

Abstract
Tau, a neuronal protein involved in neurodegenerative disorders such as Alzheimer disease, which is primarily described as a microtubule-associated protein, has also been observed in the nuclei of neuronal and non-neuronal cells. However, the function of the nuclear form of Tau in neurons has not yet been elucidated. In this work, we demonstrate that acute oxidative stress and mild heat stress (HS) induce the accumulation of dephosphorylated Tau in neuronal nuclei. Using chromatin immunoprecipitation assays, we demonstrate that the capacity of endogenous Tau to interact with neuronal DNA increased following HS. Comet assays performed on both wild-type and Tau-deficient neuronal cultures showed that Tau fully protected neuronal genomic DNA against HS-induced damage. Interestingly, HS-induced DNA damage observed in Tau-deficient cells was completely rescued after the overexpression of human Tau targeted to the nucleus. These results highlight a novel role for nuclear Tau as a key player in early stress response.
Keywords: DNA Damage, DNA-Protein Interaction, Nuclear Translocation, Oxidative Stress, Protein Phosphorylation, Tau
Introduction
Tau was first described as an essential factor for cytoskeletal microtubule assembly (1). Since then, Tau has been primarily described as a regulator of microtubule dynamics. However, due to its diverse cellular distribution, Tau likely has multiple functions. Furthermore, although Tau is primarily seen as a cytosolic protein, the nuclear localization of Tau has been described in neuronal (2, 3) and non-neuronal cells (4, 5). In mitotic HeLa cells and fibroblasts, Tau is localized to the nucleolus and is associated with the nucleolar organizer regions, and it has been suggested that Tau plays a role in the nucleolar organization and/or heterochromatization of rRNA genes (6).
Interestingly, in vitro studies have shown that purified Tau directly binds to polynucleotides with a preference toward AT-rich DNA compared with GC-rich DNA sequences. However, contradictory in vitro results have shown a protective or deleterious role of Tau in DNA integrity (7–9). In addition, a recent study reported chromosomal aberrations in fibroblasts and lymphocytes from patients carrying a Tau mutation (10).
Nevertheless, although Tau has been detected in brain nuclei (11), the function of neuronal nuclear Tau has not yet been elucidated. Furthermore, unlike other proteins present in both cellular compartments, nucleocytoplasmic shuttling of Tau has not yet been reported. The protection of genomic integrity is a major challenge for living cells that are continuously exposed to DNA-damaging injuries, especially in the brain. However, whether endogenous Tau has the capacity to protect neuronal DNA in situ has remained an unanswered question. In this study, we aimed to investigate the potential protective effects of Tau against DNA damage in central neurons.
EXPERIMENTAL PROCEDURES
Primary Embryonic Neuronal Culture
Wild-type and knock-out Tau mouse primary cortical cultures were prepared as described previously (12).
Adenovirus Growth and Labeling
HAdV-5-hTau44Wt (wild-type Tau isoform 2-3-10-) and HAdV-5-hTau44-NLS were constructed using the gateway system (Invitrogen), and they were amplified and purified in our laboratory as described previously (13). HAdV-5-hTau44-NLS was obtained by insertion of a nuclear localization signal (NLS)2 from the Epstein-Barr virus mRNA export factor EB2 (14) to the N-terminal part of human Tau. After standard virus purification by ultracentrifugation in CsCl gradient, viral genomes were quantified by measuring UV absorption at 260 nm, and the virus titer was expressed as viral physical particles per ml.
HAdV Infection
Primary cultured cells were seeded in six-well culture plates at a density of 1.28 × 106 cells per well. Cells were then infected with 2000 physical particles/cell of HAdV-5-hTau44 or HAdV-5-hTau44NLS vectors for 2 h at 37 °C in minimum volume. Culture medium was added following infection for 24 h at 37 °C.
Cell Treatment
At 10 days in vitro, cells were maintained at 37 °C (control condition(C)) or exposed to 44 °C (HS) in a 5% CO2 incubator for 1 h, or to 1 mm H2O2 for 1 h at 37 °C. Netropsin dihydrochloride (Sigma Aldrich) or methyl green (Sigma Aldrich) was added 1 h before treatment and was not removed during treatment.
Antibodies
Anti-Tau antibodies have been described previously (12, 15). The anti-lamin B and anti-Hsc70 antibodies were obtained from Santa Cruz Biotechnology, the anti-Tau3R from Sigma and the anti-human Tau antibody HT7 from Thermo Scientific.
Cell Fractionation
Cell fractionation was prepared as described previously (16).
Electrophoresis and Immunoblotting
Aliquots of cytoplasmic or nuclear extracts were processed as described previously (12). Immunolabeling was observed with an Image Reader LAS3000 (Fujifilm) and quantified by densitometry. Phosphorylated and total Tau levels were normalized against synaptophysin for the cytoplasmic fraction and lamin B for the nuclear fraction. Phosphorylated Tau levels were further normalized against the level of total Tau to measure the exact phosphorylation state.
Lactate Dehydrogenase Assay
Lactate dehydrogenase assay was assayed as described by the manufacturer (Promega).
Immunofluorescence
Cell cultures were fixed in cold 4% paraformaldehyde for 30 min at room temperature. Permeabilization was carried out in 0.2% Triton X-100 in phosphate-buffered saline for 10 min. After a 30-min saturation in 2% bovine serum albumin, immunostainings were carried out using Tau antibodies. Tau staining was revealed with goat anti-mouse or goat anti-rabbit IgG (Heavy and Light chains) antibodies coupled to Alexa Fluor® 488 (Molecular Probes). DAPI was present in the Vectashield mounting medium for fluorescence (Vector). Slides were analyzed with a Zeiss LSM710 confocal laser scanning microscope (60× magnification). Images were collected in the z direction at 0.50-μm intervals. For Tau1 labeling, cells were incubated with 1 μm ToPro3 (Molecular Probe) for 30 min and analyzed with a Leica-DMRBE microscope with a TCS 4D confocal head. Alternatively, permeabilization (0.2% Triton X-100 in phosphate-buffered saline for 10 min) was carried out before 10 min of ice-cold methanol fixation to remove soluble proteins.
Electron Microscopy
Cell cultures were fixed with 0.05% glutaraldehyde and 4% paraformaldehyde in 0.2 m Pipes buffer for 30 min at 4 °C. Cells were then incubated in phosphate-buffered saline containing 10% fetal calf serum, scraped, and centrifuged at 15,000 × g. Pellets were soaked overnight in phosphate-buffered saline containing 2.3 m sucrose and 20% polyvinylpyrrolidone. Cells were rapidly frozen in liquid nitrogen. Frozen ultrathin sections were made with a cryo-ultramicrotome (Leica) at a thickness of 85 nm. The sections were picked up on formvar-carbon-coated nickel grids. After a 30-min saturation in 2% bovine serum albumin, immunostaining was carried out using Tau5, an anti-total Tau antibody. Tau5 staining was revealed with a goat anti-mouse IgG gold conjugate (12 nm in diameter) (Jackson ImmunoResearch Laboratories). Negative staining of the ultrathin sections was carried out using 0.4% uranyl acetate and 1.8% methyl cellulose. Labeling was observed under a Zeiss 902 electron microscope.
Chromatin Immunoprecipitation
Chromatin immunoprecipitation was carried out as described previously (18). Genomic DNA from control and HS-treated cells were immunoprecipitated either with Tau1 antibody or with a polyclonal antibody directed against the nonstructural NSs protein encoded by Rift Valley Fever Virus (a generous gift from Dr. Michèle Bouloy). Immunoprecipitated and input (corresponding to 20 μg of total nonimmunoprecipitated genomic DNA) DNAs were 5′-end radioactively labeled using T4 polynucleotide kinase (exchange reaction) and [γ-32P]ATP (3000 Ci/mmol). 32P-labeled DNA was quantified after migration on a nondenaturing polyacrylamide gel using a PhosphorImager.
Comet and Fast Halo Assays
Comet and fast halo assays (four cells/slide) were embedded in a layer of 0.5% of low melting point agarose (Bio-Rad) and kept at 37 °C previously coated upon slides. Slides were replaced on a slide tray on ice packs for 3 to 5 min. Four slides (two for the nondenaturing fast halo assay and two for the Comet assay) were prepared for each cell suspension.
Nondenaturing Fast Halo Assay
The fast halo assay was used to identify DNA fragmentation due to apoptosis and/or necrosis and/or true genotoxicity. It was performed under alkaline conditions following the procedure of Sestili et al. (19). The essential steps of the fast halo assay consisted of the following. Slides were immersed in the pH 10.1 lysis solution for 10 min at +4 °C in the dark. The slides were then rinsed in PBS for <30 s and neutralized for 15 min in PBS (pH 7.4) containing 0.1 mg/ml RNase. The DNA was then exposed for 5 min to absolute ethanol to preserve all the halo assay slides.
Comet Assay
After the top layer of agarose had solidified, the slides were immersed for at least 1 h at +4 °C in the dark in a lysis solution consisting of 2.5 m NaCl, 100 mm EDTA, 10 mm Tris, pH 10, to which 1% Triton X-100 and 10% dimethyl sulfoxide were freshly added. The slides were then removed and placed on a horizontal gel electrophoresis unit, and the unit filled was with freshly prepared alkaline buffer (1 mm EDTA and 300 mm NaOH, pH > 13) to ∼0.25 cm above the slides. To reduce the variability associated with gel box slide position or multiple electrophoresis runs, slides were randomly distributed. The cells were exposed to the alkaline solution for 20 min to allow DNA unwinding and expression of single-strand breaks and alkali-labile sites. Next, electrophoresis was conducted for 20 min at 0–4 °C by applying an electric current of 0.7 V/cm (25 V/300 mA). All of these steps were conducted in the absence of daylight to prevent additional DNA damage. After electrophoresis, the slides were neutralized with 0.4 m Tris (pH 7.5), and the DNA was exposed for 5 min to absolute ethanol to preserve all the comet assay samples. Subsequently, the slides were air-dried and then stored at room temperature until scored for DNA migration (20).
Scoring
Just prior to scoring, the DNA was stained using propidium iodide (20 μg/ml distilled water; 25 μl/slide). Slides were coded and examined at 200× magnification using a fluorescent microscope (Leica Microsystems SAS-DM 2000, Heerbrugg, Switzerland), equipped with an excitation filter of 515–560 nm and a barrier filter of 590 nm, connected through a gated monochrome CCD IEEE1394 FireWire video camera (Allied Vision Technologies) to a Comet Assay IV Image Analysis System (version 4.11) with Windows XP Pro Software (Perceptive Instruments Ltd, Suffolk, UK). Images of at least 50 randomly selected cells were analyzed from the two slides prepared, i.e. 100 cells per culture. For the fast Halo assay, 100 hundred cells per slide were also randomly scored.
Choice of DNA Damage Parameters
The Olive tail moment (OTM), developed by Olive (21), was used to evaluate DNA damage. The OTM, expressed in arbitrary units, is calculated by multiplying the percent of DNA (fluorescence) in the tail by the length of the tail in micrometers (22). The tail length is measured between the edge of comet head and the end of the comet tail. A major advantage of using the OTM as an index of DNA damage is that both the amount of damaged DNA and the distance of migration of the genetic material in the tail are represented by a single number (23).
Subcellular Fractionation from Brain Slices
Brain slices were prepared as described previously (17). Mouse brains were quickly removed and placed in ice-cold artificial cerebrospinal fluid containing: 117 mm NaCl, 4.7 mm KCl, 1.2 mm NaH2PO4, 23 mm NaHCO3, 2.5 mm CaCl2, 1.2 mm MgCl2, and 25 mm glucose, continuously oxygenated with 95% O2, 5% CO2 (pH 7.4). Transverse brain slices (400 μm) were cut using a Vibratome (Leica, Wetzlar, Germany). Slices were obtained from 1.70 mm anterior to the bregma. For allowing recovery from damage, the slices were maintained for 30 min at room temperature in homemade incubation chambers containing artificial cerebrospinal fluid and continuously bubbled with 95% O2, 5% CO2. The slices were then transferred to oxygenated artificial cerebrospinal fluid at 37 °C for 1 h. For each brain, one of the two batches was then transferred to oxygenated artificial cerebrospinal fluid at 44 °C for 1 h, and the other one was kept at 37 °C. Slices were then dissected, and the appropriate quantity of cortex was removed.
Analysis and Statistics
Results are expressed as means ± S.D. of at least three independent experiments. ImageJ software was used for quantification. The Mann-Whitney test and the two-way analysis of variance test were used to statistically analyze the expression of proteins visualized by Western blots. The Student's t test was used to statistically analyze the OTM values in the Comet assay.
RESULTS
Oxidative Stress and Heat Stress Induce Nuclear Accumulation of Tau
Mild hyperthermia shares common mechanisms with oxidative stress such as that induced by exposure to H2O2 and reactive oxygen species production (24, 25). H2O2 and HS were previously reported to reduce the phosphorylation of Tau in neuronal cultures through protein phosphatase 2A activation (12, 26). Given that neuronal nuclear Tau has primarily been described under a dephosphorylated state, based on Tau1 labeling using an antibody (27), we examined whether oxidative stress or HS could modulate the cellular localization of Tau. The influence of H2O2 and HS on the cellular localization of Tau was evaluated in neurons by exposing cortical primary cultures of neurons to 1 mm H2O2 or hyperthermia (44 °C) for 1 h. H2O2 (Fig. 1A) and HS (Fig. 1B) induced a strong increase in Tau level in the nuclear fraction. A slight but significant decrease in Tau level was also detected in the cytosol after both stresses. Low molecular weight bands, indicative of Tau degradation, were not detected in the nucleus. HS-induced dephosphorylation of Tau was analyzed by immunoblotting with Tau1 antibody, which recognizes Tau only when serine residues (195–202) are dephosphorylated. Dephosphorylated Tau, detected by Tau1, increased after oxidative and heat treatment in both cytosolic and nuclear fractions. Taken together, our results suggest that both stresses induced Tau dephosphorylation and increased the nuclear localization of dephosphorylated Tau in cultured neurons.
FIGURE 1.

Cellular distribution of Tau in stress-treated cortical neurons. Western blot analysis of Tau expression in primary neuronal cultures of mouse embryonic cortex using an anti-total Tau antibody recognizing the C-terminal Tau sequence and anti-nonphospho-Tau (Tau1) antibody. Cortical cultures were maintained at 37 °C (C), treated with H2O2 (A) or heated to 44 °C (HS; B) for 1 h. Cells were subjected to cellular fractionation as described under “Experimental Procedures.” Distribution of Tau was analyzed in cytosolic and nuclear fractions. H2O2 and HS increased the nuclear Tau level in cultured neurons. Densitometric analysis of Western blot using anti-total Tau antibody. Synaptophysin (SYP) and lamin B were respectively used as specific markers of the cytoplasmic and the nuclear fractions. Results are expressed as a percentage of the control. Data shown are the mean ± S.D. of four different experiments using separate cultures.
H2O2 Induces Cytotoxicity in Neuronal Cultures
A lactate dehydrogenase assay was used to test the effect of 1 h H2O2 and HS treatments on cell viability in cultured cortical neurons and after a recovery of 24 or 48 h at 37 °C (Fig. 2). Only H2O2 significantly increased lactate dehydrogenase release 24 h after treatment. These results show that an acute stress induced by H2O2 induces cell death in neuronal cultures. Contrary to H2O2, stress induced by mild hyperthermia was not cytotoxic, demonstrating that HS-induced accumulation of Tau in the nuclei of neurons did not induce cell death. Mild hyperthermia was chosen in the following study to characterize and study the function of stress-induced nuclear Tau accumulation, free from interfering cell death mechanisms.
FIGURE 2.

Effects of stress treatments on the lactate dehydrogenase level in neuronal cultures. The influence of 1 mm H2O2 and HS for 1 h or after 24- or 48-h recovery at 37 °C on lactate dehydrogenase level was measured in cultured cortical neurons. H2O2 but not HS induced lactate dehydrogenase leakage in medium 24 h after treatment.
Heat Stress-induced Nuclear Accumulation of Tau Is Reversible
To study the consequences of HS-induced accumulation of nuclear Tau, cells were allowed to recover for 24 h at 37 °C after HS. The results shown in Fig. 3 (A and B) demonstrate that the level of nuclear Tau, as detected by anti-total Tau and Tau1 antibody, was strongly reduced after heat-stressed cells recovered at 37 °C, with the amount of nuclear Tau returning to a level similar to that observed before HS. The absence of lower molecular weight bands indicates that Tau was not degraded. These results suggest that stress-induced Tau dephosphorylation and nuclear accumulation is reversible.
FIGURE 3.

Transient nuclear accumulation of Tau in heat stress-treated neurons. A, Western blotting analysis using phospho-independent (anti-C-terminal Tau) and dephospho-Tau (Tau1) antibodies. Cortical cultures were heated to 44 °C (HS) or kept at 37 °C (C) for 1 h and then maintained at 37 °C for 24 h. Total Tau and dephosphorylated Tau labeling were analyzed both in cytosolic and nuclear fractions. A loss of nuclear Tau staining was observed 24 h after HS. B, densitometric analysis of Western blot using anti-total Tau antibody. Results are expressed as percentage of the control. Data shown are the mean ± S.D. of three different experiments. ***, p = 0.0001; *, p < 0.05.
Nuclear Tau under Heat Stress Is Mainly Dephosphorylated
The results obtained with Tau1 antibody in Fig. 1B suggested that HS induced the dephosphorylation of Tau. HS-induced dephosphorylation of Tau was further confirmed by immunoblotting with the phosphodependent Tau antibodies anti-pT212-Tau, AT180, 12E8, and AD2. Fig. 4A illustrates that HS led to a strong decrease of phosphorylated Tau. None of these phosphorylated forms of Tau were detected in the nuclear fraction under control or HS conditions, confirming that the dephosphorylated state of the nuclear form of Tau accumulated after HS.
FIGURE 4.

Heat stress modulates Tau phosphorylation in cortical neurons. A, representative Western blot analysis using anti-total Tau antibody (anti-C-terminal Tau) and phospho-dependent Tau antibodies (Tau1, PT212, AT180, 12E8, and AD2). Cortical cultures were untreated (C) or treated with HS. Tau phosphorylation at various epitopes was analyzed both in cytosolic and nuclear fractions. In the cytosol, HS induced dephosphorylation. In nuclei from HS-treated neurons, Tau accumulated in the nucleoplasm was in a dephosphorylated state. B, representative Western blot analysis using anti-total Tau antibody (anti-C-terminal Tau) and phospho-dependent antibodies (Tau1, PT212, AT180, 12E8, AD2) in brain slices from adult mouse cortex. Cortical slices were untreated or treated with HS. In adult neurons, HS induced nuclear dephosphorylation of all Tau epitopes except for Ser262–356 (12E8).
Heat Stress-induced Nuclear Accumulation of Tau Is Reproduced in Adult Neurons
In primary neuronal cultures, neurons were studied after 10 days in vitro and thus were mainly differentiated. Nevertheless, to overcome any potential artifactual effects related to the embryonic origin of the neurons, we also tested the effect of HS in the adult mouse brain. Ex vivo slices from the cerebral cortex of adult mice were incubated for 1 h at 37 or 44 °C, and the cytosolic and nuclear fractions were analyzed under the same conditions as in Figs. 1B and 4A. Hyperthermia induced an increase in the level of nuclear Tau in the adult mouse brain, as previously observed in embryonic neuronal cultures. The phosphorylation state of stress-induced nuclear Tau was analyzed in brain slices from adult mice (Fig. 4B) using the same phospho-specific antibodies as in Fig. 4A. Similar results were obtained with Tau1 (as in Fig. 1B), p-T212, AT180, and AD2 antibodies, but not with the 12E8 antibody. At 37 °C as well as after HS, nuclear Tau was detected not only with Tau1 but also, surprisingly, with 12E8. Under HS, a specific increase of Tau1 and 12E8 labeling was observed. Overall, these data indicate that in the ex vivo model of cortical slices, nuclear Tau was mainly dephosphorylated both in the control condition and under HS, except for at Ser262 and Ser356 ((Lys-X-Gly-Ser) motifs). This is consistent with previous findings that Tau is mainly dephosphorylated, except at Ser262, in cerebral extracts from heat-shocked rats (28). Interestingly, although the majority of Ser/Thr-Pro sites that flank the microtubule-binding repeats are phosphorylated by so-called Ser/Thr-Pro kinases such as Cdk5 and GSK3 (15), Ser262 and Ser356 are located within microtubule-binding domains and form KXGS motifs that are preferentially phosphorylated by MARK (Microtubule Affinity-Regulating kinase)/Par-1 kinase, CamKII (Calmodulin-Dependent Protein kinase II) or ChK2 (29–31). A differential expression and/or regulation of these kinases between embryonic and adult neurons could explain the differences observed here with the Tau 12E8 antibody.
Microscopic Visualization of Heat Stress-induced Accumulation of Tau in Neuronal Cells
The accumulation of Tau in the nuclei of neuronal cultures after HS was further analyzed by immunofluorescence and confocal microscopy analysis (Fig. 5, A–C). Under the control condition, total Tau antibody clearly stained the cellular body and neurites but only weakly labeled the nucleus (Fig. 5A, upper panel). Under the HS condition, however, accumulation of Tau was clearly observed in the nuclei of cortical primary cultured neurons (Fig. 5A, lower panel).
FIGURE 5.

Confocal microscopy analysis of Tau localization in cultured neurons. A, immunofluorescence staining of total Tau (Tau3R) in cortical neuronal cultures. Tau3R antibody specifically recognizes Tau isoforms presenting three repeat domains, this being the case of the embryonic Tau isoform, independently of their phosphorylation state. Immunolabeling of Tau and DAPI nuclear counterstaining were analyzed by confocal laser scanning microscopy. HS strongly increased Tau staining in neuronal nuclei. Scale bars indicate 20 μm. B, immunofluorescence staining of total Tau (TauC17). Cells were permeabilized before ice cold methanol fixation to only visualize nuclear bound Tau. Arrowheads indicate neuronal nuclei. Nuclear fluorescence was analyzed by confocal laser scanning microscopy. C, immunofluorescence staining of nonphospho-Tau (Ser195–202) using Tau1 antibody in cortical culture of neurons. Immunolabeling of Tau1 and ToPro3 nuclear counterstaining were analyzed by confocal laser scanning microscopy. Heat shock strongly increased Tau1 staining in neuronal cytosol and nucleus. Scale bars indicate 10 μm.
The location of Tau was also studied after removing soluble proteins by permeabilization of the cells before ice-cold methanol fixation (Fig. 5B). In this condition, Tau tightly bound to insoluble cell structures (“bound” Tau) was immunolabeled using an anti-total Tau antibody. Nuclear fluorescence was quantified using a Zeiss confocal microscope program. The average ratio of bound Tau under the HS versus the control condition was 2.06 ± 0.17 (p < 0.001).
The accumulation of dephosphorylated Tau in the nuclei of neuronal cultures after HS was illustrated using Tau1 antibody (Fig. 5C). Under control conditions, no labeling was detected in neuronal nuclei, and only weak labeling was observed in the cellular body (Fig. 5C, upper panel). Conversely, clear staining of dephosphorylated Tau was detected on the entire cell body and the nucleus after HS treatment (Fig. 5C, lower panel).
The accumulation of nuclear Tau under HS was also visualized by electron microscopy (Fig. 6). Tau was detected in the nuclei of frozen sections derived from cortical neuronal cultures only under stress conditions (Fig. 6, C (I, II, and III) associated with DNA (light gray regions in Fig. 6, C (II) as well as with nucleoli (Fig. 6, C (III)). This nucleolar localization is consistent with previous studies describing nucleolar localization of Tau in various cell lines (4, 6, 10).
FIGURE 6.
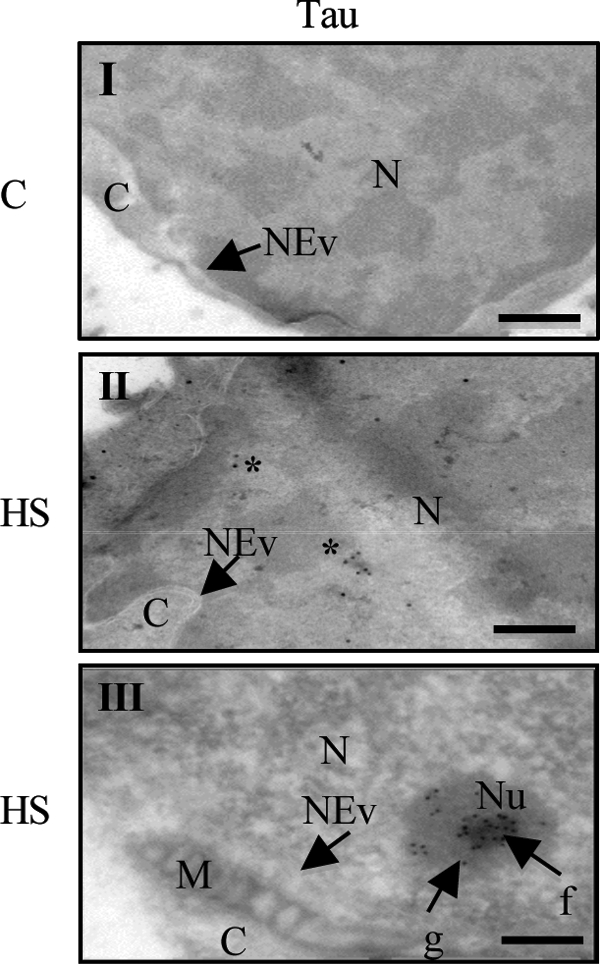
Electron microscopy analysis of Tau localization in cultured neurons. Immunogold electron microscopy of total Tau (Tau5 antibody) in primary neuronal cultures of mouse embryo cortex. M, mitochondria; C, cytosol; N, nucleus; Nu, nucleolus; f, fibrillar area; g, granular area; NEv, nuclear envelope. Cortical cultures were untreated (C, I) or treated with HS (I, III). Asterisks indicate Tau staining. Scale bars indicate 100 nm.
Heat Stress Potentiates Tau-DNA Complex Formation
The ability of Tau to form protein-DNA complexes in vitro has been previously reported by different groups (6, 8, 32, 33). To analyze the ability of Tau to interact in situ with neuronal DNA before and after HS treatment, we carried out ChIP experiments (Fig. 7). During these experiments, genomic DNA from control and HS-treated cells was immunoprecipitated with either Tau1 antibody or an antibody directed against the nonstructural NSs protein encoded by the Rift Valley fever virus, which was used here as an arbitrary negative control. The total amount of immunoprecipitated DNA was labeled at the 5′-end with [γ-32P]ATP and submitted to nondenaturing polyacrylamide electrophoresis (Fig. 7). These results indicate that the capacity of Tau protein to interact with DNA, previously described in vitro, could also be observed in situ in a dose-dependent manner. In addition, we found that the total amount of DNA interacting with nuclear Tau was ∼2-fold higher in heat-stressed cells compared with control cells (Fig. 7). The difference was even more significant considering that overall labeling of input DNA, corresponding to total genomic DNA before immunoprecipitation, was slightly weaker after HS compared with the control condition. As expected, no DNA immunoprecipitated with the anti-NSs antibody in control or in HS cells.
FIGURE 7.

Interaction of nuclear Tau with genomic DNA is enhanced after HS treatment. Equal amounts (20 or 10 μg) of genomic DNA from control (C) and heat-shocked (HS) primary cultured neurons were immunoprecipitated with Tau1 antibody or an antibody directed against the viral nonstructural NSs protein (anti-NSs used here as a negative control). Immunoprecipitated and input DNA (corresponding to nonimmunoprecipitated genomic DNA) as well as pBR322/AluI DNA were 5′-end 32P-radioactively labeled and submitted to nondenaturating electrophoresis. Tau interacted with DNA in a dose-dependent manner. HS increased the Tau-DNA interaction. Data shown are representative of three independent experiments using separate cultures.
In parallel, the concentration of DNA that immunoprecipitated with the anti-Tau1 antibody under control or HS conditions was measured using a Nanodrop spectrophotometer. The average ratio of immunoprecipitated DNA (% of input) under HS versus control conditions was 1.8 ± 0.07 (p < 0.001). This result was correlated with the increase in nuclear bound Tau observed under HS (Fig. 5B). Taken together, these data show that in addition to enhancing the nuclear fraction of Tau, HS also enhanced the amount of Tau interacting with DNA, suggesting that under the HS condition, the majority of nuclear Tau forms protein-DNA complexes with endogenous cellular DNA.
Agent That Competitively Binds to DNA Minor Groove Partially Prevents Nuclear Tau Accumulation
Previous studies demonstrated that, in vitro, purified Tau binds to synthesized DNA specifically through the minor groove (8). To assess this specificity in situ, competitor experiments were performed using netropsin, a naturally occurring antibiotic that binds to the minor groove of DNA in spots rich in AT, and methyl green, a DNA major groove binding drug. Competitive effects were tested on Tau nuclear accumulation (Fig. 8, A and B). Netropsin significantly decreased the level of nuclear Tau under heat stress conditions. Concentrations from 10 to 25 μm induced a similar effect on nuclear Tau accumulation (data not shown). Identical concentrations of methyl green did not alter heat shock-induced Tau accumulation in the nucleus.
FIGURE 8.

Netropsin modulates HS-induced nuclear Tau accumulation. (Bottom), Western blot analysis of effects of netropsin on Tau, Hsc70, and lamin B expression in the nuclear fraction using, respectively, anti-total Tau (C-terminal Tau), anti-Hsc70 and anti-lamin B antibodies. (Top), densitometric analysis of Western blot using an anti-total Tau (C-terminal Tau) antibody. Results are expressed as a percentage of the control. Data shown are the mean ± S.D. of three different experiments. *, p < 0.05.
As heat shock is known to induce rapid Hsc70 accumulation into cellular nuclei independent of minor groove binding, the detection of Hsc70 was used first to check the lack of effect of netropsin on the integrity of nuclear shuttling mechanisms and second as a negative control to check the specificity of netropsin binding to DNA at the minor groove. As expected, netropsin did not modify the heat shock-induced nuclear increase of Hsc70. Netropsin partially prevented stress-induced nuclear Tau accumulation. These data suggest that the interaction of nuclear Tau with DNA is partly mediated through interactions with the A/T-rich DNA minor groove and that minor groove-unbound Tau would not remain in the nuclear fraction. Interestingly, if minor groove-unbound Tau does not remain in the nuclear fraction, it suggests that accumulation of Tau into the nucleus under heat stress conditions is subordinate to DNA binding.
Tau Deficiency Generates Heat Stress-induced DNA Damage
Previous in vitro studies have shown that Tau protein is able to prevent DNA from damage induced by thermal denaturation and peroxidation (7, 8). To test the role of Tau in neuronal DNA in situ, we studied the effect of Tau deficiency on DNA integrity under HS using the single-cell gel electrophoresis assay (the Comet assay) (Fig. 9). The Comet assay under alkaline conditions is considered the most sensitive quantitative method for measuring damage to genomic DNA of eukaryotic cells on a single-cell basis (21). It detects DNA damage as single-stranded and/or double-stranded DNA breaks and DNA-DNA and/or DNA-protein cross-links at the level of the genome of eukaryotic cells, alkali-labile sites, incomplete DNA repair sites, and changes in chromosome structural conformations (19). The OTM was used to evaluate DNA damage in single cells by measuring DNA-specific fluorescent staining. The effect of HS on DNA integrity was assessed in both wild-type and Tau-deficient cortical neuronal cultures. Fig. 9 shows the ratio of the OTM of HS-treated cells over control cells. DNA migration was strongly reduced in Tau-deficient cells, suggesting DNA damage. Delayed migration of DNA observed in Tau-deficient cells under HS is considered to reflect either DNA cross-linking or bulk adduct formation (34). Conversely, HS did not significantly induce DNA damage in wild-type neuronal cultures, suggesting that Tau can protect neuronal DNA integrity under HS conditions.
FIGURE 9.
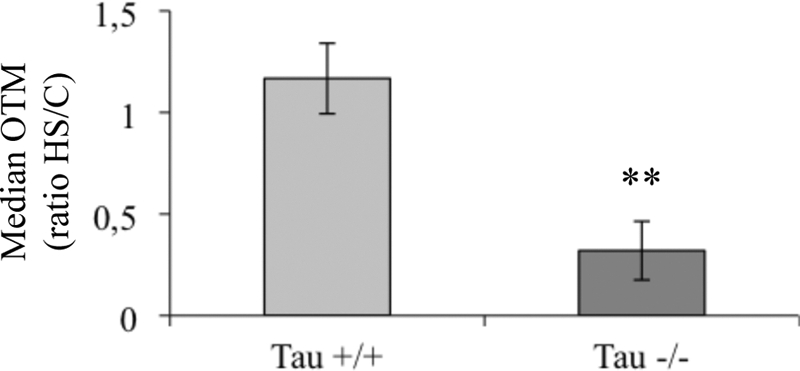
Heat stress selectively impairs DNA integrity in Tau-deficient neuronal cultures. The effect of HS on DNA from wild-type (Tau+/+) or Tau-deficient cells (Tau−/−) was measured by Comet assay. Results are presented as the OTM ratio from heat-shocked cells (HS) over control (C). Each OTM value is a median value from 150 to 200 cells. Data shown are the means from three different experiments using separate cultures. **, p < 0.01.
To test whether the DNA damage detected in Tau-deficient cultures by the Comet assay (Fig. 9) reflected cell death, the highly sensitive nondenaturing fast halo assay was used to selectively quantify damaged DNA associated with apoptosis and necrosis (19). As shown in Table 1, HS did not induce apoptotic or necrotic features in either wild-type or Tau-deficient cultures, indicating HS-induced DNA damage observed in Tau-deficient neurons was not a consequence of cell death.
TABLE 1.
Tau deficiency-related DNA damage is not related to cell death
Shown is a representative fast halo assay measuring the percentage of apoptotic and necrotic cells from wild-type and Tau-deficient cultures untreated or treated with HS. Apoptotic or necrotic cells were detected in a population of 100 cells (p < 0.001).
| Ratio HS/C |
||
|---|---|---|
| Apoptotic cells | Necrotic cells | |
| Tau+/+ | 0.66 | 1.01 |
| Tau−/− | 1.05 | 1.10 |
Overexpression of Nuclear Tau Rescues Heat Stress-induced DNA Damage in Tau-deficient Neurons
To confirm the role of Tau in protecting DNA against HS-induced damage, Tau-deficient cultures were infected with an adenoviral vector encoding human wild-type Tau (HAdV-5hTau44Wt) and were then subjected to HS. To accurately determine the specific role of nuclear Tau in DNA protection, an NLS was fused to the hTau sequence (HadV-5- hTau44-NLS) (Fig. 10A). hTau (human Tau), which was predominantly localized in the cytosolic fraction under control conditions, accumulated into the nucleus after HS treatment, whereas most of hTau-NLS was present in the nucleus under both control and stress conditions (Fig. 10A). The integrity of DNA after HS was analyzed using the Comet assay in wild-type and Tau-deficient cultures overexpressing hTau or hTau-NLS. As shown in Fig. 10B, expression of hTau or hTau-NLS fully prevented HS-induced DNA damage in Tau-deficient cultures. The complete prevention of DNA damage by nuclear-targeted Tau demonstrated that nuclear localization of Tau was, by itself, sufficient to protect DNA from HS-induced damage and that DNA protection was not related to an indirect cytosolic Tau-induced effect.
FIGURE 10.

Heat stress-induced DNA damage induced in Tau-deficient neurons is prevented by hTau-NLS or hTau overexpression. A, Western blotting analysis using a specific antibody raised against human Tau (HT7). Tau-deficient cortical cultures were infected for 24 h with adenoviral vectors coding for human Tau (HAdV-5-hTau44Wt) or human Tau-NLS (HAdV-5-hTau44NLS), heated to 44 °C (HS) or maintained at 37 °C (C) for 1 h and subjected to subcellular fractionation. hTau-NLS was specifically expressed in the nuclei independently of temperature treatment. hTau was mostly localized in the cytosolic fraction in the control condition. Nuclear hTau level increased after HS. B, Comet assay showing the effect of human Tau and human Tau-NLS overexpression in Tau-deficient cells. Data shown are the means ± S.D. of 100 cells measured for each condition. **, p < 0.005. SYP, synaptophysin.
DISCUSSION
Here, we report that oxidative or hyperthermic stress induces accumulation of dephosphorylated Tau in the nuclei of embryonic and adult neurons. The presence of Tau in the nuclei of heat-stressed neurons was necessary to protect DNA from HS-induced damage. Tau-mediated genomic DNA protection was correlated with an increase of Tau-DNA binding. This study is the first in situ demonstration that oxidative and HS insults modulate nuclear Tau translocation and that nuclear Tau is essential to protect neuronal DNA from HS-induced damage.
Although the presence of Tau in the nuclei of neurons was reported more than 15 years ago (11), this is the first demonstration that environmental conditions can modulate neuronal cytoplasmic-nuclear transport of Tau in a developmentally independent manner. Both nuclear import and export of proteins are highly regulated processes (35). Movement of proteins larger than ∼40 kDa into the nucleus requires specific transport receptors, the use of the nuclear pore complex and the recognition of specific signals in the cargo protein by a transport receptor. Given that Tau sequences do not include a canonical NLS or a nuclear export signal, cytoplasmic/nuclear translocation of Tau likely requires the interaction of Tau with one or more other proteins to pass through the nuclear pore complex.
Phosphorylation is a biological process that regulates nucleo-cytoplasmic transport (36, 37). In this work, we observed that stress-induced dephosphorylation of cytosolic Tau was correlated with nuclear accumulation of Tau. On the other hand, at 24 h after HS, increased cytosolic Tau phosphorylation was associated with decreased nuclear Tau. Rephosphorylation of Tau after HS has been previously reported in a heat shock rat model (28). Taken together, these data suggest that phosphorylation might be a major mechanism to control nuclear Tau shuttling. Further investigation is necessary to decode the mechanisms involved in the nucleo-cytoplasmic trafficking of Tau in stressed neurons.
Nevertheless, given the strong increase of Tau levels in the nuclear fraction, changes in Tau transcription within the nucleus cannot be fully ruled out. However, as observed with changes in Tau phosphorylation and reversibility, it seems that nucleo-cytoplasmic shuttling of Tau proteins is mainly responsible for the increase in nuclear Tau.
Interestingly, oxidative stress and hyperthermia quickly induce nuclear translocation of molecular chaperones (38, 39) as well as proteins involved in the etiopathology of neurodegenerative diseases, such as α-synuclein (40), the amyloid precursor protein C-terminal fragment (41) or amyloid precursor protein partner Fe65 (42) and ataxin-3 (43).
Neurons are highly differentiated postmitotic cells that cannot divide and cannot survive after irreversible damage; therefore, neurons are highly susceptible to insult. Because neurons should survive as long as the organism does, they need to employ powerful defense mechanisms to ensure their functionality and long term survival (44). Altogether our results demonstrate that nuclear Tau emerges as a novel neuronal genome caretaker in stress condition as illustrated in Fig. 11.
FIGURE 11.

Tau-mediated DNA protection in postmitotic neurons. Shown is a scheme illustrating the nuclear Tau protection of DNA integrity in heat-stressed neurons.
The capacity of Tau to protect neurons in situ from HS-induced DNA damage was correlated with an increased interaction of Tau with genomic DNA. To further characterize Tau-DNA complex formation, we predicted the DNA-binding residues (3) in the Tau sequence (supplemental Fig. 1). This analysis suggested that the proline-rich domain of Tau is highly susceptible to interacting with DNA, especially at the epitopes specifically recognized by Tau1, PT212, and AT180 antibodies. Interestingly, in vitro results from EMSA previously implicated the proline-rich domain of Tau in DNA association (8).
Results obtained with netropsin indicates that in situ Tau interacted with DNA through the A/T-rich minor groove, as previously demonstrated in vitro (8). DNA protection by minor groove-binding proteins is a mechanism common to various types of cells for efficient preservation of the integrity of the genome. Histone H1 (45) and Hmgb1 (high mobility group box 1 protein) (46) are chromatin architectural proteins that bind to the minor groove of double-stranded DNA and confer protection to DNA against radiation damage. In prokaryotes, spores of Bacillus bacteria represent an astonishing example of a powerful DNA protection mechanism mediated by minor groove-binding proteins. Dormant spores of Bacillus can survive for hundreds of years. Spores of Bacillus are extremely resistant to a wide array of extreme insults, including heat, desiccation, toxic chemicals, enzymes, and radiation. This spore resistance is mainly due to protection against DNA damage by the binding of small acid-soluble proteins to the DNA minor groove, inducing protective changes in the structure of DNA (47, 48). Interestingly, as small acid-soluble proteins, Tau is an acid-soluble molecule. Tau has been previously purified from brain extracts based on heat stability and acid solubility derived from a histone purification protocol (49). It is tempting to speculate that DNA safeguards mediated by Tau might reproduce an ancestral mechanism conserved during evolution from prokaryotic cells to highly specialized eukaryotic cells such as neurons to provide powerful resistance to stress insults. Thus, interaction of Tau with DNA through the minor groove might be important for neuronal longevity in the brain.
Interest in Tau arose when different laboratories discovered that this protein was the main component of paired helical filaments that compose neurofibrillary tangles. Neurofibrillary tangles are a hallmark of Alzheimer disease (AD) and other neurodegenerative disorders (the so-called tauopathies) (15). In AD, aberrant modifications of Tau, including hyper-and abnormal phosphorylation, oxidation, truncation, and conformational changes, induce filamentous aggregation and neurofibrillary tangle formation in selectively vulnerable neurons. Accumulation of nuclear DNA damage in neurons has been suggested to be a major form of damage involved in AD (50). Interestingly, the neurons that show neurofibrillary tangle formation in AD are the same that show age-related accumulation of nuclear DNA damage (51). An attractive hypothesis would be that pathological alterations of Tau, e.g. hyperphosphorylation, might impair its ability to shuttle between the cytoplasm and the nucleus and/or affect its affinity for DNA. Thus, altered forms of Tau would fail to efficiently protect DNA from insults such as oxidative stress, an early mechanism involved in AD etiopathology. Impairment of DNA protection by altered Tau would contribute to functional failure of neurons and might represent an essential mechanism of AD etiopathology.
In conclusion, this study highlights a new function for Tau as a key player in the neuronal early stress response and demonstrates the dramatic DNA safeguarding activity of nuclear Tau. The demonstration of a new function for neuronal nuclear Tau may have important implications for a better understanding of neuronal biology and AD etiopathology.
Supplementary Material
Acknowledgments
We thank Pr. Joris Winderickx for fruitful discussions and Dr. Marie-Laure Caillet for helpful suggestions. We are grateful to Hélène Tran, Katia Lecolle, Elodie Marciniak for technical assistance and Meryem Tardivel for confocal microscopy assistance. We are grateful to the IMPRT (Institut de Médecine Prédictive et de Recherche Thérapeutique, Lille) for access to the electron microscopy platform and the confocal microscopy platform.
This work was supported by Inserm, CNRS, IMPRT, University Lille 2, Région Nord/Pas-de-Calais, Fonds Européen de Dévelopment Régional, DN2M, as well as grants from ANR-08-MNPS002/AMYTOXTAU and from the European Community MEMOSAD (FP7 Contract 200611).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
- NLS
- nuclear localization signal
- HS
- heat stress
- hTau
- human Tau
- AD
- Alzheimer disease
- OTM
- Olive tail moment
- HAdV
- human adenovirus
- NS
- nonstructural protein.
REFERENCES
- 1. Weingarten M. D., Lockwood A. H., Hwo S. Y., Kirschner M. W. (1975) Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Loomis P. A., Howard T. H., Castleberry R. P., Binder L. I. (1990) Proc. Natl. Acad. Sci. 87, 8422–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y., Loomis P. A., Zinkowski R. P., Binder L. I. (1993) J. Cell Biol. 121, 257–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thurston V. C., Zinkowski R. P., Binder L. I. (1996) Chromosoma 105, 20–30 [DOI] [PubMed] [Google Scholar]
- 5. Cross D. C., Muñoz J. P., Hernández P., Maccioni R. B. (2000) J. Cell. Biochem. 78, 305–317 [PubMed] [Google Scholar]
- 6. Sjöberg M. K., Shestakova E., Mansuroglu Z., Maccioni R. B., Bonnefoy E. (2006) J. Cell Sci. 119, 2025–2034 [DOI] [PubMed] [Google Scholar]
- 7. Hua Q., He R. Q. (2003) Biochim. Biophys. Acta 1645, 205–211 [DOI] [PubMed] [Google Scholar]
- 8. Wei Y., Qu M. H., Wang X. S., Chen L., Wang D. L., Liu Y., Hua Q., He R. Q. (2008) PLoS One 3, 2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Padmaraju V., Indi S. S., Rao K. S. (2010) Neurochem. Int. 57, 51–57 [DOI] [PubMed] [Google Scholar]
- 10. Rossi G., Dalprà L., Crosti F., Lissoni S., Sciacca F. L., Catania M., Di Fede G., Mangieri M., Giaccone G., Croci D., Tagliavini F. (2008) Cell Cycle 7, 1788–1794 [DOI] [PubMed] [Google Scholar]
- 11. Brady R. M., Zinkowski R. P., Binder L. I. (1995) Neurobiol. Aging 16, 479–486 [DOI] [PubMed] [Google Scholar]
- 12. Galas M. C., Dourlen P., Bégard S., Ando K., Blum D., Hamdane M., Buée L. (2006) J. Biol. Chem. 281, 19296–19304 [DOI] [PubMed] [Google Scholar]
- 13. Rogée S., Grellier E., Bernard C., Loyens A., Beauvillain J. C., D'halluin J. C., Colin M. (2007) Mol. Ther. 15, 1963–1972 [DOI] [PubMed] [Google Scholar]
- 14. Hiriart E., Farjot G., Gruffat H., Nguyen M. V., Sergeant A., Manet E. (2003) J. Biol. Chem. 278, 335–342 [DOI] [PubMed] [Google Scholar]
- 15. Buée L., Bussière T., Buée-Scherrer V., Delacourte A., Hof P. R. (2000) Brain Res. Rev. 33, 95–130 [DOI] [PubMed] [Google Scholar]
- 16. Hamdane M., Bretteville A., Sambo A. V., Schindowski K., Bégard S., Delacourte A., Bertrand P., Buée L. (2005) J. Cell Sci. 118, 1291–1298 [DOI] [PubMed] [Google Scholar]
- 17. Kimura R., Devi L., Ohno M. (2010) J. Neurochem. 113, 248–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lemay J., Maidou-Peindara P., Bader T., Ennifar E., Rain J. C., Benarous R., Liu L. X. (2008) Retrovirology 5, 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sestili P., Martinelli C., Stocchi V. (2006) Mutat. Res. 607, 205–214 [DOI] [PubMed] [Google Scholar]
- 20. Tice R. R., Agurell E., Anderson D., Burlinson B., Hartmann A., Kobayashi H., Miyamae Y., Rojas E., Ryu J. C., Sasaki Y. F. (2000) Environ. Mol. Mutagen. 35, 206–221 [DOI] [PubMed] [Google Scholar]
- 21. Olive P. L., Banáth J. P., Durand R. E. (1990) Radiat. Res. 122, 86–94 [PubMed] [Google Scholar]
- 22. Hellman B., Vaghef H., Boström B. (1995) Mutat. Res. 336, 123–131 [DOI] [PubMed] [Google Scholar]
- 23. Ashby J., Tinwell H., Lefevre P. A., Browne M. A. (1995) Mutagenesis 10, 85–90 [DOI] [PubMed] [Google Scholar]
- 24. Bruskov V. I., Malakhova L. V., Masalimov Z. K., Chernikov A. V. (2002) Nucleic Acids Res. 30, 1354–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao Q. L., Fujiwara Y., Kondo T. (2006) Free Radic. Biol. Med. 40, 1131–1143 [DOI] [PubMed] [Google Scholar]
- 26. Goldbaum O., Richter-Landsberg C. (2002) Glia 40, 271–282 [DOI] [PubMed] [Google Scholar]
- 27. Binder L. I., Frankfurter A., Rebhun L. I. (1985) J. Cell Biol. 101, 1371–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shanavas A., Papasozomenos S. C. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 14139–14144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trinczek B., Biernat J., Baumann K., Mandelkow E. M., Mandelkow E. (1995) Mol. Biol. Cell 6, 1887–1902.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshimura Y., Ichinose T., Yamauchi T. (2003) Neurosci. Lett. 353, 185–188 [DOI] [PubMed] [Google Scholar]
- 31. Iijima-Ando K., Zhao L., Gatt A., Shenton C., Iijima K. (2010) Hum. Mol. Genet. 19, 1930–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greenwood J. A., Johnson G. V. (1995) Exp. Cell Res. 220, 332–337 [DOI] [PubMed] [Google Scholar]
- 33. Hua Q., He R. Q. (2002) Protein Pept. Lett. 9, 349–357 [DOI] [PubMed] [Google Scholar]
- 34. Merk O., Speit G. (1999) Environ. Mol. Mutagen. 33, 167–172 [DOI] [PubMed] [Google Scholar]
- 35. Terry L. J., Shows E. B., Wente S. R. (2007) Science 318, 1412–1416 [DOI] [PubMed] [Google Scholar]
- 36. Crampton N., Kodiha M., Shrivastava S., Umar R., Stochaj U. (2009) Mol. Biol. Cell 20, 5106–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mutoh S., Osabe M., Inoue K., Moore R., Pedersen L., Perera L., Rebolloso Y., Sueyoshi T., Negishi M. (2009) J. Biol. Chem. 284, 34785–34792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Daniel S., Bradley G., Longshaw V. M., Söti C., Csermely P., Blatch G. L. (2008) BBA 1783, 1003–1014 [DOI] [PubMed] [Google Scholar]
- 39. Kodiha M., Chu A., Lazrak O., Stochaj U. (2005) Am. J. Physiol. Cell Physiol. 289, C1034–1041 [DOI] [PubMed] [Google Scholar]
- 40. Xu S., Zhou M., Yu S., Cai Y., Zhang A., Uéda K., Chan P. (2006) Biochem. Biophys. Res. Commun. 342, 330–335 [DOI] [PubMed] [Google Scholar]
- 41. Karuppagounder S. S., Xu H., Pechman D., Chen L. H., DeGiorgio L. A., Gibson G. E. (2008) Neurochem. Res. 33, 1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakaya T., Kawai T., Suzuki T. (2008) J. Biol. Chem. 283, 19119–19131 [DOI] [PubMed] [Google Scholar]
- 43. Reina C. P., Zhong X., Pittman R. N. (2010) Hum. Mol. Genet. 19, 235–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barzilai A., Biton S., Shiloh Y. (2008) DNA Repair 7, 1010–1027 [DOI] [PubMed] [Google Scholar]
- 45. Roque A., Orrego M., Ponte I., Suau P. (2004) Nucleic Acids Res. 32, 6111–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Giavara S., Kosmidou E., Hande M. P., Bianchi M. E., Morgan A., d'Adda di Fagagna F., Jackson S. P. (2005) Curr. Biol. 15, 68–72 [DOI] [PubMed] [Google Scholar]
- 47. Setlow P. (2007) TRENDS Microbiology 15, 172–180 [DOI] [PubMed] [Google Scholar]
- 48. Lee K. S., Bumbaca D., Kosman J., Setlow P., Jedrzejas M. J. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 2806–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lindwall G., Cole R. D. (1984) J. Biol. Chem. 259, 12241–12245 [PubMed] [Google Scholar]
- 50. Coppedè F., Migliore L. (2009) Curr. Alzheimer Res. 6, 36–47 [DOI] [PubMed] [Google Scholar]
- 51. Rutten B. P., Schmitz C., Gerlach O. H., Oyen H. M., de Mesquita E. B., Steinbusch H. W., Korr H. (2007) Neurobiol. Aging 28, 91–98 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.