (Fig. 8). No PCSK9 cleavage was observed in the absence of overexpressed furin or PC5/6A, demonstrating that no other endogenous protease(s) is involved in this cleavage. Furin was by far more efficient than PC5/6A to cleave WT or F216L PCSK9, whereas furin and PC5/6A did not process the R218S mutant. Interestingly, when the cleavage site was mutated into an optimized PC substrate, RRRR218EL (21), another hepatocyte PC was now able to process the protein, suggesting that PC7 and/or PACE4 can only process PCSK9 at an optimized processing site but not at the wild type site (Fig. 8). Co-expression of PCSK9 and furin led to the appearance of another N-terminally truncated form, still detected by its C-terminal V5 tag (Fig. 8, PCSK9-ΔNx). Its apparent molecular mass was reduced by 3–4 kDa compared with that of PCSK9ΔN218. This second cleavage was not observed upon R218S overexpression, suggesting that it requires prior cleavage at RFHR218↓ (Fig. 8). Cleavage at Arg218 could lead to a partial unfolding resulting in the exposure of the second site. Whether PCSK9-ΔNx is due to cleavage by a PC at the potential downstream site RSLR251↓ or by another furin-activated protease at any nearby site, is yet to be determined. In conclusion, furin is the best convertase to process, and hence inactivate, human PCSK9 at Arg218, and by analogy mouse PCSK9 at Arg221.
FIGURE 8.

Hepatocyte furin is the major PCSK9 cleaving enzyme at Arg218. Primary hepatocytes lacking both PC5/6 and furin were transiently cotransfected with plasmids expressing no protein (V), V5-tagged human PCSK9 (WT, F216L, R218S, and RRRR218EL) and no protein (V), furin (Fur), or PC5/6A. After 48 h, conditioned media were directly analyzed by Western blotting using a V5 antibody. Note the presence of a faster migrating PCSK9 species (PCSK9-ΔNx).
PCSK9 Cleavage Is Dependent on Furin and PC5/6 Membrane-bound Activities
To determine whether PCSK9 inactivation occurs intracellularly or in the medium, we compared the activity on PCSK9 of full-length and soluble shed furin, a form lacking the transmembrane and cytosolic domains and actively secreted (29). Although PC5/6A has no transmembrane domain, its cysteine-rich C-terminal domain anchors the protein at the cell surface via heparan sulfate proteoglycans (30). We thus compared the activities of overexpressed PC5/6A and PC5/6A-ΔC (Fig. 9A). Although PCSK9-ΔN218 was reduced by ∼2-fold in the presence of PC5/6AΔC, it was barely detectable in the presence of shed furin (sFur), suggesting that PCSK9 inactivation is primarily due to intracellular and/or cell surface membrane-bound activities. To determine whether secreted PCSK9 can be cleaved at the hepatocyte cell surface, conditioned media from [Fur-PC5/6]hKO hepatocytes overexpressing PCSK9 were incubated with HEK293 cells or WT, PC5/6-hKO and Fur-hKO primary hepatocytes (Fig. 9B) for 24 or 48 h. PCSK9 was partially cleaved by both HEK293 cells and primary hepatocytes in a time-dependent manner. PCSK9 inactivation was greatest when it was incubated with WT hepatocytes, intermediate with PC5/6-hKO hepatocytes and poor with Fur-hKO ones. This indicates that a limited, but significant, fraction of exogenous PCSK9 can be cleaved at the cell surface, preferentially by furin and to a lesser extent by PC5/6.
FIGURE 9.

PCSK9 inactivation at the cell surface of hepatocytes. A, [Fur-PC5/6]hKO hepatocytes were co-transfected with a vector expressing no protein (V; first lane) or human PCSK9 (lanes 2–6) and with vectors expressing no protein (V), furin (Fur), or PC5/6A, or their truncated versions shed furin (sFur) or PC5/6ΔC. B, a 48-h conditioned medium from [Fur-PC5/6]hKO hepatocytes overexpressing human PCSK9 was applied onto no cells (cell-free), HK293 cells, or WT, PC5/6-hKO, and Fur-hKO hepatocytes for 24 or 48 h. PCSK9 cleavage was investigated by Western blotting using a V5 antibody.
DISCUSSION
This study shows unambiguously that, in vivo, human PCSK9 is cleaved at the RFHR218↓ site, which is localized in an exposed and flexible loop of the catalytic domain of PCSK9 (18). This cleavage leads to loss of activity due to unfolding of the protein and detachment of its prosegment (21). In WT human subjects, PCSK9-ΔN218 represents 15–40% of total circulating PCSK9 (Fig. 1) (21). The most direct evidence for cleavage at RFHR218↓ is the >50% drop in PCSK9-ΔN218 levels observed in the plasma of heterozygous human patients exhibiting either the F216L or R218S gain of function mutations (Fig. 1). In agreement, PCSK9 carrying the F216L or R218S mutations was respectively poorly or not cleaved at all in primary hepatocytes (Fig. 7). Moreover, the R218S mutant remained uncleaved even upon overexpression of furin (Fig. 8). Thus, abrogation of PCSK9 inactivation likely leads to increased circulating PCSK9 levels from birth and to hypercholesterolemia. In agreement, upon overexpression in HepG2 cells of PCSK9 carrying the third known human mutation associated with hypercholesterolemia and affecting the RSLR218↓ site, i.e. R215H, LDL uptake dropped by ∼14% as compared with that of WT PCSK9, indicating lower cell surface LDLR levels (22).
Mouse plasma is also characterized by the presence of 30–50% of PCSK9-ΔN221 (equivalent to human PCSK9-ΔN218) (16). We have shown in this study that although the inactivation of PC5/6 in hepatocytes (PC5/6-hKO) had no effect on mouse plasma PCSK9-ΔN221, that of furin (Fur-hKO) led to its almost complete disappearance. This indicates that cleavage of the secreted PCSK9 at RFHR221↓ is primarily achieved by furin from hepatocytes (Fig. 3). In agreement, double [Fur-PC5/6]hKO mice also lack the inactivated form of PCSK9 (data not shown). The limited contribution of PC5/6 to PCSK9 inactivation in vivo may be related to its ∼90-fold lower expression in hepatocytes, as compared with furin (Fig. 2).
The analysis of endogenous PCSK9 inactivation in isolated hepatocytes showed that secreted PCSK9-ΔN221 was reduced by 74 and 44% in Fur-hKO and PC5/6-hKO hepatocytes, respectively (Fig. 6). Similarly, overexpression of human PCSK9 in the same hepatocytes revealed that the loss of endogenous furin or PC5/6 reduced the level of PCSK9-ΔN218 by 61 or 22%, respectively (Fig. 7). Thus, our data revealed a poor impact of PC5/6 on PCSK9-ΔN221 levels in vivo (Fig. 3), but a significant one in isolated primary hepatocytes (Figs. 5 and 6). This suggests that removal of hepatocytes from their natural environment enhances the ability of PC5/6A to process PCSK9, likely due to a change in extracellular matrix proteins that bind PC5/6A (2, 31).
No production of PCSK9-ΔN218 was observed in double [Fur-PC5/6]hKO hepatocytes overexpressing human PCSK9. Moreover, overexpression of PCSK9 with PACE4 or PC7 in these cells did not generate any cleaved form (data not shown). This is taken as evidence for the unique implication of furin and PC5/6 in cleavage at Arg218 (Fig. 8; second lane). Endogenous PC5/6A has a significant activity on PCSK9, even though it is ∼90-fold less expressed than furin in hepatocytes. Surprisingly, upon overexpression of PC5/6A, PCSK9 was not as well cleaved as with furin (Figs. 7 and 8), suggesting that PCSK9 is not a better substrate for furin, but that it better colocalizes with furin at the cell surface and/or in endosomes where processing likely occurs.
The membrane-bound furin cycles between the trans Golgi network and the cell surface, and some of it gets shed into the medium (32). Our data showed that a soluble form of furin, which traffics to the trans Golgi network and is then secreted, was not effective to inactivate PCSK9 (<10%, Fig. 9A), even though it was highly abundant in the medium. This suggests that cleavage at RFHR218↓ occurs mostly at the cell surface and/or endosomes. This conclusion was supported by the fact that exogenous PCSK9 could be partially processed if added onto HEK293 cells or primary hepatocytes that express furin endogenously (Fig. 9). This processing does not occur in the absence of furin and PC5/6 (Fig. 9B, first lane). Altogether, our data suggest that in vivo cleavage of PCSK9 by furin occurs primarily at the cell surface and/or endosomes of hepatocytes. In addition, the cleavage of PCSK9 by furin seems to be LDLR-independent as overexpressed PCSK9 was cleaved at Arg218 with the same efficiency in WT and Ldlr−/− (lacking the LDLR) primary hepatocytes (data not shown).
Thanks to our mouse models, we unambiguously identified furin from hepatocytes as the main PC involved in the cleavage of PCSK9 at RLHR218↓. However, because the loss of furin, which is expected to generate higher levels of active full-length PCSK9, also resulted in higher PCSK9 mRNA levels, it remains difficult to assess the relative contribution of the cleavage to the regulation of PCSK9 activity in hepatocytes. Although LDLR levels were reduced by 26% in Fur-hKO mice, no significant change was observed in circulating cholesterol (data not shown). There may be at least two reasons for this: first, because only 15–20% of the total cholesterol is associated to LDL particles in mice (versus >70% in humans), only a major change in LDL cholesterol can be assessed. Second, although our transgenic mice (low expressors) (16) expressed 3-fold more (+200%) liver PCSK9 mRNA as compared with nontransgenic mice, they only exhibited a nonsignificant +15% increase in circulating cholesterol, suggesting that the ∼26% increase in PCSK9 mRNA observed in Fur-hKO mice may have little or no detectable physiological effect.
From a therapeutic point of view, statins that inhibit 3-hydroxy-3-methyl-glutaryl-CoA reductase reduce cholesterol synthesis but also up-regulate the mRNA levels of both the LDLR and PCSK9 (26). Thus, inhibitors or repressors of PCSK9 are promising. An approach would be to combine a statin with an inducer of furin expression in vivo. Monotherapy with fibrates resulted in a significant ∼8.5% down-regulation of human plasma PCSK9 (33). Fibrates are agonists of the peroxisome proliferator-activated receptor α and also decrease PCSK9 transcription (34). Interestingly, fenofibrates also significantly increase the mRNA levels of both furin and PC5/6 (34). A combination of a statin and fibrates would thus be expected to increase the percentage of cleaved PCSK9-ΔN218 and enhance the response to statin therapy. Finally, circulating PCSK9 levels closely match those of LDL cholesterol with a similar diurnal rhythm with fasting reducing PCSK9 levels (35). Based on the present study, it would be informative to correlate these levels with those of furin in hepatocytes and whether changes in circulating PCSK9 levels during the diurnal rhythm affect both the full-length and the PCSK9-ΔN218 forms.
In conclusion, our data showed that, in vivo, furin is the best PC to process and inactivate PCSK9 by cleavage at RLHR218↓ at the cell surface and/or endosomes of hepatocytes and therein enhancing LDLR levels. Future work on the evaluation of various pharmacological strategies to decrease the level of PCSK9 would also benefit from measurements of the plasma levels of the furin cleaved form, as higher levels would be expected to enhance the therapeutic index of a PCSK9 down-regulating compound.
Supplementary Material
Acknowledgments
We are grateful to C. Toulouse for excellent technical help, B. Mary for efficacious editorial assistance, and to professor Gerald Luc (Lille, France) for providing the human F216L and R218S plasma. We thank all of the members of the Seidah laboratory for helpful discussions.
This work was supported by Canadian Institutes of Health Research Grants MOP-102741 and CTP-82946 (to N. G. S.), a Strauss Foundation grant (to N. G. S.), Canada Chair 201652 (to N. G. S.), Fonds voor Wentenschappelijk Vlaanderen (to J. W. C.), and Geconcerteerde Onderzoeks Actie 2008/16 Grant (to J. W. C.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- PC
- proprotein convertase
- LDLR
- low density lipoprotein receptor
- QPCR
- quantitative RT-PCR.
REFERENCES
- 1. Seidah N. G., Chrétien M. (1999) Brain Res. 848, 45–62 [DOI] [PubMed] [Google Scholar]
- 2. Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., Prat A. (2008) Int. J. Biochem. Cell Biol. 40, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 3. Roebroek A. J., Umans L., Pauli I. G., Robertson E. J., van Leuven F., Van de Ven W. J., Constam D. B. (1998) Development 125, 4863–4876 [DOI] [PubMed] [Google Scholar]
- 4. Essalmani R., Zaid A., Marcinkiewicz J., Chamberland A., Pasquato A., Seidah N. G., Prat A. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 5750–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Constam D. B., Robertson E. J. (2000) Genes Dev. 14, 1146–1155 [PMC free article] [PubMed] [Google Scholar]
- 6. Villeneuve P., Feliciangeli S., Croissandeau G., Seidah N. G., Mbikay M., Kitabgi P., Beaudet A. (2002) J. Neurochem. 82, 783–793 [DOI] [PubMed] [Google Scholar]
- 7. Seidah N. G., Prat A. (2007) J. Mol. Med. 85, 685–696 [DOI] [PubMed] [Google Scholar]
- 8. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 9. Maxwell K. N., Breslow J. L. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang D. W., Lagace T. A., Garuti R., Zhao Z., McDonald M., Horton J. D., Cohen J. C., Hobbs H. H. (2007) J. Biol. Chem. 282, 18602–18612 [DOI] [PubMed] [Google Scholar]
- 11. Maxwell K. N., Fisher E. A., Breslow J. L. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2069–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., Seidah N. G. (2007) Traffic. 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 13. Zhao Z., Tuakli-Wosornu Y., Lagace T. A., Kinch L., Grishin N. V., Horton J. D., Cohen J. C., Hobbs H. H. (2006) Am. J. Hum. Genet. 79, 514–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hooper A. J., Marais A. D., Tanyanyiwa D. M., Burnett J. R. (2007) Atherosclerosis 193, 445–448 [DOI] [PubMed] [Google Scholar]
- 15. Rashid S., Curtis D. E., Garuti R., Anderson N. N., Bashmakov Y., Ho Y. K., Hammer R. E., Moon Y. A., Horton J. D. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5374–5379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., Prat A. (2008) Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 17. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Nat. Struct. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 19. McNutt M. C., Lagace T. A., Horton J. D. (2007) J. Biol. Chem. 282, 20799–20803 [DOI] [PubMed] [Google Scholar]
- 20. Li J., Tumanut C., Gavigan J. A., Huang W. J., Hampton E. N., Tumanut R., Suen K. F., Trauger J. W., Spraggon G., Lesley S. A., Liau G., Yowe D., Harris J. L. (2007) Biochem. J. 406, 203–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Benjannet S., Rhainds D., Hamelin J., Nassoury N., Seidah N. G. (2006) J. Biol. Chem. 281, 30561–30572 [DOI] [PubMed] [Google Scholar]
- 22. Cameron J., Holla O. L., Laerdahl J. K., Kulseth M. A., Ranheim T., Rognes T., Berge K. E., Leren T. P. (2008) J. Intern. Med. 263, 420–431 [DOI] [PubMed] [Google Scholar]
- 23. Allard D., Amsellem S., Abifadel M., Trillard M., Devillers M., Luc G., Krempf M., Reznik Y., Girardet J. P., Fredenrich A., Junien C., Varret M., Boileau C., Benlian P., Rabès J. P. (2005) Hum. Mutat. 26, 497–506 [DOI] [PubMed] [Google Scholar]
- 24. Roebroek A. J., Taylor N. A., Louagie E., Pauli I., Smeijers L., Snellinx A., Lauwers A., Van de Ven W. J., Hartmann D., Creemers J. W. (2004) J. Biol. Chem. 279, 53442–53450 [DOI] [PubMed] [Google Scholar]
- 25. Postic C., Shiota M., Niswender K. D., Jetton T. L., Chen Y., Moates J. M., Shelton K. D., Lindner J., Cherrington A. D., Magnuson M. A. (1999) J. Biol. Chem. 274, 305–315 [DOI] [PubMed] [Google Scholar]
- 26. Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N. G., Bernier L., Prat A. (2004) Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 27. Dubuc G., Tremblay M., Paré G., Jacques H., Hamelin J., Benjannet S., Boulet L., Genest J., Bernier L., Seidah N. G., Davignon J. (2010) J. Lipid Res. 51, 140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Postic C., Magnuson M. A. (2000) Genesis. 26, 149–150 [DOI] [PubMed] [Google Scholar]
- 29. Decroly E., Wouters S., Di Bello C., Lazure C., Ruysschaert J. M., Seidah N. G. (1996) J. Biol. Chem. 271, 30442–30450 [DOI] [PubMed] [Google Scholar]
- 30. Nour N., Mayer G., Mort J. S., Salvas A., Mbikay M., Morrison C. J., Overall C. M., Seidah N. G. (2005) Mol. Biol. Cell 16, 5215–5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mayer G., Hamelin J., Asselin M. C., Pasquato A., Marcinkiewicz E., Tang M., Tabibzadeh S., Seidah N. G. (2008) J. Biol. Chem. 283, 2373–2384 [DOI] [PubMed] [Google Scholar]
- 32. Thomas G. (2002) Nat. Rev. Mol. Cell Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mayne J., Dewpura T., Raymond A., Cousins M., Chaplin A., Lahey K. A., Lahaye S. A., Mbikay M., Ooi T. C., Chrétien M. (2008) Lipids Health Dis. 7, 22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kourimate S., Le May C., Langhi C., Jarnoux A. L., Ouguerram K., Zaïr Y., Nguyen P., Krempf M., Cariou B., Costet P. (2008) J. Biol. Chem. 283, 9666–9673 [DOI] [PubMed] [Google Scholar]
- 35. Persson L., Cao G., Stahle L., Sjoberg B., Troutt J. S., Konrad R. J., Galman C., Wallen H., Eriksson M., Hafstrom I., Lind S., Dahlin M., Amark P., Angelin B., Rudling M. (2010) Arterioscler. Thromb. Vasc. Biol. 30, 2666–2672 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.