

Abstract
Protein secretion in bacteria is driven through the ubiquitous SecYEG complex by the ATPase SecA. The structure of SecYEG alone or as a complex with SecA in detergent reveal a monomeric heterotrimer enclosing a central protein channel, yet in membranes it is dimeric. We have addressed the functional significance of the oligomeric status of SecYEG in protein translocation using single molecule and ensemble methods. The results show that while monomers are sufficient for the SecA- and ATP-dependent association of SecYEG with pre-protein, active transport requires SecYEG dimers arranged in the back-to-back conformation. Molecular modeling of this dimeric structure, in conjunction with the new functional data, provides a rationale for the presence of both active and passive copies of SecYEG in the functional translocon.
Keywords: ATPases, Membrane Proteins, Protein Structure, Protein Translocation, Single Molecule Biophysics, SecY Complex
Introduction
The heterotrimeric SecYEG complex forms the protein channel of a ubiquitous protein translocation machinery responsible for secretion and membrane protein insertion. Transport is coupled either directly to protein synthesis from bound ribosomes or post-translationally to specialized energy transducers. Bacteria utilize SecA for the latter role, wherein cycles of ATP hydrolysis induce conformational changes that are translated into a directional mechanical drive (1, 2).
Despite the elucidation of several structures of SecYEG in isolation, as well as bound to SecA or ribosomes (3–7), the oligomeric status and arrangement of subunits in the active translocon remain unresolved. The SecYEG complex visualized in the presence of high detergent concentrations is monomeric (3), irrespective of the presence of its physiological partners SecA (4) or ribosome nascent chain complexes (7, 8). However, at low concentrations of detergent or in the presence of phospholipids, particularly cardiolipin (CL),3 SecYEG forms dimers (9, 10). The structure of the membrane-bound SecYEG complex is a dimer in a so-called back-to-back orientation, in which transmembrane segments (TMS) from two adjacent SecE subunits form the interface (6, 11).
Given that the protein channel is formed by a single copy of SecYEG (3, 12), the role of dimerization in SecYEG function remains unclear. A genetically fused tandem of SecY retains translocation activity (13), although this requires only one of the copies to be functional (14). It appears therefore, that in the dimeric form of the complex, one SecYEG protomer is engaged in protein translocation and is associated with a passive non-translocating partner. However, it has yet to be determined whether or not a monomeric SecYEG complex is sufficient for successful translocation alone, or if the dimeric arrangement is obligatory. The function of the passive protomer in the latter has so far not been addressed.
The structure of SecA comprises a pre-protein cross-linking domain (PPXD), an α-helical scaffold domain (HSD), an α-helical wing domain (HWD), and two RecA-like nucleotide-binding domains (NBD1 and NBD2), between which ATP is bound and hydrolyzed (15). Crystal structures have been obtained both for dimers of SecA (15) and for monomers of the enzyme (16), but dimers predominate in solution (17, 18). The structure of SecA bound to SecYEG in a 1:1 stoichiometry in the presence of an ATP analog (ADP-BeFx or ADP-AlFx), reveals a major conformational change in the PPXD, which rotates away from the HWD to contact NBD2, in comparison with the structure of SecA alone (4). Another consequence of the interaction is the insertion of a two-helix finger motif, part of the HSD of SecA, into the entrance of the channel at the center of the SecY subunit, consistent with a direct role for the motif in pre-protein transport (4). Its close proximity to the inserted protein substrate, which extends from SecA into SecYEG, has been confirmed by thiol cross-linking and mutagenesis studies (19). Therefore, the structure of the SecA-SecYEG complex represents SecA associated with the active translocating SecYEG protomer.
To better understand the translocation reaction, we sought to address unresolved questions on the oligomeric state and arrangement of the channel within the active translocon. Single molecule fluorescence imaging revealed that SecYEG monomers can bind the pre-protein substrate proOmpA, but cannot transport it across the membrane; dimers of SecYEG are necessary to complete transport. The arrangement of the physiological dimer was examined by generation of cross-linked species trapped in the back-to-back arrangement, followed by assessment of its behavior in functional assays and single molecule fluorescence experiments. The results were used to guide the construction of an atomic model of the membrane-bound Escherichia coli SecA-(SecYEG)2 complex, examination of which rationalizes the necessity for an additional passive SecYEG protomer within a back-to-back dimer.
EXPERIMENTAL PROCEDURES
Expression and Purification of the Translocation Components
Full-length proOmpA, a truncated version (proOmpAΔ176) and SecA were purified as previously described (20). The mutant SecA795C was stored in higher concentrations of DTT (10 mm) to prevent disulfide bond formation between cysteine residues that otherwise readily formed at the dimer interface.
Mutant and wild-type SecYEG were overexpressed and purified as previously reported (20, 21). To obtain sufficient quantities of the tandem dimeric version of SecYEG (13), 20 liters of the appropriate strain were grown to a A600 nm of 1 in a 30-liter Bio Bench bioreactor (Applikon Biotechnology), induced with 1 mm IPTG and harvested after 3.5 h.
Inner membrane vesicles (IMVs) harboring overexpressed SecYEG were made by resuspending the total membrane fraction from 1–2 liters of starting liquid culture into 5–10 ml of 20 mm Tris-HCl, pH 8.0, and 50 mm KCl (TK), followed by loading onto a sucrose step gradient of 36, 45, 51, and 56% (w/v) and centrifugation at 180,000 × g for 2 h at 4 °C. The brown IMV fraction in the middle of the tubes was collected, diluted in TK and re-centrifuged. The pellet was resuspended in TK buffer.
Photo-inducible Cross-linking of SecYEG
Lipid-depleted SecYEG (10) was reconstituted into liposomes composed of total E. coli polar lipids as previously described (21). The resultant proteoliposomes (containing 5.4 μm SecYEG), and detergent-solubilized SecYEG (4 μm) in the absence or presence of 60 μm CL, were cross-linked by visible light radiation in the presence of 2 mm ammonium persulfate and either 0.1 mm or 0.8 mm Tris-bipyridylruthenium(II) for 45 s or 3 min, respectively (10, 22). IMVs (8 mg/ml protein) were cross-linked with either 1 mm or 2 mm Tris-bipyridylruthenium(II) for 1 or 2 min. After irradiation, the reaction was quenched with 0.1 m DTT, and the cross-linked products were identified by SDS-PAGE and mass spectrometry, or by Western blotting using antibodies to SecY and SecE.
Cysteine Labeling of Specific Translocation Components
Single cysteine mutants (SecYEL106CG and SecYA103CEG) were generated in a cysteine-less background of SecYEG (23). SecY103-AF488EG (Y*EG) and SecYY97-AF488EG (YY*EG) were produced by respectively labeling SecYA103CEG and the tandem dimer engineered with a unique cysteine on the second copy of SecY, SecYYV97CEG, with Alexa Fluor 488 C5-maleimide (Molecular Probes, Invitrogen), using a 2-fold molar excess of dye over the mutant complex. The mixtures were incubated on ice for 1 h and purified by size-exclusion chromatography (PD10 and/or Superdex 200 HR, XK 16/60 or a Superose 12HR column, GE Healthcare). The bound dye was almost exclusively associated with the SecY or SecYY subunits and present at the expected 1:1 molar ratio, as judged by absorption spectroscopy.
proOmpAΔ176 containing 2 cysteine residues (S296C and C311) was similarly labeled with Alexa Fluor 488 C5-maleimide in 50 mm K-HEPES, pH 7.5, containing 6 m urea. The resultant fluorescent protein, designated proOmpAA488, was purified by size-exclusion and reverse-phase chromatography. The fully labeled product eluted from a C8-column in a linear gradient of 5–95% (v/v) acetonitrile, 0.1% (v/v) trifluoroacetic acid and was dried in a SpeedVac system and re-dissolved in 50 mm K-HEPES, pH 7.5, containing 6 m urea.
SecA795Fl was produced by labeling SecA795C with 5-iodoacetamidofluorescein (Molecular Probes, Invitrogen). 5-Iodoacetamidofluorescein (60 μm; dissolved in DMSO), and 30 μm SecAA795C in 20 mm Tris, pH 7.5, 100 mm KCl, 10 mm DTT were combined such that DMSO was diluted 1 in 20. Reactions were incubated at 25 °C for 1.5 h, and quenched with 50 mm DTT. The unreacted probe was separated from labeled protein by size exclusion chromatography (Superose 10/300 column, G.E. Healthcare) in 20 mm Tris, pH 7.5, and 100 mm KCl.
Formation of Covalently Cross-linked SecYEG Dimers by Oxidation of Specific Single Cysteine Mutants
Total membranes overexpressing SecYEL106CG were resuspended in 20 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 10% (v/v) glycerol containing 2 mm CuPh and incubated for 1 h at 4 °C. The oxidizing agent was removed by dialysis and the cross-linked SecYEG was purified in the usual way (21). SecYEL106CG was fully cross-linked, and so uncross-linked material, for the purpose of comparison, was derived from non-oxidized membranes. The purified material was analyzed by non-reducing SDS-PAGE and size-exclusion experiments conducted on a Superose 6 HR column in SecYEG isolation buffer.
Measurement of the Stimulation of SecA ATPase Activity
Steady-state SecA ATPase measurements were monitored at 25 °C using a pyruvate kinase (PK)/lactate dehydrogenase (LDH)-coupled assay as previously described (18). Data were fitted to a one-site quadratic tight ligand-binding equation with background, Equation 1 defined as,
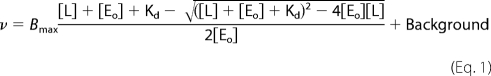 |
where v is equal to enzyme velocity, Bmax is the total capacity of SecA-ligand, [L] is the total ligand (i.e. SecYEG or proOmpA) concentration, [Eo] is the total SecA concentration, and Kd is the dissociation constant for SecA ligand. The Background is the ATPase activity without added ligand.
Measurement of the Affinity of the Interaction between SecYEG and SecA
SecA795Fl fluorescence was monitored using a Jobin Yvon Fluorolog (Horiba Scientific) at excitation and emission wavelengths of 495 nm and 515 nm, respectively. Assays were performed in 20 mm Tris, pH 8; 130 mm NaCl; 10% glycerol; 2 mm MgCl2; 0.1% dodecyl nonaoxyethylene ether (C12E9); 1 mm AMPPNP, and 2 nm SecA795Fl. Where appropriate, 40 μm CL was included, and SecYEG stocks were pre-incubated with 40 μm CL for 1 h. The specificity of the interaction was assessed by competition with 1 μm wild-type SecA. Where titrations were performed in weak binding conditions (i.e. when the Kd was greater than the SecA795Fl concentration), data were fitted to the one-site weak binding Equation 2, defined as,
 |
where [L] is the total concentration of ligand (SecYEG), F is the signal change, Fmax is the maximum signal change, and Kd is the dissociation constant. When titrations were performed under tight binding conditions (i.e. when the Kd was equal to or lower than the SecA795Fl concentration), data were fitted to the tight ligand-binding Equation 3,
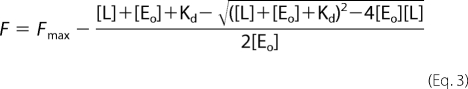 |
where [Eo] is the SecA795Fl concentration.
In Vitro Translocation
Protein translocation assays were performed as previously described (20). The translocated product was analyzed by SDS-PAGE and where indicated detected by Western blotting using an antibody against proOmpA or by fluorescence using a Typhoon 9400 (Molecular Dynamics) with laser excitation at 488 nm and a 526 nm emission filter.
Proteoliposome Immobilization for Single Molecule Fluorescence Imaging
SecYEG or SecYA103CEG selectively labeled with Alexa Fluor 488 (SecY103-AF488EG) were reconstituted into vesicles as described previously (20) with lipid vesicles containing a 98.5:1.5 (mol/mol) mixture of E. coli polar extract lipids and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) (biotin-DOPE). Extruded SecYEG proteoliposomes (100 nm) were tethered by streptavidin-biotin chemistry onto a supported lipid bilayer (supplemental data).
Single Molecule ProOmpAA488 Translocation Reaction
Substrate binding and fluorescence quenching experiments employed proOmpAΔ176 labeled with Alexa Fluor 488 (proOmpAA488) and were conducted at 25 °C. Where indicated, the coverslip bearing immobilized proteoliposomes was exposed to 20 μg/ml SecA and 2.5 mm ATP. proOmpAA488 was added in a 50-fold molar excess with respect to the total amount of unlabeled SecYEG added to the surface, incubated for 20 min, and washed five times with translocation buffer to remove excess proOmpAA488. To distinguish binding from transport, 30 mm tryptophan (quencher) was added to the SecYEG vesicles/SecA/ATP solution for 20 min before visualization using a custom-built TIRF microscope (24) (supplemental data).
Molecular Modeling of the E. coli Membrane-bound SecA-(SecYEG)2 Complex
The active copy of E. coli SecYEG was created by standard homology modeling of the E. coli sequences on to the T. maritima template structure (PDB accession 3DIN) (4). The passive copy of SecYEG was modeled on the basis of the M. jannaschii SecYEβ structure (PDB accession 1RH5) (3). Both copies were then combined by fitting onto the membrane-bound dimeric structure (11). Where possible, the known structures of E. coli SecA were fitted domain by domain (25), also according to the T. maritima SecA-(SecYEG)1 structure followed by a refinement protocol. Further details are provided in supplemental data and Fig. S1.
RESULTS
Reconstituted and Native Membrane Vesicles Harbor SecYEG Dimers Arranged in the Back-to-Back Conformation
To assess the oligomeric status and arrangement of the SecYEG complex in membranes harboring an active translocon, we employed nonspecific photoactivated chemical cross-linking. Proteoliposomes reconstituted with SecYEG, as well as IMVs capable of SecA- and ATP-dependent transport of proOmpA (20), were irradiated with light in the presence of the cross-linker Tris-bipyridylruthenium(II) and analyzed by SDS-PAGE and additionally, for IMVs, by Western blotting. In both cases products were formed with the same mobility as a tandem dimer of SecY produced by genetic fusion (13) (Fig. 1, a and b; YY), identifying them as a cross-linked dimer of SecY subunits (Fig. 1, a and b; SecY-Y). A second band resulting from cross-linking exhibited the same mobility as the disulfide linked dimer of SecE (SecE-E) produced by the oxidation of membranes expressing SecYEL106CG (26) (106X; Fig. 1, a and c). Mass spectrometry confirmed the identities of the cross-linked species (indicated by ‡ in Fig. 1a) as indicated SecY-Y and SecE-E.
FIGURE 1.

Probing the oligomeric state of SecYEG in active reconstituted and native membranes. a, photo-cross-linking of detergent-solubilized SecYEG (YEG-soluble) in the absence and presence of 60 μm CL, or in reconstituted proteoliposomes (YEG-membrane), was performed by exposure to light radiation. Proteins were subsequently separated by SDS-PAGE and visualized by Coomassie Blue staining. A marker for cross-linked SecY-Y was provided by a genetically-fused, tandem dimer of SecY (YY) (13) and for SecE-E by SecYEL106CG (106X) (26). * denotes samples that have been irradiated. The predicted compositions indicated for the cross-linked species are consistent with their observed mobilities, and the bands indicated by ‡ were confirmed by mass spectrometry to contain accordingly SecY or SecE. b and c, IMVs overexpressing SecYEG (YEG-IMVs) were photo-cross-linked, separated by SDS-PAGE, transferred to nitrocellulose for Western blotting and probed with monoclonal antibodies against SecY (b) and SecE (c). − denotes no irradiation; samples in lanes 1 and 2 were irradiated for 90 s with 1 mm or 2 mm cross-linker, respectively, and likewise, lanes 3 and 4 for 180 s with 1 mm or 2 mm.
The extent of cross-linking was greatly reduced when SecYEG was destabilized by high concentrations of detergent (9), when primarily cross-links between SecY and SecE, likely to represent intra-protomer cross-linking, were detected (SecY-E, Fig. 1a). However, as expected, the SecY-Y and SecE-E cross-links were largely reinstated in solution in the presence of cardiolipin (CL) (Fig. 1a) (10). Therefore, this lipid appears to be stabilizing the membrane-bound form of the complex. The identification of these cross-links in membranes highly active in protein translocation assays (20), indicates that the membranes harbor SecYEG dimers in contact via adjacent SecE and SecY subunits, characteristic of the back-to-back orientation (6, 11).
SecYEG Dimers Arranged in the Back-to-Back Configuration Are Required for a Productive, High Affinity Interaction with SecA
A further series of experiments were designed to test the functional characteristics of the SecYEG dimeric back-to-back orientation identified in membranes. The ATPase activity of SecA is partially activated, or primed, upon the interaction with SecYEG; further stimulation is then associated with pre-protein binding and translocation (20, 27). The former response can be used to measure the apparent association behavior of SecYEG with SecA in detergent solution (10, 20). This association is tightened by the presence of lipids (particularly CL), which stabilize the SecYEG dimer, resulting in a higher affinity binding site for SecA. In addition, CL bound to SecYEG confers an increase in the turnover of ATP by SecA (10). We have used these two properties, which are separate and distinct from one another, to assess the functionality of SecYEG dimers in the back-to-back configuration.
To generate a stable, detergent-soluble version of the back-to-back SecYEG dimer we exploited the mutant SecYEL106CG, which has been shown to form cross-linked dimers in native IMVs (26). Membranes overexpressing the mutant were incubated with copper-phenanthroline (CuPh) to generate covalently bound dimers associated at the back-to-back interface (Fig. 2a). As was previously shown, the cross-linking procedure was highly efficient. The resulting dimer was then purified from aggregated material using gel filtration, enabling its functional characteristics to be unequivocally defined. Analysis by non-reducing gel electrophoresis revealed the purified complex to be fully cross-linked (Fig. 2b; note the absence of uncross-linked SecE). A comparison with the uncross-linked version of the mutant and with the wild-type complex by analytical size-exclusion chromatography (Fig. 2c), showed that the cross-linked version was exclusively dimeric.
FIGURE 2.

The back-to-back form of the SecYEG cross-linked in the membrane is stable and dimeric in solution. a, schematic side view and ribbon representation of the back-to-back (6) dimer of SecYEG: SecY subunits (dark and light gray), SecE (TMS 3 and the amphipathic cytosolic helix, pink), SecG (tan). In the scheme, SecG is not shown, and the channel and lateral gate (for TMS insertion (3), dashed lines) are closed. The cross-link between the two cysteines is shown by a blue bar. In the ribbon diagram, the cysteine residues employed for cross-linking, SecEL106C (26) (blue), and for labeling with AF488, SecYA103C (red; see Fig. 5), are shown in space-fill. b, non-reducing SDS-PAGE and c, size-exclusion chromatography analysis of the purified wild-type complex (YEG), and uncross-linked (106) and cross-linked (106X) forms of SecYEL106CG, the latter as a result of CuPh treatment of membranes prior to extraction and purification. The mobilities of the individual subunits and the cross-linked dimers of SecE (SecE-E) are indicated.
The ability of these back-to-back SecYEG dimers to interact productively with SecA and stimulate its ATPase activity was tested and compared with the uncross-linked version, which is predominantly monomeric when diluted in detergent solution without lipids (Fig. 2c) (9, 10, 21). As before (10), the monomeric form of SecYEG interacts weakly with SecA (Kd ∼ 910 nm) while the dimeric version, stabilized by CL, binds more tightly (Kd ∼ 65 nm) (Fig. 3, a and b and Table 1). In contrast, the back-to-back cross-linked species associates tightly with SecA even in the absence of CL (Kd ∼ 78 nm) (Fig. 3, a and b and Table 1).
FIGURE 3.

The back-to-back version of the SecYEG dimer provides a high affinity platform required for the productive association of SecA. a, analysis of SecA ATPase stimulation by monomeric (YEG) and back-to-back dimeric forms (106X) of SecYEG. The data obtained in the absence of CL are shown again, using an expanded scale in b. ATPase activity of SecA (0.3 μm) was measured in detergent solution (0.03% C12E9) in the presence of 40 μm CL, with increasing concentrations of either wild-type SecYEG (YEG, black solid line, open circles) or SecYEL106CG (106X, gray solid line, open circles), or in the absence of CL (black and gray dashed lines and filled circles, respectively). With the exception of SecYEL106CG +CL (106X + CL), which showed decreased activities at high concentrations, probably as the result of aggregation, the data were fitted according to a tight binding equation (“Experimental Procedures”). Error bars represent S.D. (n > 3). c, estimation of the affinity of the monomeric (YEG) and back-to-back dimeric forms (106X) of SecYEG for SecA by fluorescence spectroscopy. Wild-type SecYEG (YEG) or cross-linked SecYEL106CG (106X) were titrated into a solution of 2 nm SecA795Fl in the presence or absence of 40 μm CL. The data were fitted to Equations 2 or 3 (“Experimental Procedures”). Controls were performed with the addition of an excess of unlabeled wild-type SecA (xs A), which competed for the binding and obliterated the fluorescence change. Kd values from the fitting procedures are shown in Table 1.
TABLE 1.
Kd values calculated from fitting the data shown in Fig. 3
Calculated Kd values for SecYEG binding to SecAADP were measured via steady-state ATPase assays, and for SecAAMPPNP via fluorescence spectroscopy. In the latter case, binding to cross-linked SecYEG (106X) gave Kd values significantly less than the enzyme concentration, and as such cannot be accurately determined from the data, but are interpreted as <1 nm.
| Kd SecAADP-SecYEG | Kd SecAAMPPNP-SecYEG | |
|---|---|---|
| nm | nm | |
| Wild-type SecYEG | 914 ± 288 | 42 ± 1.9 |
| Wild-type SecYEG + CL | 65 ± 45 | 2.3 ± 0.22 |
| SecYEL106CG cross-linked (106x) | 78 ± 49 | <1 nm |
| SecYEL106CG cross-linked (106x) + CL | Not fitted | <1 nm |
The additional ability of CL to stimulate the ATPase activity of SecA (10), was shown again here for the wild-type, as well as for the back-to-back cross-linked form (Fig. 3a). The reduction in activity induced in the latter instance at higher concentrations of SecYEG is most likely caused by its increased susceptibility to CL-induced aggregation, also observed in the SecYYEG genetically conjoined tandem (10, 13). Nevertheless, the effect was clear; that is, the back-to-back dimer forms a high affinity and productive complex with the steady-state complex of SecA, the state bound to ADP (20).
Further experiments were performed to test the interaction of SecYEG dimers with SecA in the ATP-bound state using the non-hydrolyzable ATP analog AMPPNP. A mutant, SecAA795C, was derivatized with fluorescein (labeled protein designated SecA795Fl) and its fluorescence was found to be quenched upon interaction with SecYEG and AMPPNP (Fig. 3c); reciprocating the behavior exhibited by SecY268FlEG when exposed to SecA and AMPPNP (23). The specificity of the response was demonstrated by successful competition with a large excess of unlabeled wild-type SecA.
Wild-type SecYEG, monomeric in detergent solution in the absence of lipids, interacted more tightly with SecAAMPPNP (Kd ∼ 42 nm) than with SecAADP (Table 1). As above, the formation of SecYEG dimers induced by CL resulted in a 10-fold increase in the affinity (Kd ∼2.3 nm). In contrast, the back-to-back cross-linked dimer bound very tightly to SecA (Kd < 1 nm), regardless of the presence of CL (Fig. 3c and Table 1). The data confirm that the back-to-back arrangement of the SecYEG dimer favorably interacts with both the ATP- and ADP-associated forms of SecA.
SecYEG Dimers Arranged in the Back-to-Back Configuration Are Fully Functional in Protein Translocation
Having established that the back-to-back SecYEG dimer productively associates with SecA, the complex was reconstituted into phospholipid vesicles and tested for its ability to fully stimulate SecA ATPase activity and drive translocation (Fig. 4). The binding parameters for proOmpA and the SecA-SecYEG complex showed that the SecYEL106CG cross-linked dimer has the same turnover and affinity for the substrate as the uncross-linked mutant and the wild type (Fig. 4a and Table 2). Likewise, the ability of the complex to couple the ATPase activity to protein transport was also unaffected by cross-linking (Fig. 4b). The results show that the membrane-bound SecYEG dimers, covalently linked through the SecE subunits (106X), are fully functional and indistinguishable from the wild-type complex. Therefore, the transport reaction is unlikely to involve very large structural rearrangements at the dimer interface.
FIGURE 4.

The SecYEG complex held in the back-to-back orientation is fully active in ATP driven translocation. a, steady-state analysis of the translocation-coupled ATPase activity stimulated by membrane-bound SecYEG and proOmpA. ATPase activity of SecA (50 nm) was measured in the presence of 1 mm ATP and 0.6 μm SecYEG in proteoliposomes with increasing concentrations of proOmpA as indicated, applied from a stock dissolved in 6 m urea. Proteoliposomes were reconstituted with wild-type SecYEG (YEG), SecYEL106CG (106), or cross-linked SecYEL106CG (back-to-back dimer, 106X). The data sets were fitted according to Equation 1 (“Experimental Procedures”) and the parameters (Kd and kcat,) are shown in Table 2. b, measurement of in vitro translocation of proOmpA (0.7 μm) into the same SecYEG proteoliposomes (YEG, 106, and 106X) in the presence of SecA. The extent of proOmpA translocation, i.e. protection from protease digestion, was detected by Western blotting and compared with a measure of 10% of the total proOmpA in each reaction (ATP +, Protease −) and a negative control performed in the absence of ATP (ATP −, Protease +).
TABLE 2.
Kd and kcat values for SecA-SecYEG binding to proOmpA, calculated from fitting the data shown in Fig. 4
| Kd[SecA-SecYEG]-proOmpA | kcat | |
|---|---|---|
| nm | s−1 | |
| Wild-type SecYEG | 72 ± 41 | 518 ± 69 |
| SecYEL106CG uncross-linked (106) | 93 ± 27 | 627 ± 45 |
| SecYEL106CG cross-linked (106x) | 92 ± 21 | 504 ± 28 |
SecYEG Dimers Arranged in the Back-to-Back Configuration Are Obligatory for Protein Translocation
The results described above indicate that the back-to-back dimeric SecYEG complex is competent for protein transport. However, the ensemble approaches used cannot reveal whether this dimeric arrangement is obligatory for translocation. To address this question, single molecule total internal reflection fluorescence (TIRF) microscopy was used to assess the functional capability of SecYEG complexes in different oligomeric forms. To this end, SecYEG harboring a single cysteine residue incorporated within a cytoplasmic loop at position 103 in SecY was labeled with Alexa Fluor 488 C5-maleimide to generate channels bearing a fluorophore at a single site (SecY103-AF488EG; Fig. 2a).
Proteoliposomes spiked with biotinylated lipid (100 nm diameter) reconstituted with SecY103-AF488EG were then immobilized onto a lipid bilayer surface that also contained biotinylated lipid, using a streptavidin linker, and visualized by single molecule TIRF microscopy (supplemental data) (Fig. 5a). The vesicle density on the surface was optimized such that ∼ 85 vesicles harboring SecY103-AF488EG could be recorded simultaneously within an evenly illuminated image area. The number of SecYEG molecules contributing to each fluorescent spot was then quantified in proteoliposome samples reconstituted at different lipid-to-protein ratios (LPRs) by recording the number of discrete photobleaching events. The results of these experiments showed that a low concentration of SecY103-AF488EG (1.5 nm) and high LPR (73000:1) resulted in more than 90% of the fluorescent vesicles containing translocons that photobleached in a single-step, indicative of the presence of a single labeled SecYEG monomer per liposome (Fig. 5, a and b). By contrast, reconstitution at lower LPR resulted in proteoliposomes that primarily contained SecYEG in dimeric and/or higher oligomeric forms.
FIGURE 5.

Single molecule fluorescence measurements of protein translocation and the oligomeric state of SecYEG. a, representative time-course of fluorescence intensity showing quantized photobleaching in a single step for a vesicle harboring a single SecY103-AF488EG molecule. The images were recorded at a video rate of 60 ms/image frame with an Andor iXon EMCCD camera. The y axis plots the photon counts (not corrected for background). Inset, representative image obtained by TIRF microscopy showing a field of tethered SecY103-AF488EG vesicles. b, distribution of different types of photobleaching events observed for 500 vesicles reconstituted at different LPRs. Trajectories that could not be clearly described as representing single, double or discrete numbers of multiple step(s) are designated “unclassified”. c, ATPase activity of 0.3 μm SecA measured in the presence of 1 mm ATP in the absence (basal) and in the presence of 1 μm SecYEG in proteoliposomes without protein substrate (membrane) or with 0.7 μm of either wild-type proOmpA, proOmpAΔ176 (proOmpA 2 cys) or Alexa Fluor 488-labeled proOmpAΔ176 (proOmpAA488). Error bars represent S.D. (n = 3). d, in vitro translocation reactions carried out with 1 μm SecYEG in proteoliposomes, 50 nm SecA, 1 mm ATP, and 0.7 μm of labeled proOmpAΔ176 (proOmpAA488). Successfully translocated proOmpA substrate was protected from proteinase K (PK) digestion and detected using a Typhoon 9400 fluorescence imager. e, comparison of the numbers of fluorescent vesicles observed following addition to proteoliposomes, harboring different forms of SecYEG, with SecA and proOmpAA488, in the presence or absence of ATP, and subsequent treatment with tryptophan. Results are shown as mean ± S.D. (n = 5). The vesicles tested contained either single copies of SecY103-AF488EG ((Y*EG)1; LPR = 73,000:1), unlabeled single copies of SecYEG monomers ((YEG)1 wild-type; LPR = 73,000:1), unlabeled multiple copies of SecYEG ((YEG)n wild-type; LPR = 730:1), labeled tandem SecY dimers ((YY*EG)1; LPR 73,000:1), or single copies of unlabeled SecYEL106CG cross-linked in the back-to-back configuration ((106X)1 back-to-back; LPR = 73,000:1).
To investigate the relationship between the oligomeric status of SecYEG and its functional capability, a substrate of the translocon, proOmpAΔ176, with cysteine residues at positions 296 and 311, was labeled with Alexa Fluor 488 C5-maleimide. To confirm that this substrate (designated proOmpAA488) is capable of translocation, the rate of ATP turnover in SecA associated with vesicles containing many copies of SecYEG was measured in the standard ensemble set-up (20). The results revealed that the rates of ATP hydrolysis stimulated during substrate translocation using labeled proOmpAA488 are similar to those obtained with the unlabeled cysteine mutant of proOmpA and to wild-type proOmpA (Fig. 5c). The concomitant protease protection of proOmpAA488, detected using a fluorescence scanner, confirmed that the labeled substrate had been successfully translocated into the proteoliposomes (Fig. 5d), albeit with a reduced efficiency compared with the unlabeled protein (20).
Building on the ability of proOmpAA488 to be translocated by wild-type SecYEG, single molecule fluorescence experiments were next employed to determine whether different oligomeric forms of SecYEG are capable of binding and/or translocating proOmpAA488. For this purpose, vesicles containing monomeric unlabeled SecYEG were prepared as described above. In addition, vesicles containing a single copy of cross-linked back-to-back dimeric SecYEL106CG were formed under the same conditions. Confirmation that the latter reconstitution conditions yielded primarily a single dimer per vesicle was provided by the observation that the number of fluorescent vesicles obtained with the fluorescently labeled tandem dimer SecYY97-AF488EG (Fig. 5e, panel (YY*EG)1) was equivalent to that obtained with SecY103-AF488EG (Fig. 5e, panel (Y*EG)1). Proteoliposomes containing many copies of SecYEG were prepared using a lower LPR (730:1) i.e. a 100-fold higher concentration of SecYEG; use of such a low LPR is known to induce the formation of dimers in the membrane (Fig. 1a) (6), although the additional presence of higher oligomeric assemblies could not be ruled out.
Addition of proOmpAA488, SecA and ATP to proteoliposomes containing unlabeled single copies of SecYEG (Fig. 5e, panel (YEG)1 wild-type), multiple copies (Fig. 5e, panel (YEG)n, wild-type) and single copies of the cross-linked dimeric form (Fig. 5e, panel (106x)1) resulted in the appearance of fluorescent vesicles, indicating that all three were able to bind proOmpAA488. Little or no fluorescence above background was observed when ATP was omitted (Fig. 5e) or if vesicles without SecYEG were used (data not shown), demonstrating the specificity of the substrate for the channel.
To differentiate between proOmpAA488 binding to SecYEG and translocation through the channel into the interior of the proteoliposome, tryptophan was employed as a membrane-impermeable fluorescence-quenching agent (28, 29). In ensemble experiments the free Alexa Fluor dye was found to undergo a 65% quench by 30 mm tryptophan. However, when the dye was encapsulated within the vesicles, it was unaffected by exposure to external tryptophan for the time period employed for the experiments, confirming its inability to cross the bilayer (data not shown).
The addition of tryptophan, following incubation of immobilized proteoliposomes with proOmpAA488, decreased the fluorescence of vesicles containing SecYEG monomers by 67 ± 8%, indicative of the substrate being bound to the outer surface of the vesicle, but being unable to be translocated (Fig. 5e, panel (YEG)1, wild-type). By contrast, the fluorescence of vesicles harboring many copies of wild-type SecYEG incubated with proOmpAA488 was not significantly decreased by tryptophan, consistent with translocation of the labeled substrate into the vesicle interior (Fig. 5e, panel (YEG)n, wild-type). Confirmation of the functionality of dimeric assemblies in transport was provided by investigation of the single copies of dimers of SecYEL106CG cross-linked in the back-to-back orientation, which were also found to yield an ATP-dependent protection from the external quencher (Fig. 5e, panel (106x)1).
Atomic Model of the Membrane-bound Translocon
The findings described indicate that the active, membrane-bound translocon is composed of SecYEG complexes arranged as a back-to-back dimer. These experimental observations validate the medium resolution structure of membrane-bound SecYEG (6, 11). To extend the analysis to higher resolution, the information obtained above was combined with various structures of SecYEG and SecA to construct an atomic model of the membrane-bound dimeric E. coli translocon active in the post-translational reaction, as described in supplemental data (Fig. 6 and supplemental Fig. S1).
FIGURE 6.

Atomic model of the E. coli membrane-bound translocon SecA-(SecYEG)2. a, cartoon representation of face-on and b, end-on side-views of dimeric SecYEG associated with SecA. The insets in the lower right are schematic representations of the model views in the back-to-back configuration, where the active and passive SecYEG protomers in the dimer are drawn with the channels respectively partially open (white and solid line ellipsoid) and closed (gray and dashed line ellipsoid); in this arrangement, the lateral gates (dashed gray lines) face away from each other. In the model, the passive and active SecY copies are colored in light gray and light blue, respectively. SecE is represented in light and dark gray for the passive and active complex, respectively. SecG is colored in tan. Similarly, differential coloring is used to highlight the individual domains of SecA: NBD1 (light blue), NBD2 (blue), HSD including the two-helix finger (black), PPXD (green), and HWD (red). The cytoplasmic loops of the passive SecY between TMS 6–7 (SecY-C4) and TMS 8–9 (SecY-C5) are shown respectively in yellow and cyan. a, SecE residue Leu-106 is shown in solid molecular representation in purple, while SecA residue Tyr-794 at the end of the two-helix finger is similarly shown in yellow. b, solid residues on NBD1 and SecY-C4 show the residues that could (purple and green), or could not (light blue and yellow) be cross-linked (14) (see supplemental Fig. S2, b and c for more detail). The insets at the top right of a and b show alternative views, rotated as indicated compared with the main figures.
In brief, the x-ray structure of the Thermotoga maritima SecA-(SecYEG)1 complex (4) was used to model the active channel while the Methanococcus jannaschii SecYEβ structure (3) provided the template for the passive copy. The two protomers were then fitted onto the medium resolution structure of the membrane-bound SecYEG dimer from E. coli (6). SecYEG-bound SecA was built primarily by domain-wise superimposition of the E. coli structure (25) onto the T. maritima counterparts (4). After the remodeling of two areas in the initial structure to remove clashes between SecA and SecY in the passive protomer, energy minimization was used to relax the resultant structure to a nearby local minimum, producing the final model shown in Fig. 6. The geometry of the refined model was assessed by PROCHECK (30), and was not significantly altered from the quality of the template structures. Analysis of the Ramachandran plot of the model showed that the percentages of residues occupying the most favored, allowed, generously allowed and disallowed regions were respectively 64, 29, 4, and 1; compared with 69, 26, 4, and 1 for the T. maritima structure.
Interactions between SecA and the Passive Protomer of the Back-to-Back SecYEG Dimer
Analysis of the molecular model of the complex between SecA and the back-to-back SecYEG dimer revealed that the presence of a second copy of SecYEG expands the available surface onto which SecA can dock. In this context, major contacts between SecA and both SecYEG protomers involve the large protruding cytoplasmic loop between TMS 6–7 (SecY-C4) and the cytoplasmic loop connecting TMS 8–9 of SecY (SecY-C5). The model reveals new information on possible contacts between SecA and the passive component of the SecYEG dimer (Fig. 6).
The passive SecYEG protomer makes major contacts with the HSD, NBD1, and the HWD of SecA, involving both the N- and C-terminal halves of SecY. The two lobes of the SecY subunit containing TMS 1–5 and 6–10 enclose the protein channel (3), which in the passive complex is non-translocating and therefore referred to as closed. The HWD contacts the relatively small loop near the N-terminal end of SecY between TMS 2–3 (SecY-C2), and an adjacent one in SecG between TMS 1–2 (SecG-C1) (supplemental Fig. S2a). In the C-terminal half, SecY-C4 interacts with the NBD1 of SecA, and SecY-C5 with the HSD and the HWD (supplemental Fig. S2b). An important confirmation of some of these proposed interactions comes from a previous study of disulfide-bridge cross-linking of SecA to the passive second copy of SecY (14). The results, which identified specific residues in the SecY-C4 loop that are close neighbors of ones in the NBD1 of SecA, are remarkably consistent with this model (supplemental Fig. S2, b and c). In this conformation, NBD2 and the PPXD of SecA do not directly contact the passive complex, but instead are intimately associated with the active counterpart in an important region connecting the catalytic center of ATP hydrolysis and the pre-protein binding site (4, 19, 31).
DISCUSSION
Despite many advances in our understanding of the structures of the components of the translocon, questions remained on the nature of its functional form in the native environment of the bilayer. The presence of detergents as solubilizing agents has destabilizing effects on the quaternary structure of many membrane protein complexes, including SecYEG (9). The extraction of certain endogenous phospholipids like CL during protein preparation may contribute to this problem. In solution, SecYEG exists in an equilibrium between monomers and dimers, where high concentrations of detergent favor the monomer (9), explaining why the structures determined by x-ray crystallography (at high detergent concentration) and single particle analysis of electron micrographs (low protein concentration) reveal only monomers.
In the presence of CL in solution, or when completely free of detergent in the membrane at sufficient concentration, SecYEG forms dimers (6, 9, 10). The experiments described here show that the back-to-back dimers, stabilized in solution by CL and visualized in two-dimensional crystals, are also present in translocation-competent membranes reconstituted with purified SecYEG, as well as in IMVs. Given the apparent requirement for only a single active channel within the dimer (14), the reason for this redundancy is unclear.
To address this question, we used a single cysteine mutant that had previously been shown to cross-link the two adjacent SecE subunits by TMS3 at the back-to-back interface (26). The fully cross-linked dimer, structurally similar to those observed in translocation-competent membranes and two-dimensional crystals, was purified and its activity characterized. Compared with the monomer, this species has a higher affinity for SecA and was capable of stimulating the ATPase activity and coupling this to protein transport. This finding contrasts with a previous study, which found that oxidized crude IMVs containing the cross-linked SecYEL106CG dimer were inactive in protein translocation (26). In fact, the oxidized membranes contain a high proportion of aggregated and inactive SecYEG complexes (data not shown). Here, by purifying and reconstituting mono-disperse cross-linked SecYEL106CG, its ability to stimulate the rate of ATP hydrolysis by SecA and to translocate substrate was revealed.
In addition to the ensemble analysis, we have harnessed the power of single molecule measurements of membrane-bound protein translocation reactions to examine the functional capability of monomeric and dimeric SecYEG. Previous studies demonstrated that fluorescently derivatized pre-protein can be driven by SecA into crude IMVs overexpressing the SecYEG complex (32, 33). Here, this phenomenon was exploited for the purposes of single molecule fluorescence detection, and in the reconstitution of transport from the purified components. The affinity of the fluorescent substrate and its ability to stimulate the translocation-associated ATPase activity of SecA was indistinguishable from that of its unlabeled counterpart.
Single molecule measurements showed unambiguously that SecYEG dimers, together with SecA and ATP, are necessary and sufficient for translocation of pre-protein. The observation that translocation activity was retained following cross-linking in a back-to-back orientation strongly supports the contention that this dimeric arrangement, observed in the structure obtained by electron cryo-microscopy (6), represents the natural state of the functional translocon. Monomers of SecYEG, while incapable of transport, nonetheless were found to support the association of proOmpA with SecYEG in a SecA- and ATP-dependent manner. This phenomenon may reflect the insertion of the signal sequence and N-terminal section of the pre-protein, leaving the fluorescent tags at the C terminus untranslocated. Therefore, at least two distinct energy-transducing steps in the translocation process have been distinguished: the binding and possible insertion event involving a single active protomer within the dimer, followed by a tracking phase through the membrane that requires a second passive copy of SecYEG.
The information on the functional form of membrane-bound SecYEG was used in the construction of an atomic model of the active translocon responsible for post-translational protein translocation. The model describes the passive and active forms of SecYEG and their interaction network with SecA. The model satisfies the functional restrictions described here, as well as the results of cross-linking experiments (14), which could not be accounted for in the x-ray structure of the complex containing only one copy each of SecA and SecYEG (4).
The dependence of post-translational transport on a dimeric SecYEG complex may result from a requirement to form a productive relationship with SecA. The dimeric complex of SecYEG, stabilized in detergent solution by CL or by the genetic fusion of two SecY subunits, interacts with and activates SecA much more effectively than its monomeric counterpart (10). The form chemically cross-linked via the back-to-back interface behaves in the same way, implicating this arrangement of the protomers as the functional state of the translocon. Hence, the second non-translocating copy of SecYEG must contribute significantly to the interaction platform with which SecA productively associates, as shown here in Fig. 6. These interactions substantially enlarge the cytosolic interface between SecA and SecYEG, consistent with its higher affinity interaction. The extensive interactions between SecA and SecYEG span the cytosolic face of the passive complex and might help to keep the inactive channel closed.
The implied major movements of the NBDs, PPXD, and the two-helix finger of SecA, in response to specific stages of the ATP hydrolytic cycle (4, 20, 23), may require more extensive stator contacts provided by the passive complex. Such connections could prevent the dissociation of SecA during a stage of the transport cycle associated with a loosening of the association between the motor and the translocating copy of SecYEG, for example when associated with ADP (20, 23).
Like the active SecYEG subunit, the passive copy also contacts regions of SecA that are otherwise involved in its homodimeric interface (15), and may therefore help to promote the translocation-dependent monomerization of SecA (34). The presence of the second copy of SecYEG could have important consequences on the structure of the associated SecA required to promote the activated forms of the complex defined kinetically (20). Furthermore, its perturbation of the HWD and PPXD could help displace the signal sequence, which binds between the two (35). Therefore, the passive SecYEG complex may well have additional roles in the activation of SecA and the initiation of translocation through the modulation of its oligomeric state, tertiary structure, and polypeptide binding capabilities.
As our understanding of protein translocation through the outer and inner membranes of Gram-negative bacteria improves, it reveals surprising levels of sophistication and diversity. Striking resemblances between the Sec-dependent translocation mechanism and the otherwise unrelated usher export pathways have been highlighted by this study, and by recent work on the assembly apparatus for type I pili (36). The latter has also been shown to involve two pore complexes working in tandem, with one protomer actively involved in translocation, assembly and polymerization, while the other remains closed, possibly facilitating substrate recruitment. This kind of co-operative process may thus define a paradigm for other multi-component translocation machines.
Supplementary Material
Acknowledgments
We thank J. E. Walker for providing the E. coli C43(DE3) strain; T. A. Rapoport for donating the plasmids carrying proOmpAΔ176, SecA795C, and pBAD22-SecEHisYYV97CG; and F. Duong for the pTrcEHisYYG expression construct. We would like to thank C. Gell for building the TIRF microscope, T. Wilkop, and K. Huard for technical assistance, and J. Clark, M. F. Engel, O. Tojira, and D. J. Brockwell for helpful discussions. We are very grateful to A. R. Clarke for assistance with kinetic analysis.
This work was supported by the BBSRC (Project Grant BB/F002343/1, to I. C.), Wellcome Trust Studentship Project Grant 080705/Z/06/Z (to K. D.) and Project Grant 084452 (to I. C.), and Equipment Grant 082140 (to I. C.) for the bioreactor.

The on-line version of this article (available at http://www.jbc.org) contains supplemental data and Figs. S1 and S2.
- CL
- cardiolipin
- AMPPNP
- adenosine 5′-(β,γ-imido)triphosphate
- TMS
- transmembrane segments
- HSD
- α-helical scaffold domain
- HWD
- α-helical wing domain
- TIRF
- total internal reflection fluorescence.
REFERENCES
- 1. Lill R., Cunningham K., Brundage L. A., Ito K., Oliver D., Wickner W. (1989) EMBO J. 8, 961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Economou A., Pogliano J. A., Beckwith J., Oliver D. B., Wickner W. (1995) Cell 83, 1171–1181 [DOI] [PubMed] [Google Scholar]
- 3. van den Berg B., Clemons W. M., Jr., Collinson I., Modis Y., Hartmann E., Harrison S. C., Rapoport T. A. (2004) Nature 427, 36–44 [DOI] [PubMed] [Google Scholar]
- 4. Zimmer J., Nam Y., Rapoport T. A. (2008) Nature 455, 936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitra K., Schaffitzel C., Shaikh T., Tama F., Jenni S., Brooks C. L., 3rd, Ban N., Frank J. (2005) Nature 438, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breyton C., Haase W., Rapoport T. A., Kühlbrandt W., Collinson I. (2002) Nature 418, 662–665 [DOI] [PubMed] [Google Scholar]
- 7. Becker T., Bhushan S., Jarasch A., Armache J. P., Funes S., Jossinet F., Gumbart J., Mielke T., Berninghausen O., Schulten K., Westhof E., Gilmore R., Mandon E. C., Beckmann R. (2009) Science 326, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ménétret J. F., Schaletzky J., Clemons W. M., Jr., Osborne A. R., Skånland S. S., Denison C., Gygi S. P., Kirkpatrick D. S., Park E., Ludtke S. J., Rapoport T. A., Akey C. W. (2007) Mol. Cell 28, 1083–1092 [DOI] [PubMed] [Google Scholar]
- 9. Bessonneau P., Besson V., Collinson I., Duong F. (2002) EMBO J. 21, 995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gold V. A., Robson A., Bao H., Romantsov T., Duong F., Collinson I. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 10044–10049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bostina M., Mohsin B., Kühlbrandt W., Collinson I. (2005) J. Mol. Biol. 352, 1035–1043 [DOI] [PubMed] [Google Scholar]
- 12. Cannon K. S., Or E., Clemons W. M., Jr., Shibata Y., Rapoport T. A. (2005) J. Cell Biol. 169, 219–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duong F. (2003) EMBO J. 22, 4375–4384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Osborne A. R., Rapoport T. A. (2007) Cell 129, 97–110 [DOI] [PubMed] [Google Scholar]
- 15. Hunt J. F., Weinkauf S., Henry L., Fak J. J., McNicholas P., Oliver D. B., Deisenhofer J. (2002) Science 297, 2018–2026 [DOI] [PubMed] [Google Scholar]
- 16. Osborne A. R., Clemons W. M., Jr., Rapoport T. A. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10937–10942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Woodbury R. L., Hardy S. J., Randall L. L. (2002) Protein Sci. 11, 875–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gold V. A., Robson A., Clarke A. R., Collinson I. (2007) J. Biol. Chem. 282, 17424–17432 [DOI] [PubMed] [Google Scholar]
- 19. Erlandson K. J., Miller S. B., Nam Y., Osborne A. R., Zimmer J., Rapoport T. A. (2008) Nature 455, 984–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robson A., Gold V. A., Hodson S., Clarke A. R., Collinson I. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 5111–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Collinson I., Breyton C., Duong F., Tziatzios C., Schubert D., Or E., Rapoport T., Kühlbrandt W. (2001) EMBO J. 20, 2462–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fancy D. A., Kodadek T. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 6020–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robson A., Booth A. E., Gold V. A., Clarke A. R., Collinson I. (2007) J. Mol. Biol. 374, 965–976 [DOI] [PubMed] [Google Scholar]
- 24. Gell C., Brockwell D., Smith D. A. (2006) A Handbook of Single Molecule Fluorescence Spectroscopy, Oxford University Press [Google Scholar]
- 25. Papanikolau Y., Papadovasilaki M., Ravelli R. B., McCarthy A. A., Cusack S., Economou A., Petratos K. (2007) J. Mol. Biol. 366, 1545–1557 [DOI] [PubMed] [Google Scholar]
- 26. Kaufmann A., Manting E. H., Veenendaal A. K., Driessen A. J., van der Does C. (1999) Biochemistry 38, 9115–9125 [DOI] [PubMed] [Google Scholar]
- 27. Lill R., Dowhan W., Wickner W. (1990) Cell 60, 271–280 [DOI] [PubMed] [Google Scholar]
- 28. Marmé N., Knemeyer J. P., Sauer M., Wolfrum J. (2003) Bioconjug. Chem. 14, 1133–1139 [DOI] [PubMed] [Google Scholar]
- 29. Chen H., Ahsan S. S., Santiago-Berrios M. B., Abruña H. D., Webb W. W. (2010) J. Am. Chem. Soc. 132, 7244–7245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laskowski R. A., Moss D. S., Thornton J. M. (1993) J. Mol. Biol. 231, 1049–1067 [DOI] [PubMed] [Google Scholar]
- 31. Bauer B. W., Rapoport T. A. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 20800–20805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Keyzer J., van der Does C., Driessen A. J. (2002) J. Biol. Chem. 277, 46059–46065 [DOI] [PubMed] [Google Scholar]
- 33. Liang F. C., Bageshwar U. K., Musser S. M. (2009) Mol. Biol. Cell 20, 4256–4266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Or E., Navon A., Rapoport T. A. (2002) EMBO J. 21, 4470–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gelis I., Bonvin A. M., Keramisanou D., Koukaki M., Gouridis G., Karamanou S., Economou A., Kalodimos C. G. (2007) Cell 131, 756–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Remaut H., Tang C., Henderson N. S., Pinkner J. S., Wang T., Hultgren S. J., Thanassi D. G., Waksman G., Li H. (2008) Cell 133, 640–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}