

Abstract
The polymorphic deletion of Glu-155 from human glutathione transferase omega1 (GSTO1-1) occurs in most populations. Although the recombinant ΔGlu-155 enzyme expressed in Escherichia coli is active, the deletion causes a deficiency of the active enzyme in vivo. The crystal structure and the folding/unfolding kinetics of the ΔGlu-155 variant were determined in order to investigate the cause of the rapid loss of the enzyme in human cells. The crystal structure revealed altered packing around the Glu-155 deletion, an increase in the predicted solvent-accessible area and a corresponding reduction in the buried surface area. This increase in solvent accessibility was consistent with an elevated Stern-Volmer constant. The unfolding of both the wild type and ΔGlu-155 enzyme in urea is best described by a three-state model, and there is evidence for the more pronounced population of an intermediate state by the ΔGlu-155 enzymes. Studies using intrinsic fluorescence revealed a free energy change around 14.4 kcal/mol for the wild type compared with around 8.6 kcal/mol for the ΔGlu-155 variant, which indicates a decrease in stability associated with the Glu-155 deletion. Urea induced unfolding of the wild type GSTO1-1 was reversible through an initial fast phase followed by a second slow phase. In contrast, the ΔGlu-155 variant lacks the slow phase, indicating a refolding defect. It is possible that in some conditions in vivo, the increased solvent-accessible area and the low stability of the ΔGlu-155 variant may promote its unfolding, whereas the refolding defect limits its refolding, resulting in GSTO1-1 deficiency.
Keywords: Crystal Structure, Enzyme Mutation, Enzyme Structure, Genetic Polymorphism, Protein Folding, GSTO1-1 Deficiency, Glutathione Transferase Omega
Introduction
The omega class glutathione transferases (GSTO)2 are among the more recently identified members of the cytosolic glutathione transferase super family (1, 2). Members of the omega class have a number of novel features and have been implicated in important metabolic pathways and in the etiology of some common neurological diseases (3–7). In contrast to the other human GSTs, which have active site tyrosine or serine residues, the omega class GSTs have an active site cysteine residue that is essential for its primary catalytic activities (1). The omega class GSTs were originally shown to catalyze thioltransferase and ascorbate reductase reactions, and other studies have shown that they also catalyze the reduction of monomethyl arsonate in the methylation pathway for the disposition of arsenic (3, 4). In addition, recent studies have shown that GSTO1-1 has S-phenacyl glutathione reductase activity and plays a significant role in the biotransformation of α-haloketones (8, 9). As well as these catalytic activities, GSTO1-1 has been implicated in the post-translational processing and activation of the proinflammatory mediator interleukin-1β (10) and has also been found to act as an inhibitor of cardiac muscle ryanodine receptor Ca2+ channels (11). Interestingly, some genetic mapping studies have strongly implicated an omega class GST as a factor influencing the age at onset of Alzheimer and Parkinson diseases (5, 6, 12). Other studies have implicated genetic variation in GSTO1 in vascular dementia, stroke, and cerebrovascular atherosclerosis (7, 13). The mechanism by which genetic variation mediates these effects has not been determined, but it could be via the effects on the activity or expression of the omega class GSTs.
Previous studies have identified a range of polymorphisms in the GSTO genes (2, 3, 14). Although a number of single nucleotide polymorphisms have been identified in non-coding regions, their effects on transcription, splicing, and mRNA stability have yet to be evaluated (15). In contrast, several polymorphisms in coding regions have been studied in more detail (2, 3, 14, 16). A polymorphic deletion of three base pairs at the junction of exon 4 and intron 4 is of particular interest. This deletion could result in the deletion of Glu-155 or a splicing abnormality. This polymorphism has been identified in a range of different ethnic groups reaching a frequency of >10% in one Chinese sample (2, 14, 17–19). The deletion occurs in two haplotypes characterized by a Glu or Lys at position 208 (19). Recombinant expression of the Glu-155 deletion variant in Escherichia coli affords an active enzyme that has decreased heat stability (2, 19). We recently reported that the T-47D breast cancer cell line is hemizygous for the Glu-155 deletion allele and is deficient in GSTO1-1 enzymatic activity (20). In addition, lymphoblastoid cell lines from individuals who were heterozygous for the Glu-155 deletion were reported to have only 50% of normal catalytic activity with 4-nitrophenacylglutathione, a specific GSTO1-1 substrate (20). These studies suggest that the Glu-155 deletion variant is very compromised in vivo.
Given the role played by GSTO1-1 in the disposition of α-haloketones (8) and the targeting of GSTO1-1 by anti-inflammatory drugs (10) as well as the proposed role of genetic variation in GSTO1 in the progress of several neurological disorders (6, 7), it is apparent that natural polymorphisms in GSTO1 that affect function are of significant pharmacogenetic and clinical interest. Consequently we have investigated the effect of the polymorphic Glu-155 deletion on the enzyme structure and stability by solving its crystal structure and studying its unfolding equilibrium.
EXPERIMENTAL PROCEDURES
Expression of Recombinant GSTO1-1 Variants
In this study several recombinant GSTO1-1 isoenzymes have been studied. There is a common polymorphism where Asp-140 substitutes Ala-140. This substitution does not appear to influence function, but because Ala-140 is the most common allele, we refer to it as the wild type and the substituted variant as A140D. In each case the Glu-155 deletion variants have alanine at residue 140. cDNAs encoding GSTO1 wild type, GSTO1-A140D, GSTO1 ΔGlu-155/Glu-208, and GSTO1 ΔGlu-155/Lys-208 were cloned into the pHUE expression vector and transfected into E. coli BL21(DE3). The expression and purification of recombinant proteins using the pHUE vector and ubiquitin cleavage techniques have been previously described in detail (21). Briefly, the recombinant His6-ubiquitin-GSTO fusion protein was purified by chromatography on nickel-agarose. The His-ubiquitin fusion moiety was removed by digestion with the catalytic core of Usp2 and re-chromatography on nickel-agarose. The purified proteins were dialyzed against 20 mm Tris, pH 8, 60 mm NaCl, 5 mm dithiothreitol for storage and crystallization experiments.
Crystallization and Structural Determination of GSTO1-1 ΔGlu-155/Glu-208
Unsuccessful attempts were made to grow crystals of GSTO1 ΔGlu-155 under identical conditions to those reported for the native enzyme (1). The protein was, therefore, subjected to the index screen (Hampton Research) to find new conditions. The vapor diffusion technique was employed using a 96-well sitting drop crystallization tray (Nunc, Roskilde, Denmark). For screening, 1 μl of protein (12 mg/ml) was mixed with 1 μl of 10 mm glutathione and 1 μl of reservoir solution. Reservoirs contained 100 μl of each screen condition. The tray was then sealed with Crystal Clear tape (Hampton Research) and allowed to equilibrate at 18 °C. After 6 days, crystals were observed in condition 62 (0.1 m Tris, pH 8.5, 0.2 m trimethyl amine N-oxide dihydrate, 20% (w/v) polyethylene glycol monomethyl ether 2000). Scaling up was achieved via a doubling of the overall crystallization volume using the same method. Crystals were flash-frozen at 100 K using an Oxford cryostream. Cryoprotection was by sequential transfer to artificial mother liquor containing 20% glycerol by 5% increments. The crystals were then transferred to liquid nitrogen and stored until subsequent data collection using a Rigaku RU-200 rotating anode x-ray generator with mirror optics and Mar345 desktop beamline. Processing of data collected from a single crystal was performed using the programs DENZO and SCALEPACK (22). Phase information was obtained by molecular replacement using the structure of the native enzyme (1) as a search model with the program MOLREP (23). Model rebuilding to match experimental density, including the deletion of Glu-155, was achieved with the programs O (24) and Coot (25) and alternated with restrained refinement by the program REFMAC (26). The program PHENIX (27) was then employed for the final stages of refinement using the simulated annealing option with default settings. Finally, the program MolProbity (28) was used to add hydrogens to the structure and diagnose Asn/Gln/His flips before validation deposition (PDB code 3LFL).
Spectroscopy and Activity Measurements
All fluorescence data were measured on PerkinElmer Life Sciences LS 50B luminescence spectrophotometer. Tryptophan fluorescence was used to monitor structural change. Excitation was at 295 nm, and fluorescence was recorded in the range of 305–430 nm. 1-Anilino-8-naphthalenesulfonate (ANS) was added to protein samples in a molar ratio of 50:1 (29) and equilibrated for 50 min for complete interaction. ANS binding spectra were recorded in the range of 405–540 nm with excitation at 390 nm. Enzymatic activity was determined with the GSTO1-1 specific substrate S-(4-nitrophenacyl)-glutathione as described previously (9).
Tryptophan Fluorescence Quenching by Acrylamide
Acrylamide (8 m) was added to 1 μm GSTO1 proteins (calculated by a molar extinction coefficient of 82,800 m−1cm−1 at 280 nm) to achieve different concentrations and mixed thoroughly. Fluorescence intensities were monitored at 336 nm. Each protein sample was repeated at least three times and then averaged. Quenching data were analyzed by the Stern-Volmer equation (30),
 |
where Fo and F are the fluorescence intensities before and after the addition of acrylamide, [Q] is the acrylamide concentration, and KSV is the Stern-Volmer constant, which can reflect the burial of tryptophan residues and, thus, indicate the protein accessibility.
Circular Dichroism Measurement
Circular dichroism (CD) was monitored on ChirascanTM Circular Dichroism Spectrometer (Applied Photophysics) using a cuvette with a 0.1-cm light path. Ellipticity at 222 nm was collected and plotted against variation of urea concentration.
Equilibrium Unfolding Experiments
Urea-induced unfolding experiments were performed in 20 mm sodium dihydrogen phosphate with 40 mm ammonium phosphate, pH 7.0, at 1 μm protein concentration. To reach equilibrium, protein samples with different urea concentrations (0–9 m) were incubated at 30 °C for 5 h and then cooled down to room temperature for intrinsic fluorescence, ANS binding, and circular dichroism measurements. For the analysis of the intrinsic fluorescence the ratio of emission intensity at 333 nm (folded protein) and 355 nm (unfolded protein) was determined, and the relative intensity at 475 nm was determined for ANS binding. Data from three-four separate replicates from two purification batches of each protein were averaged.
Urea unfolding curves were fitted to a two-state (N2 ↔ 2U) unfolding model or a three-state model (N2 ↔ I2↔ 2U) (31, 32) using SigmaPlot Version 10.0. The equilibrium fractions of native, intermediate, and denatured state were calculated by a previously described method (32).
Protein Refolding
Refolding of unfolded proteins was detected using S-(4-nitrophenacyl)-glutathione reduction assay and tryptophan fluorescence recovery. For activity reversibility, proteins were brought to a concentration of 1 μm in 6 m urea and allowed to unfold for 5 h at 30 °C. The proteins were then diluted 6 times and allowed to refold at room temperature. Enzyme activity was determined after 1 h. When refolding was monitored by intrinsic fluorescence, 30 μm proteins were fully unfolded in 7.4 m urea. The enzymes were then diluted 10 times, and the emission intensities were recorded every minute. Curve fitting was undertaken in SigmaPlot Version 10.0.
RESULTS
Crystal Structure of GSTO1 ΔGlu-155/Lys-208
To determine whether the deletion of Glu-155 caused any significant structural alterations, we crystallized and solved the structure of the GSTO1 ΔGlu-155/Glu-208 variant. The crystals were found to be of a different space group (P21212) compared with the native enzyme (P3121) and diffracted to 2.1 Å. Table 1 summarizes the crystallographic data and model statistics.
TABLE 1.
X-ray data collection and refinement statistics
r.m.s.d., root mean square deviation.
| Space group | P21212 |
| Unit cell parameters (Å) | a = 73.0, b = 201.9, c = 53.4 |
| Reflections measured/unique | 389,373/44,498 |
| Resolution range (Å) | 36.51-2.10 (2.18-2.10)a |
| R-merge (%) | 12.8 (37.0)a |
| Completeness | 94.4 (80.7)a |
| Mean I/σI | 21.4 (3.5)a |
| R/Rfree (%) | 19.91/26.05 |
| r.m.s.d. bonds (Å) | 0.008 |
| r.m.s.d. angles | 1.120° |
| PDB accession code | 3LFL |
a The highest resolution bin.
Molecular replacement in MOLREP (23) revealed three monomers in the asymmetric unit. Monomer A sits adjacent to a crystallographic 2-fold axis so as to form a physiological dimer with its crystallographic partner (A-A). Monomers B and C form a second dimer (B-C). The overall topology is similar to that of the wild type enzyme, except for the following; the deletion of residue Glu-155 results in the premature termination of helix α5 and a conformational change in the helix α5 to α6 loop (Fig. 1A). Its subsequent removal from the search model required significant rebuilding of the associated residues, a process coupled with restrained refinement until local aberrations in the mFo − DFc electron density map were removed. As shown in Fig. 1B, several perturbations in the surrounding structure contribute to this enzyme altered phenotype. The hydrophobic interactions (not shown in this figure) of adjacent residues Val-156 and Leu-157 have been disrupted by Cα translations of 5.3 and 8.0 Å, respectively. This has caused Val-156 to be exposed to solvent. The translocation of Leu-157, on the other hand, has produced a new interaction with Leu-119 and, of particular interest, an interaction with the side chain of Pro-68 from the other subunit in the physiological dimer, effectively creating a new interaction at dimer interface. In addition, the Thr-158 Cα has shifted by 6.4 Å, allowing its side chain to occupy the position previously inhabited by that of Val-156. Such changes have contributed to an increase in the water accessibility of Leu-157 to >100 Å2 and an increase in the buried area at the interface of twice this magnitude. In total there are only 7 contacts ≤3.5 Å between residues 150–164 of α5 and residues 192–202 of loop-α7 in the ΔGlu-155/Glu-208 enzyme, 18 fewer than in the wild type enzyme. The altered packing has likely played a role in the observed refolding anomaly of Glu-155 deletion variants discussed below. The alterations in the dimer interface do not appear to preclude the formation of heterodimers between wild type and deletion-containing monomers.
FIGURE 1.

Structural changes induced by the deletion of Glu-155. A, a view perpendicular to the crystallographic 2-fold axis of the native structure (translucent yellow), the ΔGlu-155 A monomer (cyan), and the non-crystallographic 2-fold axis of the ΔGlu-155 B and C monomers (magenta), which produce the GSTO1-1 physiological dimer. The position of Glu-155 and GSH is shown in stick representation. B, a closer inspection of the deletion site within the ΔGlu-155 A-monomer (cyan) compared with the wild type enzyme (yellow) is shown. Changes to the associated residue side chain conformations are in stick representation. The new interaction formed at the dimer interface is also visible. The adjacent monomer is shown in magenta. The figures were generated using PyMol.
The potential flexibility of the C-terminal region can be observed in the ΔGlu-155/Glu-208 structure. Comparison of the active site of monomer A with the wild type enzyme reveals a rotation of the putative “H-site” residue, Trp-222, resulting in the removal of its indole nitrogen from the binding pocket, increasing its hydrophobic character. As shown in Fig. 2A, the entrance to the binding pocket has also been partially occluded by the movement of Arg-183 and Tyr-229. Helix α9, of monomer A, which forms part of the active site, has shifted away from the GSH binding pocket (G-site), possibly to accommodate movements in residues Tyr-229 and Trp-222 (Fig. 2A). Although the B and C monomers display similar structural changes relating to the deletion site, it is the conformational shift of their C termini that is most remarkable. As shown in Fig. 2B helix α9 of monomer B has moved toward the G-site and rotated so that Leu-228 and Tyr-229 block the H-site entrance. There is a significant rearrangement of Trp-222, which has been excluded from the H-site and is stacked against helix α6, exposed to bulk solvent. The net effect is to make the “H-site” smaller. Furthermore, the last 10–14 residues forming helix α10 and its connecting loop fail to show substantial mFo − DFc density in monomers B and C. These significant changes do not appear to be a consequence of crystal packing, as the crystal contacts in each monomer are different. The different conformations seen in the two dimers (A-A and B-C) may, therefore, reflect the conformational space sampled in solution. Such helix α9 movements may be crucial to the binding of ligands of variable size and shape, for example monomethyl arsonate and dehydroascorbate.
FIGURE 2.

The flexibility of helix α9 observed in the ΔGlu-155 structure. A, a closer view of the A monomer active site reveals several key differences compared with the native enzyme. Of most interest is the different rotamer adopted by Trp-222, dramatically increasing the hydrophobicity within the conserved H-site. The ΔGlu-155 A-monomer is shown in cyan compared with the wild type enzyme in yellow. B, comparison of the native structure to that of the B monomer reveals much more pronounced structural change. Helix α9 has shifted toward the site of glutathione binding by several angstroms, dramatically reducing the size of the H-site. The ΔGlu-155 B-monomer is shown in magenta with the overlaid native structure in yellow. The figures were generated using PyMol.
Equilibrium Unfolding/Folding of GSTO1 ΔGlu-155/Glu-208
Initially, to determine whether the deletion of Glu-155 caused subtle changes to the enzyme conformation in solution, the Stern-Volmer constant (KSV) was calculated by quenching the natural tryptophan fluorescence using acrylamide. Linear Stern-Volmer plots revealed a clear difference between the variant enzymes with the Glu-155 deletion and the enzymes with A or D at residue 140 (Fig. 3). This difference is reflected numerically in KSV for each enzyme (5.258 (R2 = 0.9989) for wild type (Ala-140), 5.840 (R2 = 0.9982) for A140D, 6.975 (R2 = 0.999) for ΔGlu-155/Glu-208, and 7.299 (R2 = 0.9979) for ΔGlu-155/Lys-208). The data suggest that the deletion of Glu-155 increases the accessibility of tryptophan residues. This is supported by calculations from the crystal structure of increased accessible surface area of total protein and of tryptophan residues in the ΔGlu-155 (Table 2).
FIGURE 3.
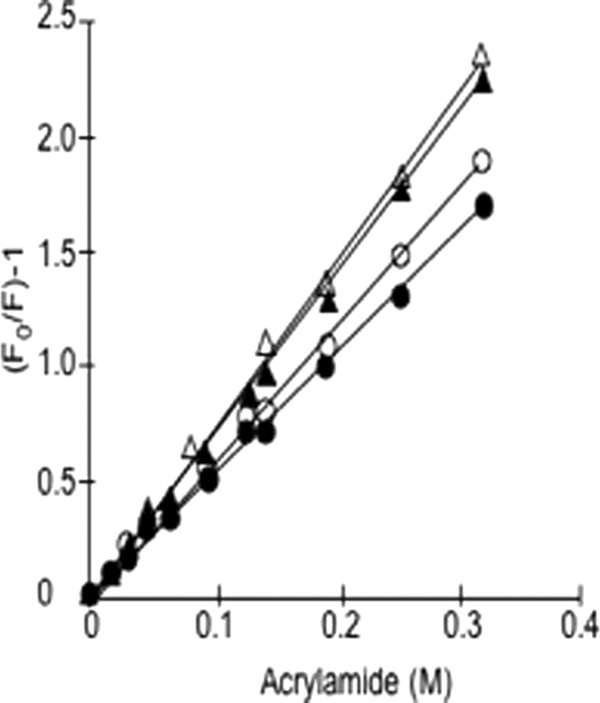
Tryptophan fluorescence quenching by acrylamide. The experimental values and trend lines are shown as wild type (●), A140D (○), ΔGlu-155/Glu-208 (▾), and ΔGlu-155/Lys-208 (▵).
TABLE 2.
Interface and surface area (SA) of ΔGlu-155/Glu-208 and wild type crystal structure
All calculations come from online PISA (35) of EBI services.
| Solved residues | SA of dimer | Total buried area of dimer | Total SA of Trp residues in dimer | Buried area of dimer interface | |
|---|---|---|---|---|---|
| Å2 | Å2 | Å2 | Å2 | ||
| GSTO1 wild type (PDB code 1EEM) | Ser-5–Leu-241 | 20,961.5 | 3,859.9 | 32.48 | 1,885.6 |
| ΔGlu-155 monomer A | Ser-5–Leu-240 | 21,639.1 | 3,317.3 | 33.74 | 2,116.6 |
Characterization of Urea-induced Unfolding
The unfolding of the GSTO1-1 enzymes in the presence of urea was examined using tryptophan fluorescence, CD, and ANS binding (Fig. 4). The fluorescence data (Fig. 4a) for the wild type and A140D enzymes indicate that they do not begin unfolding before the urea concentration exceeds 3.7 m and by visual inspection appear to exhibit a single transition phase. In contrast, the equilibrium denaturation of the ΔGlu-155 enzymes start to show an unfolding signal below 1 m urea and clearly have a biphasic transition profile. The difference in the unfolding profiles between the Glu-155-containing and ΔGlu-155 enzymes is also evident in the CD data (Fig. 4b) where the ΔGlu-155 enzymes start losing their helical secondary structure content at the lowest concentrations of urea. It is also of interest to note that at the starting point of the unfolding curve in Fig. 4b the ΔGlu-155 variants have a lower helical content than the wild type enzymes, which is consistent with the ΔGlu-155/Glu-208 structure. The data from the ANS binding measurements (Fig. 4c) provide the clearest evidence that the unfolding profiles of the ΔGlu-155 enzymes are different from the Glu-155-containing enzymes. Using tryptophan fluorescence, protein concentration dependence was observed for the transition phase of the wild type enzyme and notably only during the second transition phase of the ΔGlu-155/Glu-208 enzyme (Fig. 5). Taken together, these data suggested that the equilibrium unfolding of the wild type and A140D enzymes might be described by a two-state model (N2 ↔ 2U), whereas the unfolding of the ΔGlu-155 enzymes might best be described by a three-state unfolding model (N2 ↔ I2 ↔ 2U), where I2 represents a partially unfolded dimeric intermediate (33, 34).
FIGURE 4.

The equilibrium unfolding of GSTO1-1 in urea. Unfolding was monitored by tryptophan fluorescence expressed as the ratio of fluorescence at 333 nm to the fluorescence at 355 nm (a), ellipticity at 222 nm of circular dichroism (b), and relative intensity of ANS binding (c). The three state curve fits for N2 ↔ I2 ↔ 2U are shown in a and c, and the panels immediately below a and c show the residuals obtained after fitting the data to either two or three state models. The calculated population of the native, intermediate, and unfolded states of GSTO1-1 as a function of urea concentration are plotted for wild type and A140D in panel d (solid line for wild type and dashed line for A140D), ΔGlu-155/Glu-208 in panel e, and ΔGlu-155/Lys-208 in panel f. The experimental values for each enzyme are shown as wild type (●), A140D (○), ΔGlu-155/Glu-208 (▾), and ΔGlu-155/Lys-208 (▵).
FIGURE 5.

The protein concentration dependence of equilibrium unfolding of GSTO1-1 variants in urea. The unfilled symbols represent determinations at 1 μm protein, and the filled symbols represent determinations at 8 μm protein. The circles in panel A represent the wild type enzyme, and the triangles in panel B represent the ΔGlu-155/Glu-208 variant.
Tryptophan fluorescence and ANS binding data from the ΔGlu-155 enzymes were fitted to a three-state model with an R2 > 0.999, whereas the fitting of fluorescence data from the wild type and A140D enzymes to a two-state model gave a lower R2 (0.9986 and 0.9974, respectively) and lower free energy values (6.41 and 5.15 kcal/mol, respectively) that were inconsistent with the previously observed lower thermal stability for ΔGlu-155 enzymes (2). When the three-state model was used to re-fit the data from the wild type and A140D enzymes, a better fit (Fig. 4a) with a higher R2 (0.9996 and 0.9993, respectively) was obtained. The distribution of residuals for a two-state and three-state fit for the wild type and A140D enzymes is shown in Fig. 4a, and the residual sums of squares for wild type and A140D enzymes were also improved from 0.0037 and 0.0062 to 0.0010 and 0.0017. Based on the fitting result, equilibrium denaturation of the wild type and A140D proteins appears to be better described by a three-state unfolding model. Given the absence of protein concentration dependence through the first transition phase of the ΔGlu-155 enzyme (Fig. 5), it appears that the transition intermediate formed by the ΔGlu-155 enzymes is likely to be a dimer (33, 34). However it is not clear whether this is the case for wild type and A140D enzymes.
The population of the transition intermediate appears to differ significantly between the Glu-155 containing and ΔGlu-155 enzymes. Based on the three-state model, the fractions of the transitional intermediate and the native and unfolded states at different urea concentrations were calculated and plotted (Fig. 4, d–f). The population of intermediate formed by the wild type and A140D enzymes is largely overlapped by the unfolded fraction, suggesting that this intermediate is very transitory and may indicate why there is no obvious intermediate state reflected in the equilibrium unfolding curves. In contrast, the intermediate of the ΔGlu-155 enzymes is clearly distinguishable at lower urea concentrations and has an increased area of exposed hydrophobic surface as illustrated by the ANS binding data (Fig. 4c).
Thermodynamic Analysis of Urea-induced Unfolding
The thermodynamic parameters for the Glu-155 containing and Glu-155 deletion enzymes are shown in Table 3. The three-state fit of tryptophan fluorescence data from the wild type and A140D enzymes provided a similar total free energy change of 14.4 and 14.3 kcal/mol, respectively, which is larger than that obtained for the ΔGlu-155 enzymes (8.6 and 8.7 kcal/mol for ΔGlu-155/Glu-208 and ΔGlu-155/Lys-208, respectively). Similar results were obtained from the ANS binding data. The main difference between the wild type and the ΔGlu-155 variant occurs in the initial unfolding to the transition intermediate. Studies using intrinsic fluorescence revealed a free energy change for this first transition of around 8.5 kcal/mol for the wild type compared with around 2.8 kcal/mol for the ΔGlu-155 variant. These data suggest that the enzymes with the Glu-155 deletion are less stable than Glu-155-containing enzymes, and the lower stability of ΔGlu-155 enzymes comes primarily from a propensity to enter the first transition phase.
TABLE 3.
Thermodynamic parameters characterizing the urea-induced unfolding transition monitored by tryptophan fluorescence and ANS binding
The data were fitted to a three-state transition model via a dimeric intermediate.
| ΔGN→I0 | mN→I | [Urea]1/2,1 | ΔGI→U0 | mI→U | [Urea]1/2,2 | ΔGN→U0 | |
|---|---|---|---|---|---|---|---|
| kcal/mol | kcal/mol/m | m | kcal/mol | kcal/mol/m | m | kcal/mol | |
| Tryptophan fluorescence | |||||||
| WT | 8.51 ± 0.46 | 1.81 ± 0.11 | 4.69 ± 0.06 | 5.91 ± 2.42 | 1.00 ± 0.36 | 5.91 ± 0.33 | 14.42 |
| A140D | 9.16 ± 0.82 | 2.02 ± 0.20 | 4.55 ± 0.09 | 5.14 ± 1.67 | 0.87 ± 0.26 | 5.94 ± 0.22 | 14.3 |
| ΔGlu-155/Glu-208 | 2.84 ± 0.79 | 1.48 ± 0.37 | 1.92 ± 0.12 | 5.78 ± 0.35 | 1.37 ± 0.08 | 4.22 ± 0.03 | 8.62 |
| ΔGlu-155/Lys-208 | 3.34 ± 0.97 | 1.80 ± 0.49 | 1.86 ± 0.11 | 5.39 ± 0.31 | 1.29 ± 0.07 | 4.19 ± 0.03 | 8.73 |
| ANS binding | |||||||
| WT | 7.65 ± 2.43 | 2.45 ± 0.80 | 3.11 ± 0.11 | 7.07 ± 0.42 | 1.44 ± 0.08 | 4.91 ± 0.03 | 14.72 |
| A140D | 5.43 ± 2.41 | 2.10 ± 0.91 | 2.58 ± 0.14 | 5.58 ± 0.57 | 1.13 ± 0.12 | 4.93 ± 0.06 | 11.01 |
| ΔGlu-155/Glu-208 | 3.51 ± 1.55 | 1.95 ± 0.77 | 1.77 ± 0.17 | 6.54 ± 0.58 | 1.55 ± 0.13 | 4.20 ± 0.04 | 10.05 |
| ΔGlu-155/Lys-208 | 3.89 ± 1.64 | 1.88 ± 0.79 | 2.06 ± 0.15 | 4.98 ± 0.62 | 1.27 ± 0.14 | 3.92 ± 0.07 | 8.87 |
Reversibility of Unfolding
The ability of the enzymes to re-fold after urea-induced unfolding was evaluated by fluorescence recovery and the restoration of catalytic activity (Fig. 6). The two methods used here to monitor the refolding gave similar results. Refolding of the wild type Glu-155-containing enzymes was essentially complete after 1 h with the recovery of around 95%. In contrast, the enzymes with the Glu-155 deletion regained only around 70% of their original activity or fluorescence intensity over this period.
FIGURE 6.

Refolding of GSTO1 variants. Symbols and solid curves represent the recovery of fluorescence. The right-hand columns show the recovery of activity with 4-nitrophenacylglutathione as a substrate (mean ± S.D.).
To identify whether the lower recovery of Glu-155 deletion enzyme came from a lower refolding rate, an exponential regression analysis was applied to the fluorescence recovery data. For the wild type and A140D enzymes, a two-phase exponential regression was better than a single-phase exponential regression as improvements in the fit were observed in R2 (0.9984 and 0.9992 compared with 0.9924 and 0.9900, respectively) in the S.E. of estimates (0.0050 and 0.0069 compared with 0.0135 and 0.0158, respectively) and in the residual sums of squares (0.0005 and 0.0002 compared with 0.0022 and 0.0030, respectively). Interestingly the refolding data for the ΔGlu-155/Lys-208 variant could be fitted to a single phase or two-phase exponential regression with the same R2 (0.9978) and residual sums of squares (0.0003) and a similar S.E. (0.0053 and 0.0058, respectively). However the two-phase exponential regression generated an unusually high k1 of over 2200 min−1 and other unusual parameters, suggesting that a single phase exponential analysis is more appropriate for the ΔGlu-155/Lys-208 variant.
Therefore, the refolding curve of wild type and A140D enzymes monitored by fluorescence was better fitted by a two-phase exponential equation, which respectively gave 3.18 ± 0.57 and 3.34 ± 0.52 min−1 for k1 and 0.22 ± 0.06 min−1and 0.27 ± 0.05 min−1 for k2, whereas the refolding of ΔGlu-155/Lys-208 variant followed single-phase exponential kinetics with a rate of 4.04 ± 0.80 min−1. This suggests that the ΔGlu-155 variants still have a similar fast-refolding phase like the wild type and A140D enzymes but have lost the second slow refolding phase as shown in Fig. 6.
DISCUSSION
Previous studies have indicated that the polymorphic deletion of Glu-155 causes a deficiency of GSTO1-1 in cell lines, but the mechanism causing this deficiency is not clear. As the deletion can potentially alter splicing at the end of exon 4, we previously undertook Northern blotting analysis to eliminate a splicing defect as a cause of the GSTO1-1 deficiency (20). That study showed that T47D breast cancer cells that are hemizygous for the Glu-155 deletion allele produce mRNA of the appropriate size, suggesting that abnormal splicing is unlikely to be the cause of the deficiency. Previous pulse-chase studies in T47D cells indicate that the Glu-155 deletion protein has a shorter than normal t½ (20). Although this variant can be expressed as an active enzyme in E. coli, the recombinant enzyme is relatively heat-sensitive (20). Given the clinical interest in GSTO1-1, we were interested in determining the mechanism by which the Glu-155 deletion mediates this deficiency.
The crystal structure indicated that the deletion of Glu-155 caused only small perturbations of the overall protein fold. Although there is a slight decrease in the volume and apparent accessibility of the H site, the distinct structural forms of ΔGlu-155 dimers observed in the crystal structures point toward increased flexibility of the protein. This may explain the previously observed increase in catalytic activity of ΔGlu-155 mutants (Table 4), as conformations suitable for binding and catalysis of diverse substrates are more accessible to the flexible mutant protein. The catalytic activity of the recombinant ΔGlu-155 enzymes and the absence of immunologically detectable GSTO1-1 in T47D cells suggest that the deletion may cause a defect in stability or folding rather than a loss of function. We found several structural factors that could have a deleterious impact on stability including the increase in the water accessible surface area, a reduction in buried hydrophobic surface area, and the large decrease in molecular contacts between helices α5 and α7. Even the increased buried area of dimer interface was not a positive factor because of the concomitant increase in the surface area of solvent-exposed hydrophobic residues.
TABLE 4.
The activity of recombinant wild type and ΔGlu-155 GSTO1-1 variants with a range of substrates
All activities are shown as μmol/min/mg, mean ± S.D. CDNB, 1-chloro-2,4,dinitrobenzene; 2,4-DCPG; S-(2,4-dichlorophenacylglutathione). All data were compiled from Refs. 2, 8, and 19.
| Enzyme | CDNB | Thiol transferase | Dehydroascorbate | Monomethyl arsonate | Dimethyl arsonate | 2,4-DCPG |
|---|---|---|---|---|---|---|
| Human GSTO1-1 | 0.047 ± 0.01 | 2 ± 0.21 | 0.13 ± 0.005 | 0.33 ± 0.037 | 0.12 ± 0.006 | 1.3 ± 0.1 |
| 0.039 ± 0.01 | 2.6 ± 0.26 | |||||
| ΔGlu-155, Glu-208 | 6 ± 0.09 | 0.25 ± 0.045 | 0.65 ± 0.007 | 0.3 ± 0.014 | 1.3 ± 0.05 | |
| ΔGlu-155, Lys-208 | 0.063 ± 0.01 | 4.8 ± 0.07 | 0.21 ± 0.03 | 0.67 ± 0.066 | 0.37 ± 0.015 | 1.1 ± 0.03 |
| 3.6 ± 0.36 |
The lower free energy change ΔGN→U0 supports the lower stability of the ΔGlu-155 enzymes. We also found evidence that at low urea concentrations the ΔGlu-155 enzymes form a transitional intermediate that is not significantly populated by the Glu-155-containing enzymes. Because their first transition is not dependent on the protein concentration, the unfolding intermediate formed by the ΔGlu-155 enzymes appears to be a dimer with more hydrophobic area exposure revealed by the ANS binding data. The nature of the intermediate formed by the Glu-155-containing enzymes is difficult to identify with the present data.
The capacity of the Glu-155 deletion variants to refold after complete unfolding in urea was limited compared with the Glu-155 containing Ala-140 and Asp-140 enzymes (Fig. 6). This limitation only occurred during the second slow phase of refolding. Because refolding is usually thought to be the reverse of unfolding, the refolding of the Glu-155 deletion enzymes is probably limited by the population of the transitional intermediate. As mentioned above, the intermediate has a larger exposed hydrophobic surface, which probably promotes precipitation or prevents further refolding of the protein resulting in the lack of the second slow phase for the ΔGlu-155 enzymes. Intracellular proteins are dynamic and may be subjected to conformational changes under different intracellular conditions. It is possible that the increased accessibility to solvent and decreased contacts in the Glu-155 deletion enzyme may promote unfolding and restrict refolding by presenting a barrier of increased hydrophobic surface exposure, leading to a dynamic equilibrium that favors an unfolded state in vivo.
We, therefore, conclude that the deletion of Glu-155 causes GSTO1-1 deficiency by its low stability, resulting from its increased propensity to unfold, and a novel mechanism that inhibits refolding. The structural characteristics of the deletion mutant, including a high accessible surface area and less buried hydrophobic area as well as altered packing around ΔGlu-155 agree with these observations. The results help us to understand the impact of the Glu-155 deletion allele on the level of GSTO1-1 activity in vivo, which may contribute to the observed association of GSTO1-1 with the age at onset of Alzheimer disease and the activation of the pro-inflammatory cytokine IL-1β.
Acknowledgments
We express our gratitude to Professor Mitchell Guss of the School of Molecular and Microbial Biosciences, University of Sydney, for the use of equipment during x-ray data collection.
This work was supported by National Health and Medical Research Council Project Grant 366731.
The atomic coordinates and structure factors (code 3LFL) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
- GSTO
- omega class glutathione transferase
- ANS
- 1-anilino-8-naphthalenesulfonate.
REFERENCES
- 1. Board P. G., Coggan M., Chelvanayagam G., Easteal S., Jermiin L. S., Schulte G. K., Danley D. E., Hoth L. R., Griffor M. C., Kamath A. V., Rosner M. H., Chrunyk B. A., Perregaux D. E., Gabel C. A., Geoghegan K. F., Pandit J. (2000) J. Biol. Chem. 275, 24798–24806 [DOI] [PubMed] [Google Scholar]
- 2. Whitbread A. K., Tetlow N., Eyre H. J., Sutherland G. R., Board P. G. (2003) Pharmacogenetics 13, 131–144 [DOI] [PubMed] [Google Scholar]
- 3. Whitbread A. K., Masoumi A., Tetlow N., Schmuck E., Coggan M., Board P. G. (2005) Methods Enzymol. 401, 78–99 [DOI] [PubMed] [Google Scholar]
- 4. Zakharyan R. A., Sampayo-Reyes A., Healy S. M., Tsaprailis G., Board P. G., Liebler D. C., Aposhian H. V. (2001) Chem. Res. Toxicol. 14, 1051–1057 [DOI] [PubMed] [Google Scholar]
- 5. Li Y. J., Scott W. K., Hedges D. J., Zhang F., Gaskell P. C., Nance M. A., Watts R. L., Hubble J. P., Koller W. C., Pahwa R., Stern M. B., Hiner B. C., Jankovic J., Allen F. A., Jr., Goetz C. G., Mastaglia F., Stajich J. M., Gibson R. A., Middleton L. T., Saunders A. M., Scott B. L., Small G. W., Nicodemus K. K., Reed A. D., Schmechel D. E., Welsh-Bohmer K. A., Conneally P. M., Roses A. D., Gilbert J. R., Vance J. M., Haines J. L., Pericak-Vance M. A. (2002) Am. J. Hum. Genet. 70, 985–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Y. J., Scott W. K., Zhang L., Lin P. I., Oliveira S. A., Skelly T., Doraiswamy M. P., Welsh-Bohmer K. A., Martin E. R., Haines J. L., Pericak-Vance M. A., Vance J. M. (2006) Neurobiol. Aging 27, 1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kölsch H., Larionov S., Dedeck O., Orantes M., Birkenmeier G., Griffin W. S., Thal D. R. (2007) Stroke 38, 2847–2850 [DOI] [PubMed] [Google Scholar]
- 8. Board P. G., Anders M. W. (2007) Chem. Res. Toxicol. 20, 149–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Board P. G., Coggan M., Cappello J., Zhou H., Oakley A. J., Anders M. W. (2008) Anal. Biochem. 374, 25–30 [DOI] [PubMed] [Google Scholar]
- 10. Laliberte R. E., Perregaux D. G., Hoth L. R., Rosner P. J., Jordan C. K., Peese K. M., Eggler J. F., Dombroski M. A., Geoghegan K. F., Gabel C. A. (2003) J. Biol. Chem. 278, 16567–16578 [DOI] [PubMed] [Google Scholar]
- 11. Dulhunty A., Gage P., Curtis S., Chelvanayagam G., Board P. (2001) J. Biol. Chem. 276, 3319–3323 [DOI] [PubMed] [Google Scholar]
- 12. Li Y. J., Oliveira S. A., Xu P., Martin E. R., Stenger J. E., Scherzer C. R., Hauser M. A., Scott W. K., Small G. W., Nance M. A., Watts R. L., Hubble J. P., Koller W. C., Pahwa R., Stern M. B., Hiner B. C., Jankovic J., Goetz C. G., Mastaglia F., Middleton L. T., Roses A. D., Saunders A. M., Schmechel D. E., Gullans S. R., Haines J. L., Gilbert J. R., Vance J. M., Pericak-Vance M. A., Hulette C., Welsh-Bohmer K. A. (2003) Hum. Mol. Genet. 12, 3259–3267 [DOI] [PubMed] [Google Scholar]
- 13. Kölsch H., Linnebank M., Lütjohann D., Jessen F., Wüllner U., Harbrecht U., Thelen K. M., Kreis M., Hentschel F., Schulz A., von Bergmann K., Maier W., Heun R. (2004) Neurology 63, 2255–2260 [DOI] [PubMed] [Google Scholar]
- 14. Paiva L., Marcos R., Creus A., Coggan M., Oakley A. J., Board P. G. (2008) Pharmacogenet. Genomics 18, 1–10 [DOI] [PubMed] [Google Scholar]
- 15. Mukherjee B., Salavaggione O. E., Pelleymounter L. L., Moon I., Eckloff B. W., Schaid D. J., Wieben E. D., Weinshilboum R. M. (2006) Drug. Metab. Dispos. 34, 1237–1246 [DOI] [PubMed] [Google Scholar]
- 16. Tanaka-Kagawa T., Jinno H., Hasegawa T., Makino Y., Seko Y., Hanioka N., Ando M. (2003) Biochem. Biophys. Res. Commun. 301, 516–520 [DOI] [PubMed] [Google Scholar]
- 17. Yu L., Kalla K., Guthrie E., Vidrine A., Klimecki W. T. (2003) Environ. Health Perspect. 111, 1421–1427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Marnell L. L., Garcia-Vargas G. G., Chowdhury U. K., Zakharyan R. A., Walsh B., Avram M. D., Kopplin M. J., Cebrián M. E., Silbergeld E. K., Aposhian H. V. (2003) Chem. Res. Toxicol. 16, 1507–1513 [DOI] [PubMed] [Google Scholar]
- 19. Schmuck E. M., Board P. G., Whitbread A. K., Tetlow N., Cavanaugh J. A., Blackburn A. C., Masoumi A. (2005) Pharmacogenet. Genomics 15, 493–501 [DOI] [PubMed] [Google Scholar]
- 20. Schmuck E., Cappello J., Coggan M., Brew J., Cavanaugh J. A., Blackburn A. C., Baker R. T., Eyre H. J., Sutherland G. R., Board P. G. (2008) Int. J. Biochem. Cell Biol. 40, 2553–2559 [DOI] [PubMed] [Google Scholar]
- 21. Baker R. T., Catanzariti A. M., Karunasekara Y., Soboleva T. A., Sharwood R., Whitney S., Board P. G. (2005) Methods Enzymol. 398, 540–554 [DOI] [PubMed] [Google Scholar]
- 22. Otwinowski Z., Minor W. (1997) Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 23. Vagin A. A., Teplyakov A. (1997) J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 24. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 27. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 28. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Erhardt J., Dirr H. (1995) Eur. J. Biochem. 230, 614–620 [PubMed] [Google Scholar]
- 30. Lakowicz J. (1986) Principles of Fluorescence Spectroscopy, Plenum Press, New York [Google Scholar]
- 31. Szpikowska B. K., Mas M. T. (1996) Arch. Biochem. Biophys. 335, 173–182 [DOI] [PubMed] [Google Scholar]
- 32. Gloss L. M., Matthews C. R. (1997) Biochemistry 36, 5612–5623 [DOI] [PubMed] [Google Scholar]
- 33. Neet K. E., Timm D. E. (1994) Protein Sci. 3, 2167–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grimsley J. K., Scholtz J. M., Pace C. N., Wild J. R. (1997) Biochemistry 36, 14366–14374 [DOI] [PubMed] [Google Scholar]
- 35. Krissinel E., Henrick K. (2007) J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]