

Abstract
A newly described glomerulotubular feedback loop may explain the relationship between glomerular damage, epitope spreading, tubulointerstitial nephritis, proteinuria as a progression factor, and the importance of the local milieu in kidney damage. It also opens the horizons for exciting innovative approaches to therapy of both acute and chronic kidney diseases.
Keywords: chronic kidney disease, glomerular disease, glomerulosclerosis, immunology and pathology, tubular epithelium
B AND T CELLS IN GLOMERULONEPHRITIS
In the beginning there were B cells. There were, of course, other cells observed in experimental and human glomerulonephritis (GN), but the dictum in the early studies of experimental autoimmune GN (EAG) demanded that the only important lymphocyte was the B cell, because of secreted immunoglobins, which fixed to antigens in the kidney and induced GN. So strong was this theory that suggestions to the contrary were soundly derided.1,2 However, increasing evidence suggested that this paradigm was only partly true. Two models were used to study GN. In the first model of nephrotoxic serum nephritis, heterologous antibodies were administered, fixed to the glomerular basement membrane (GBM), and induced an inflammatory response. Although this was a planted-antigen model and not truly an autoimmune GN, and was not directly pertinent to the human condition, nonetheless, it provided an excellent vehicle to examine glomerular inflammation. Interestingly, this disease model with antibodies only on the GBM was also associated with tubulointerstitial nephritis (TIN). The other model, described by Steblay, involved induction of an autoimmune response to self-antigens in the GBM. This was most analogous to Goodpasture’s syndrome in man. Severe disease could be transferred to monkeys by eluates from a Goodpasture patient’s kidney, but only transient mild disease was transferred by circulating antibodies.3 The ability of the cellular immune system alone to induce EAG was first documented by studies that we performed in an avian model. Chickens were rendered antibody deficient by bursectomy and demonstrated a normal intact delayed hypersensitivity (DTH) system.4 Upon immunization with GBM, florid GN developed with no antibody response and no antibody deposits on the GBM. The disease could be transferred by cells but not antibodies.
Few tools were present in those days to examine the types of cells involved, the pathogenic mechanisms or the true interrelationship between cells and antibodies. Over the ensuing decades, the availability of monoclonal antibodies, recombinant proteins, knock-out and knock-in mice, transgenic animals, and a host of other sophisticated tools have resulted in an increasingly complicated, yet clearer picture of pathogenic processes. It is clear that B and T cells, indeed many cells, are involved in the pathogenesis of autoimmune GN, and in many other glomerular diseases (Figure 1).5–7 Biopsies from humans with GN contain macrophages, CD4 +, and CD8 + cells within the interstitial compartment but also in the glomeruli, with dendritic cells (DCs) isolated to the interstitium.8–10 Indeed, antibody-negative GN, first described by Stilmant et al.11 (pauci-immune disease), is phenotypically indistinguishable from many other types of GN with antibody deposits and was postulated to be a cell-mediated process. Identification of the responsible antigen of Goodpasture’s syndrome, the non-collagenous domain of α3 type IV collagen (Col4A3) with resultant production of recombinant proteins of Col4A3, provided yet further opportunities to investigate the role of T cells in the pathogenesis of GN. Work from many laboratories demonstrated that immunization with Col4A3 could result in GN without antibodies, that multiple interventions with monoclonal antibodies or immunosuppressive drugs that targeted T cells could suppress the disease, and that tolerance could be induced, all suggesting a significant role of T cells.7,12–14 The disease could be transferred with antibodies, but could also be transferred with CD4 + T cells derived from nephritic animals.15,16 The latter disease took a long time to develop, and was marked by an intense TIN followed at a later time by GN. No antibodies were detected in these rats and none of the transferred cells elaborated anti-GBM antibody. Identification of the nephritogenic peptides in this model allowed the demonstration that a pure T-cell epitope was capable of inducing GN without antibody. This also allowed the demonstration that both intra- and intermolecular epitope spreading occurred consequent to T-cell-induced injury.12,17,18 The nephritogenic epitope was restricted to MHC Class II and epitope spreading occurred through local antigen presentation within the kidney draining lymph nodes (KDLNs).19,20 These studies were thus consistent with initiation of a T-cell response to antigen, resulting in GBM damage, release of additional epitopes from the GBM with consequent autoimmunization, epitope spreading, and amplification of the disease process. The exact mechanism of how that might occur was not clear, nor was attention paid to the intense TIN seen in these animals as seen in human disease, consisting of macrophages, CD4 +, and CD8 + cells.21
Figure 1. Cells in GN.
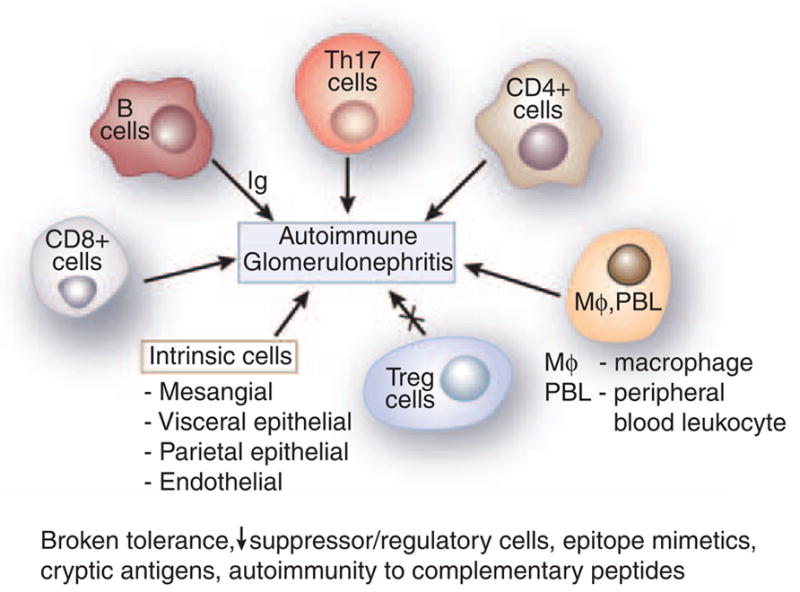
Figure 1 represents an aggregate of data derived from animal models. B cells were classically considered to be involved in the pathogenesis of GN by elaboration of immunoglobulin (Ig). Th-17, CD4 +, and CD8 + cells have a significant role as shown by abrogation of activity leading to amelioration of GN. Macrophages and peripheral blood leukocyte (PBL) are essential in the histological changes of GN. All result in a variable increase in the mesangial matrix, and involvement of the visceral and parietal epithelial cells. T regulatory cells downregulate disease. Experimental autoimmune glomerulonephritis presumably arises secondary to various etiologies, including broken tolerance, a decrease in suppressor/regulatory cells, epitope mimetics, exposure of cryptic antigens, and possibly autoimmunity to complementary peptides.
GLOMERULOSCLEROSIS AND TIN WITH GN
Glomerulosclerosis and TIN are observed in many human and animal models of immune and nonimmune glomerular damages. Indeed, the severity of chronic TIN is the best correlate of decline in kidney function and long-term outcome rather than the severity of glomerular histological damage.22 This correlates with proteinuria, and the animal models induced with protein-overload proteinuria, as well as human clinical studies, document a pivotal role of proteinuria in disease progression.23 In fact, proteinuria reduction is now a mainstay of slowing progression of chronic kidney disease (CKD). For decades renal pathologists assured us that glomerulosclerosis and TIN in kidney diseases are the normal consequence of any type of damage and is simply a ‘mopping-up’, scarring-down process as the kidney attempts to remodel. The mechanism for developing TIN is not clear, but many postulates have been provided to suggest pivotal roles for upregulation of cytokines/chemokines in response to filtered proteins that gain access to the interstitial space.
Several recent papers now bring into sharp focus an exciting model of pathogenesis, which ties together the concept of glomerular damage with epitope spreading, TIN, proteinuria-induced disease amplification, and communication with the glomerulus through tubular mechanisms—a new form of glomerulotubular feedback. The common denominators for this model are CD8 + T-lymphocytes and DCs.
CD8 + CELLS
CD8 + T cells are important in various types of GN.24 They are found infiltrating the glomerulus, in the interstitium, and as periglomerular or tubule-associated infiltrates. In animal models, anti-CD8 monoclonal antibodies administration can prevent diseases, including EAG, and other glomerulopathies. Despite the demonstrated importance of CD8 + T cells in GN, little information is available regarding their target molecules and cell types, their activation by accessory cells, and their interaction with other kidney cells during pathogenesis. In two recent studies, the role of CD8 + T-cells in renal disease has been addressed by the use of a model antigen delivered to kidney tubules either as naturally occurring components or as shed transgene products.25,26 The study of Macconi et al.25 considers the contribution of autoantigens, in this case albumin, to the activation of CD8 + T cells during kidney injury induced by five-sixths nephrectomy, renal mass reduction (RMR). CD8 + T cells are classically considered to respond only to peptides derived from intracellular antigens processed by cytosolic proteosomes and loaded onto MHC-I by a MHC-I assembly and loading pathway in the endoplasmic reticulum. The significance of DC cross-presentation in which peptides from extracellular antigens such as viral particles, apoptotic cells, and soluble secreted proteins are endocytosed, processed, and loaded onto MHC-I molecules for presentation to CD8 + T cells has been increasingly appreciated and the process mechanism is better understood.27 In their recent study, Macconi et al. proposed that kidney injury results in the activation of interstitial renal DCs, which have received albumin protein fragments captured, degraded, and transported from the tubular lumen by tubular epithelial cells (TECs). These activated and albumin-loaded DCs migrate to KDLNs and prime CD8 + T cells to become effectors specific for the albumin peptide, which can be seen as MHC class I (MHC-I)-peptide complexes on renal cell surfaces. The renal cells then become potential cytotoxicity targets and contribute to further kidney injury because of CD8 + T-cell killing. By 4 weeks after RMR, significantly more albumin peptide (a.a. 1–24)-specific CD8 + T cells were found in KDLNs. Albumin-specific CD8 + T-cell induction was blocked by inhibitors of proteosomes, which also reduced TIN and glomerular damage in vivo in RMR rats. The results suggest that proteins in the urinary space, transported by TECs, can activate CD8 + T cells during kidney inflammation and the DC has a key role in presenting these exogenous proteins by a proteosome-dependent cross-presentation mechanism. It will be important in future experiments to identify the renal DC subsets responsible for the cross-presentation of exogenous antigen to CD8 + DC and further confirm the role of proteosomes in this proposed scheme.
In the second study, Heymann et al.26 used a transgenic mouse host that expresses ovalbumin (OVA) on glomerular podocytes. Transfer of naïve ovalbumin-specific transgenic CD8 + (OT-I) or CD4 + (OT-II) T cells alone or together caused no disease. However, co-injection of naïve OT-I cells with activated OT-II cells induced inflammation of the parietal epithelial cells and periglomerular infiltration of lymphocytes, DCs, and macrophages. Repeated injections of cells caused progression of TIN, glomerular sclerosis, proteinuria, and impaired renal function. Finally, the authors showed that recruited DC, derived from proinflammatory circulating monocytes rather than resident kidney DC, infiltrated the kidney, and that DCs were required for the periglomerular infiltration. This showed not only that CD8 + effector cells are critical in inducing chronic GN, but also that glomerular antigens released from podocytes are captured by DCs and presented to CD8 + T cells for activation, that transferred antigen-specific CD4 + T cells are required for CD8 + T-cell-mediated disease induction, and that DC, but not other kidney MHCII + cells are responsible for CD8 + T-cell activation.
DC IN PATHOGENESIS
The results of these studies touch upon four critical issues that require further studies. The first is the relative contributions of TEC’s transcellular transport versus direct DC uptake of antigens from the tubular lumen for antigen presentation to T cells. The second issue is whether specific subsets of DC are responsible for cross-presentation and, if so, what the subsets are. The third issue concerns the pathway of extracellular antigen processing and cross-presentation in DC. The last concerns the role of CD4 + T cells in CD8 + T-cell activation and GN pathogenesis. To address these questions, we will briefly review DC in the kidney, lymphoid organs, and other tissues, with emphasis on antigen transport and cross-presentation in the mouse (Figure 2).
Figure 2. Processing of tubular proteins by renal dendritic cells.

Uptake and presentation of tubular proteins by renal DCs. The figure shows the potential pathways by which tubular antigens are internalized, processed, and presented to CD8 + and CD4 + T-cells in the kidney draining lymph nodes. DC subsets in the interstitium internalize antigens in the tubular lumen either directly by inserting pseudopods into the lumen (CD103 + DCs) or indirectly by internalizing antigen transported and/or processed by transcellular antigen transport through tubular epithelial cells (TECs). The presence of all DC types shown has been published, except for CD103 + DCs, which have been identified in preliminary studies (Sung SJ). For cross-presentation, antigens can be directly processed in the endosomes and the MHC-I molecules are loaded by the released peptides and presented to CD8 + T cells. Other possible pathways of exogenous antigen loading—by transit through the cytosol with processing by proteosomes and MHC-I loading in the endoplasmic reticulum, and processing and MHC-I loading in the phagosomes—are not shown. In cross-presentation in the KDLNs, CD4 + T-cell help is shown. Cross-presentation by CD11b + DCs has been shown to be much inferior to other DC types and is not shown. The main functions of CD8 + T cells and CD4 + helper cells, but not those of Treg cells, are shown.
DC possesses the distinguishing features that make them versatile and proficient antigen-presenting cells. These features are the expression of MHC class II, costimulation molecules, cytokines for T-cell stimulation, the ability to mobilize and migrate to visceral and lymphoid organs in response to antigenic and pathogenic stimuli, maturation from an antigen-internalizing cell type to an antigen-presenting cell type, and differentiation into subsets with differential functional specialization.28 In mouse lymphoid organs, DCs are classified into three functionally and phenotypically distinct subsets, the conventional CD8 + and CD11b + DC subsets, and the plasmacytoid DC (PDC) subset. Two additional subsets, the Langerhans cells and the CD103 + DCs, are associated with epithelial tissues. These DC subsets are specialized to perform varied antigen-presentation functions, such as presentation of soluble antigens to CD4 + T cells, cross-presentation of exogenous antigens to CD8 + T cells, antiviral functions including viral antigen presentation and interferon production, and the secretion of effector molecules comprising chemokines, cytokines, active oxygen species, NO, and arachidonate metabolites. Lymphoid CD11b + DCs, which also express the 33D1 antigen DCIR2, mediate the CD4 + T-cell activation by soluble antigens through MHC II.29 Lung CD11b +/high DCs also produce high levels of cytokines and chemokines and differentially express mRNA of enzymes responsible for active oxygen species, NO, prostaglandins, and leukotrienes production (Sung SJ, GEO accession #GSE17322). Splenic CD8 + DCs that express DEC205 are responsible for the uptake of apoptotic cells, cross-presentation to CD8 + T cells, and stimulation of Foxp3 + regulatory T cells.28,30 CD103 + DCs occur as a major population primarily in epithelial tissues.31,32 They are functionally and phenotypically similar to CD8 + DCs.32–34 In the intestines, CD103 + DCs mediate regulatory T-cell induction in a TGF-β-and retinoic acid-dependent manner.32 In lungs, in which no CD8 + DCs are present, CD103+ DC mediate cross-presentation.35 Similar to some gut DCs, CD103 + DC express tight junction proteins that enable them to traverse the lung epithelium to capture airway antigens.31,32 Plasmacytoid DCs in mice have a significant role in viral disease by secreting IFN-α and by cross-presenting viral antigens.36 They are also important in inducing regulatory T cells to suppress allograft rejection in transplant models. Human DC populations are not as well studied and therefore seem less complex. Blood DCs are comprised of two major populations, the BDCA-1 + (CD1c +) myeloid DC, which is similar to the mouse CD11b + DC, and the BDCA-2 + BDCA-4 + PDC, which exhibits the properties of both mouse PDC and CD8 + conventional DCs. In the kidney, activation of CD8 + T cells in GN would be expected to require PDCs in humans and PDC, CD8 + DC, or CD103 + DC in mouse for cross-presentation (Figure 2).
Regarding DC subset functions, important gaps remain in the understanding of CD8 + and CD4 + T-cell activation in the kidney. Despite the overarching conclusion that CD8 + T cells have significant roles in GN,26 further detailed, mechanistic studies are needed. The first concerns the DC type capable of mediating this cross-presentation. Kidney DC has been reviewed recently.37 The kidney contains a large population of CX3CR1 + F4/80 + DCs, which lines the outer medullary tubules and is prevalent elsewhere. These DCs are CD8− and seem to correspond to the CD11b + splenic DCs. In gut and lungs CX3CR1 + DCs are CD11b +, whereas CD8 + and CD103 + DCs are CX3CR1−. Thus, it is likely that the CX3CR1 + DCs lining the kidney tubules are responsible only for CD4 + T-cell activation, whereas CD8 + or CD103 + DC populations that are likely to cross-present to CD8 + T cells have not been studied. CD103+ DCs have been identified in the kidney as a significant DC population (Sung SJ, unpublished). It is reasonable to speculate that CD103 + DCs along with PDCs and CD8 + DCs are the principal CD8 + T-cell-stimulating DC populations and this hypothesis needs to be examined. CD103+ DCs have the additional advantage of being able to traverse the epithelial tight junctions as in lungs31,32 and directly capture tubular proteins for CD8 + T-cell cross-presentation (Figure 2). This direct access of DC to the tubular lumen raises the second question of the relative roles TECs and DCs have in accessing antigens in the tubular space. DC types other than CD103 + DCs may also be able to upregulate tight junction proteins upon activation to access the tubular lumen directly. The third question on the pathway of MHC-I loading by peptides to form exogenous proteins has been addressed in human PDCs and mouse bone-marrow-derived DCs.38,39 In these DCs, exogenous proteins are processed and loaded onto MHC-I molecules directly in the endosomes without transversing the cytosol and without the involvement of cytosolic proteosomes. For particulate antigens such as apoptotic cells and large debris, phagosomes have also been shown to be an important site for MHC-I loading of peptides from ingested particles.27 Thus, proteosomes may have some role in albumin presentation to CD8 + T cells, as suggested by Macconi.27 However, it should not be surprising that direct endosome loading of MHC-I by tubular proteins is found to be the predominant mechanism. Receptor trafficking may also contribute to cross-presentation. Antigens taken up by DEC205 and mannose receptors induce robust cross-presentation,28,40 which may be a result of the receptors targeting antigens either to the correct cross-presenting DC type or to a MHC-I processing and targeting compartment. The existence of an intracellular receptor targeting pathway for cross-presentation, if proven, can explain the functional specificities of DC subsets in cross-presentation by their preferential expression of MHC-I pathway targeting receptors such as DEC205. Regarding the last question on the role of CD4 + T cells in CD8 + transgenic OT-I T-cell activation, it has been shown that CD4 + T-cell help is needed for secondary CD8 + T-cell responses.41 This explains the requirement for activated OT-II cells in GN induction in the model of Heymann et al.26 What is not addressed is the mechanism by which antigenic tolerance in the kidney is broken by the transgenic T-cell transfers. Recent data indicate that T-cell-mediated tolerance in peripheral organs is mostly regulated by either IL-10-producing Tr1 or Foxp3 + CD4 + regulatory T cells.42,43 In the study of Heymann et al., the balance between conventional T cells and regulatory T-cell function is upset by the repeated injection of activated OT-II cells, resulting in a more favorable T helper cell effect. In GN, the imbalance of T helper versus regulatory T-cell function in the diseased state may be small. Thus, manipulating regulatory T-cell numbers in the kidney may constitute a significant strategy for disease intervention.
THE GLOMERULOTUBULAR FEEDBACK LOOP
The studies from Macconi and Heymann provide significant insight into the relationship between glomerular injury, TIN, and damage (Figure 3). A common link in both the RMR and transgenic podocyte models is the presence of protein in the ultrafiltrate, which can be degraded by TECs and possibly directly processed by DCs, and presented to T cells. These proteins consist of multiple self-antigens (albumin) and innumerable other proteins and peptides (i.e., fragments of GBM and other cryptic glomerular antigens).44 These peptides are likely involved in a continuing state of tolerance maintenance, or, under certain circumstances, can initiate an autoimmune state. A vicious cycle would be perpetuated with a continuing supply of antigen to the immune system and the interstitium with a de facto delayed hypersensitivity reaction in the interstitium. However, as clinicians, we have seen many patients with minimal change disease, membranous nephropathy, and other proteinuric states with no interstitial disease. A clue to this conundrum may lie in the fact that in the normal state, DC uptake of antigen and cross-presentation results in apoptosis of CD8 + cells without production of inflammatory cytokines, with induction of tolerance.45 On the other hand, in the presence of injured cells, the process of cross-presentation to CD8 + cells results in active immunity as postulated by Matzinger, secondary to so-called danger signals or damage-associated molecular patterns (DAMPs).46 In this latter scenario, the presence of DAMPs results in CD4 + help and CD8 + CTL activation and initiation of tissue injury and autoimmunity. Emerging evidence has shown that endogenous cytoplasmic and nuclear constituents, likely released from necrotic rather than apoptotic cell death, have the potential to engage toll-like receptors and augment inflammatory damage.47–49 These various danger signals thus amplify the original immunological or non-immunological damage and drive progression of injury-induced organ damage. Other questions remain from both the Macconi and Heymann studies. In the Heymann study, what happened to the animals after the injection of OT-I and OT-II cells stopped? Did suppressor mechanisms restore normalcy to the animals or was a chronic process initiated? How did ovalbumin become expressed in parietal epithelial cells? How did ovalbumin-specific T cells congregate outside of Bowman’s capsule and elicit glomerular damage as opposed to being inside Bowman’s capsule as described by Wu et al.16 for GBM antigen-specific T cells? How does proteinuria result in or contribute to TIN in some situations but not others? If it is DAMPs, how is that involved and what are the specifics? Why is chronic TIN not associated with significant glomerular damage? Why do DCs endogenous to the kidney not participate in TIN and instead circulating DCs are recruited in the model described by Heymann et al.?
Figure 3. The glomerulotubular feedback loop.

The figure depicts the glomerulotubular feedback loop involving new paradigms as described by Macconi and Heymann. Under normal circumstances the glomerular filtrate contains many peptides, proteins, and other substances that are filtered or shed into the urinary space. These are taken up by dendritic cells directly from the tubular lumen or after processing by tubular epithelial cells (TECs). Normally, cell death is affected by apoptosis with the presentation of these antigens to T cells without inflammatory cytokines, resulting in tolerance and suppression. Even proteinuria in this circumstance, consisting of a large amount of self-antigens, is not associated with damage sans an inflammatory process. On the other hand, damaged kidney, whether by reduced mass or by autoimmune injury, induces cell necrosis rather than apoptosis, with the release of damage-associated molecular patterns (DAMPs), recruitment of exogenous cells, and stimulation of endogenous cells to proliferate and elaborate the matrix and inflammatory cytokines. The ultrafiltrate contains normal constituents, but also now contains constituents released by renal injury. This inductive phase may progress, depending on regulatory feedback. However, when DCs process and present antigen to T cells in this inflammatory milieu, the result is tubulointerstitial nephritis (TIN) and feedback to the glomerulus, either directly or through a periglomerular infiltrate, which somehow communicates with the glomerulus across Bowman’s capsule. Thus, a glomerulotubular feedback loop is established in the setting of inflammation. This leads to further glomerular damage with CD8 + CTL cross-presentation of previously irrelevant or self-antigens and progression of TIN and glomerulosclerosis.
In conclusion, the pivotal studies in the Macconi and Heymann papers show a link between the glomerulus and the tubulointerstitium in the development and progression of chronic kidney disease. They provide a mechanism whereby glomerular damage, whether by GN, RMR, or other glomerular injury, can initiate a glomerulotubular feedback loop resulting in amplified acute disease and progression of chronic kidney disease. It seems likely that even though the initial GN comes under control, the ‘second wave’ will continue to increase interstitial and glomerular damage. Many steps need to be elaborated to better understand the pathogenesis and the interrelationship between these compartments and how intervention can have an impact on the clinical course of these diseases in humans. A better understanding of these issues provides the opportunity for exciting, novel therapeutic interventions, which may be broadly applicable to many different glomerular processes, and even to the enormous problem of chronic kidney disease.
Acknowledgments
This work was supported by USPHS Grants DK55801 from the NIDDKD (WKB) and AI079906 from the NIAID (SJS).
Footnotes
DISCLOSURE
The authors declared no competing interests.
References
- 1.Rocklin R, Lewis E, David J. In-vitro evidence for cellular hypersensitivity to glomerular-basement membrane antigens in human glomerulonephritis. N Engl J Med. 1970;283:497–501. doi: 10.1056/NEJM197009032831001. [DOI] [PubMed] [Google Scholar]
- 2.Dixon FJ. What are sensitized cells doing in glomerulonephritis? N Engl J Med. 1970;283:536–537. doi: 10.1056/NEJM197009032831011. [DOI] [PubMed] [Google Scholar]
- 3.Lerner RA, Glassock RJ, Dixon FJ. The role of anti-glomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J Exp Med. 1967;126:989–1004. doi: 10.1084/jem.126.6.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolton WK, Tucker FL, Sturgill BC. New avian model of experimental glomerulonephritis consistent with mediation by cellular immunity. J Clin Invest. 1984;73:1263–1276. doi: 10.1172/JCI111328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikolic-Paterson DJ, Lan HY, Atkins R. Macrophages in immune renal injury. In: Neilson EG, Couser WG, editors. Immunologic Renal Diseases. Lippincott-Raven; Philadelphia: 1997. pp. 575–592. [Google Scholar]
- 6.Dean EG, Wilson GR, Li M, et al. Experimental autoimmune Goodpasture’s disease: a pathogenetic role for both effector cells and antibody in injury. Kidney Int. 2005;67:566–575. doi: 10.1111/j.1523-1755.2005.67113.x. [DOI] [PubMed] [Google Scholar]
- 7.Li S, Holdsworth SR, Tipping PG. Antibody independent crescentic glomerlonephritis in m chain deficient mice. Kidney Int. 1997;51:672–678. doi: 10.1038/ki.1997.97. [DOI] [PubMed] [Google Scholar]
- 8.Nolasco FE, Cameron JS, Hartley B, et al. Intraglomerular T cells and monocytes in nephritis: Study with monoclonal antibodies. Kidney Int. 1987;31:1160–1166. doi: 10.1038/ki.1987.123. [DOI] [PubMed] [Google Scholar]
- 9.Bolton WK, Innes DJ, Sturgill BC, et al. T-cells and macrophages in rapidly progressive glomerulonephritis: clinicopathologic correlations. Kidney Int. 1987;32:869–876. doi: 10.1038/ki.1987.288. [DOI] [PubMed] [Google Scholar]
- 10.Segerer S, Heller F, Lindenmeyer M, et al. Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int. 2008;74:37–46. doi: 10.1038/ki.2008.99. [DOI] [PubMed] [Google Scholar]
- 11.Stilmant MM, Bolton WK, Sturgill BC, et al. Crescentic glomerulonephritis without immune deposits: clinicopathologic features. Kidney Int. 1979;15:184–195. doi: 10.1038/ki.1979.24. [DOI] [PubMed] [Google Scholar]
- 12.Bolton WK, Chen L, Hellmark T, et al. Epitope spreading and autoimmune glomerulonephritis in rats induced by a T cell epitope of Goodpasture’s antigen. J Am Soc Nephrol. 2005;16:2657–2666. doi: 10.1681/ASN.2004100823. [DOI] [PubMed] [Google Scholar]
- 13.Hopfer H, Maron R, Butzmann U, et al. The importance of cell-mediated immunity in the course and severity of autoimmune and anti-glomerular basement membrane disease in mice. FASEB J. 2003;17:860–868. doi: 10.1096/fj.02-0746com. [DOI] [PubMed] [Google Scholar]
- 14.Reynolds J, Tam FWK, Chandraker A, et al. CD28-B7 blockade prevents the development of experimental autoimmune glomerulonephritis. J Clin Invest. 2000;105:643–651. doi: 10.1172/JCI6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohda T, Okada S, Hayashi A, et al. High nephritogenicity of monoclonal antibodies belonging to IgG2a and IgG2b subclasses in rat anti-GBM nephritis. Kidney Int. 2004;66:177–186. doi: 10.1111/j.1523-1755.2004.00719.x. [DOI] [PubMed] [Google Scholar]
- 16.Wu J, Hicks J, Borillo J, et al. CD4+ T cells specific to a glomerular basement membrane antigen mediate glomerulonephritis. J Clin Invest. 2002;109:517–524. doi: 10.1172/JCI13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Arends J, Borillo J, et al. A self T cell epitope induces autoantibody response: mechanism for production of antibodies to diverse glomerular basement membrane antigens. J Immunol. 2004;172:4567–4574. doi: 10.4049/jimmunol.172.7.4567. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Hellmark T, Pedchenko V, et al. A nephritogenic peptide induces intermolecular epitope spreading on collagen IV in experimental autoimmune glomerulonephritis. J Am Soc Nephrol. 2006;17:3076–3081. doi: 10.1681/ASN.2006070688. [DOI] [PubMed] [Google Scholar]
- 19.Robertson J, Wu J, Arends J, et al. Characterization of the T-cell epitope that causes anti-GBM glomerulonephritis. Kidney Int. 2005;68:1061–1070. doi: 10.1111/j.1523-1755.2005.00498.x. [DOI] [PubMed] [Google Scholar]
- 20.Robertson J, Wu J, Arends J, et al. Activation of glomerular basement membrane-specific B cells in the renal draining lymph node after T cell-mediated glomerular injury. J Am Soc Nephrol. 2005;16:3256–3263. doi: 10.1681/ASN.2005040421. [DOI] [PubMed] [Google Scholar]
- 21.Bolton WK, May WJ, Sturgill BC. Proliferative glomerulonephritis in rats: A model for autoimmune glomerulonephritis in humans. Kidney Int. 1993;44:294–306. doi: 10.1038/ki.1993.244. [DOI] [PubMed] [Google Scholar]
- 22.Risdon RA, Sloper JC, De Wardener HE. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. The Lancet. 1968;292:363–366. doi: 10.1016/s0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- 23.Eddy AA, McCulloch L, Adams J, et al. Interstitial nephritis induced by protein-overload proteinuria. Am J Pathol. 1989;135:719–733. [PMC free article] [PubMed] [Google Scholar]
- 24.Kurts C, Heymann V, Lukacs-Kornek V, et al. Role of T cells and dendritic cells in glomerular immunopathology. Semin Immunopathol. 2007;29:317–335. doi: 10.1007/s00281-007-0096-x. [DOI] [PubMed] [Google Scholar]
- 25.Macconi D, Chiabrando C, Schiarea S, et al. Proteasomal processing of albumin by renal dendritic cells generates antigenic peptides. J Am Soc Nephrol. 2009;20:123–130. doi: 10.1681/ASN.2007111233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heymann F, Meyer-Schweisinger C, Hamilton-Williams EE, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest. 2009;119:1286–1297. doi: 10.1172/JCI38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen L, Rock KL. Priming of T cells by exogenous antigen cross-presented on MHC class I molecules. Curr Opin Immunol. 2006;18:85–91. doi: 10.1016/j.coi.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 28.Steinman RM. Understanding immunogenicity. Eur J Immunol. 2007;37:S53–S60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 29.Dudziak D, Kamphorst AO, Heidkamp GF, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315:107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 30.den Haan JM, Lehar SM, Bevan MJ. CD8(+) but not CD* (−) dentritic cells cross-prime cytotoxic T cells in vivo. J Exp Med. 2000;192:1685–1696. doi: 10.1084/jem.192.12.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sung SS, Fu SM, Rose CE, Jr, et al. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176:2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 32.Coombs JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bedoui S, Whitney P, Waithman J, et al. Cross-presentation of viral and self antigens by skin-derived CD103+dentritic cells. Nat Immunol. 2009;10:488–495. doi: 10.1038/ni.1724. [DOI] [PubMed] [Google Scholar]
- 34.HIildner K, Edelson B, Purtha W, et al. Batf3 deficiency reveals a critical role for CD8alpha+dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.del Rio M, Rodriguez-Barbosa J, Kremmer E, et al. CD1033 and CD103+bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+and CD8+T cells. J Immunol. 2007;178:6861–6866. doi: 10.4049/jimmunol.178.11.6861. [DOI] [PubMed] [Google Scholar]
- 36.Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dentritic cells. Immunity. 2008;29:352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 37.John R, Nelson PJ. Dendritic cells in the kidney. J Am Soc Nephrol. 2007;18:2628–2635. doi: 10.1681/ASN.2007030273. [DOI] [PubMed] [Google Scholar]
- 38.DiPucchio T, Chatterjee B, Smed-Sorensen A, et al. Direct proteasome-independent cross-presentation of viral antigen by plasmacytoid dendritic cells on major histocompatibility complex class I. Nat Immunol. 2008;9:551–557. doi: 10.1038/ni.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burgdorf S, Scholz C, Kautz A, et al. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 40.Burgdorf S, Lukacs-Kornek V, Kurts C. The mannose receptor mediates uptake of soluble but not of cell-associated antigen for cross-presentation. J Immunol. 2006;176:6770–6776. doi: 10.4049/jimmunol.176.11.6770. [DOI] [PubMed] [Google Scholar]
- 41.Janssen E, Lemmens E, Wolfe T, et al. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2004;421:852–856. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 42.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 43.Awasthi A, Carrier Y, Peron JP, et al. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 44.Pendergraft WF, III, Preston GA, Shah RR, et al. Autoimmunity is triggered by cPR-3(105–201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10:72–79. doi: 10.1038/nm968. [DOI] [PubMed] [Google Scholar]
- 45.Lukacs-Kornek V, Burgdorf S, Diehl L, et al. The kidney-renal lymph node-system contributes to cross-tolerance against innocuous circulating antigen. J Immunol. 2008;180:706–715. doi: 10.4049/jimmunol.180.2.706. [DOI] [PubMed] [Google Scholar]
- 46.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 47.Babelova A, Moreth K, Tsalastra-Greul W, et al. Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem. 2009;36:24035–24048. doi: 10.1074/jbc.M109.014266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lang A, Benke D, Eitner F, et al. Heat shock protein 60 is released in immune-mediated glomerulonephritis and aggravates disease: in vivo evidence for an immunologic danger signal. J Am Soc Nephrol. 2005;16:383–391. doi: 10.1681/ASN.2004040276. [DOI] [PubMed] [Google Scholar]
- 49.Pan H, Wu G, Li X, et al. High Mobility Group Box 1: a potential therapeutic target for systemic lupus erythematosus. Mol Biol Rep. 2009 doi: 10.1007/s 11033-009-9485-7. [DOI] [PubMed] [Google Scholar]