

Abstract
Hepatitis C virus (HCV) is a liver-tropic blood-borne pathogen that affects more than 170 million people worldwide. Although acute infections are usually asymptomatic, up to 90% of HCV infections persist with the possibility of long-term consequences such as liver fibrosis, cirrhosis, steatosis, insulin resistance, or hepatocellular carcinoma. As such, HCV-associated liver disease is a major public health concern. Although the currently available standard of care therapy of pegylated interferon α plus ribavirin successfully treats infection in a subset of patients, the development of more effective, less toxic HCV antivirals is a health care imperative. This review not only discusses the limitations of the current HCV standard of care but also evaluates upcoming HCV treatment options and how current research elucidating the viral life cycle is facilitating the development of HCV-specific therapeutics that promise to greatly improve treatment response rates both before and after liver transplantation.
Keywords: interferon, STAT-C, combination therapy, personalized therapy, emerging drug targets, viral life cycle
Hepatitis C virus (HCV) belongs to the Flaviviridae family, which includes other medically relevant viruses such as yellow fever, West Nile, dengue, and tickborne encephalitis viruses, but two characteristics that distinguish HCV are its hepatotropism and tendency to establish chronic infection. Although HCV persists in most infected individuals and is a leading cause of liver disease globally, the development of disease is highly variable, and the mechanisms that mediate disease are not understood. In the absence of a sufficient understanding of HCV infection, we have been left to rely clinically on suboptimal therapeutics leaving more than 170 million people worldwide infected. This review discusses not only the limitations of the current HCV standard of care (SOC), but more importantly the exciting advancements and ongoing research efforts that have the potential to revolutionize HCV clinical care in the foreseeable future.
HCV standard of care
With a worldwide prevalence of 2.2% and no vaccine available, interferon (IFN)-based therapy continues to be the primary intervention option available for the treatment of HCV In 1991, IFN-α monotherapy was approved for HCV treatment; however, the sustained virological response (SVR) rate was below 20%.1 In 1998, addition of ribavirin (RBV) was found to improve SVR rates to 34%–42%. In 2001, the development of stabilized pegylated-IFN (PegIFN) further increased the SVR rates to ~50%. Although combination PegIFN/RBV therapy achieves SVR rates of~80% in individuals infected with genotype (GT) 2 or GT3 and 40%–50% in individuals infected with GT1 or GT4,2,3 these rates are still far from ideal, are affected by numerous host specific factors, and the therapy itself has a spectrum of adverse effects (reviewed in Glue et al4).5
To maximize treatment effectiveness while minimizing toxicity, the optimal duration of IFN/RBV therapy is determined by viral GT and the decision to continue treatment depends on treatment response. Patients with GT1 and GT4 are generally treated for 48 weeks with PegIFN-α-2a (180 μg/wk) or PegIFN-α-2b (1.5 μg/kg body weight/wk) in combination with weight-based RBV dosing of 1,000–1,400 mg/d. Patients with GT2 and GT3 are treated for 24 weeks with PegIFN-α-2a or PegIFN-α-2b and RBV Response parameters that determine whether treatment should be continued include early viral response (EVR) at week 12 and rapid viral response (RVR) at week 4 (reviewed in Poordad et al).6 EVR, defined as undetectable or ≥2-log10 decrease in serum HCV RNA by week 12 after the initiation of therapy, has long been considered a successful predictor of treatment failure (negative predictive value of 97%–100% for SVR);7,8 however, the positive predictive value of EVR to identify patients that will attain SVR is less accurate.2,3,6 As such, RVR, defined as undetectable serum HCV RNA by week 4 after the initiation of therapy, is also being adopted as an additional earlier predictor of treatment outcome.
Limitations of HCV SOC
Therapy-associated toxicity represents a major hurdle facing the current treatment. PegIFN/RBV treatment results in significant side effects, which necessitates dose reduction in 35%–42% of those treated with one-third of this group requiring discontinuation of therapy. The major side effects include influenza-like symptoms (eg, fever, fatigue, headache, myalgias, and arthralgias), hematologic abnormalities (eg, anemia and neutropenia), and neuropsychiatric symptoms (eg, depression and anxiety).9
Suboptimal efficacy is also a major limitation of PegIFN/RBV SOC, with viral, host, and environmental factors having a significant impact on treatment success. For example, differences in SVR rates indicate that HCV GT1 and GT4 are more resistant to PegIFN/RBV treatment than GT2 and GT3. Independently, high viral load is also associated with poorer response to therapy.
Host factors, such as male gender,10 older age,11,12 higher body mass index,12,14 coinfections,15,16 insulin resistance,17–19 and more advanced liver disease20,21 all contribute to poorer treatment response. Host ethnicity, in particular, appears to be a major determinant of IFN-based HCV treatment outcome. Most notably, African Americans have significantly lower response rates to PegIFN/RBV compared with other racial groups.6,7 Although not as disparate, Hispanics also have poorer response rates to PegIFN/RBV compared with non-Hispanic whites (34% vs 49%, P < 0.001) despite similar treatment adherence rates.22 Although the reasons for these host-related differences in response rates are not completely understood, they highlight that HCV patients may require individualized treatment and monitoring to reach SVR.
Many environmental factors that reduce HCV treatment response are related to adherence. In particular, self-administration of IFN injections and daily RBV dosing can make long-term adherence challenging, especially when the side effects associated with SOC severely impair a patient’s quality of life. Although individualized treatments in conjunction with management of side effects can help maintain adherence, doses often need to be reduced or terminated and failure to meet the assigned dose or duration is associated with lower SVR.7,23 Importantly, the variable efficacy of IFN treatment, which is dependent on host-specific immune responses, and other adherence-limiting issues could theoretically be reduced by the development of more specifically targeted HCV inhibitors and combination treatment strategies.
Emerging treatments and combination therapies
Currently, new potential HCV inhibitors are being tested clinically in combination with PegIFN and RBV. This not only suggests that IFN, likely in combination with RBV, will remain the backbone of HCV therapy for some time, but also reflects the consensus opinion that, like the highly active retroviral therapy used for the treatment of human immunodeficiency virus (HIV), the future of anti-HCV therapy will take a “cocktail” approach allowing for reduced dosing and treatment duration of toxic compounds while avoiding the viral escape that can occur during monotherapy with single inhibitors. Therefore, new emerging therapies include improved IFN-α formulations (to improve efficacy and ease of administration) and alternative RBV-like molecules (to reduce toxicity); however, the major focus has switched to more targeted inhibitors.
Interferon α
Several modified IFN-α products are under clinical development.
Albinterferon α-2β (AlbIFN; Human Genome Sciences, Rockville, MD) is IFN covalently bound to albumin, yielding increased half-life.24 In Phase II clinical trials, AlbIFN dosed every 2 weeks (BIW) or monthly with daily RBV yielded SVR similar to SOC (which requires weekly PegIFN dosing), with comparable side effects between groups.25 The Phase III ACHIEVE-1 trial compared AlbIFN BIW plus RBV with SOC in treatment-naive GT1 patients. Patients receiving 1,200 μg AlbIFN discontinued therapy because of pulmonary toxicity, but the patients receiving 900 μg AlbIFN achieved SVR rates comparable to that of SOC (48.7% vs 50.6%), with similar side effects.26,27 The Phase III ACHIEVE-2/3 trial compared 900 μg AlbIFN/RBV to SOC in GT2 or GT3 treatment-naive patients and showed comparable SVR in the non-Asian group (79.8% vs 80.5%). However, Asians exhibited higher SVR with SOC when compared with AlbIFN/RBV (95% vs 79%).28
Locteron (BLX-883; Biolex Therapeutics, Pittsboro, NC) consists of recombinant IFN α-2β in a biodegradable, polymeric drug delivery system that allows for biweekly dosing.29 The Phase IIa SELECT-1 trial evaluated Locteron plus RBV in 32 GT1 treatment-naive patients and found SVR and side effect profiles comparable to historical SOC data.30 A Phase II SELECT-2 trial is in progress.
Other IFN-a products in clinical development include Belerofon (Nautilus Biotech, Evry, France), which contains a mutation to lower protease sensitivity as a means of increasing stability,31 and oral IFN (Amarillo Biosciences, Amarillo, TX), which is administered as lozenges (http://www.amarbio.com/clinical-trials.html). In the DIRECT trial high daily dosing of one of the original unmodified IFNs, alfacon-1 (Infergen, CIFN), was also recently tested in combination with RBV in patients who were previously nonresponders, but overall, patients showed SVR rates similar to that of other trials involving nonresponders, with only a selected subset of 10 patients with low fibrosis scores (F0–F2) and a > 2-log decrease in virus level achieving a higher SVR of 30% (3/10).32
RBV analogs and alternatives
Clinically, the inosine monophospate dehydrogenase inhibitors, mycophenolate mofetil and merimepodib (VX-497), had little HCV-antiviral efficacy as RBV alternatives; however, liver targeting of RBV is also being tested as a means to reduce RBV-associated toxicities. The RBV prodrug taribavirin (TBV; formerly viramidine; Valeant Pharm., Aliso Viejo, CA), which is activated only in the liver, was developed in an attempt to reduce RBV-induced anemia. Originally, the Phase III VISER-1 and VISER-2 trials reported that TBV was associated with less anemia than RBV and also significantly lower SVR; however, post hoc analysis revealed that patients receiving higher than 18 mg/kg TBV had SVR rates comparable to RBV.33,34 Consistent with this finding, a subsequent Phase IIb study of higher TBV doses (20, 25, or 30 mg/kg/d) vs weight-based RBV in 275 GT1 treatment-naive patients showed similar SVR compared with SOC.35 Notably, the 20 and 25 mg/kg/d TBV groups had less anemia than the RBV group (13.4% and 15.7% vs 35%), but had nearly twice the incidence of diarrhea.35
Targeted treatments
As optimized versions of IFN and RBV promise less frequent, more convenient dosing with decreased toxicity, the largest improvements in treatment efficacy in the near future are expected to be achieved by increasing the complexity of the anti-HCV regimen to include more compounds, particularly compounds directly targeting the virus and/or critical virus–host interactions. Specifically targeted antiviral therapy for hepatitis C (STAT-C) is a term originally coined to describe inhibitors that target HCV proteins.36 Although issues of viral escape will have to be managed, these targeted therapies in theory should be less dependent on host-specific responses and avoid side effects associated with host immune stimulation. The two types of STAT-C molecules that have progressed furthest and are expected to be the first available clinically are NS3/4a protease and NS5B polymerase inhibitors.
NS3/4a protease inhibitors
The HCV NS3 serine protease and NS4a cofactor assemble to cleave the viral polyprotein into four nonstructural proteins (Figure 1); blocking this cleavage, therefore inhibits viral replication. Inhibition of NS3/4a also restores host innate immune signaling that is otherwise suppressed when NS3/4a cleavages the Toll-IL-1 receptor domain (TRIF) and IFN-β promoter stimulator (IPS-1, CARDIF, VISA), a mechanism by which HCV prevents dsRNA-induced IFN signaling.37–40 Initial proof of concept protease inhibitor clinical studies were conducted with BILN2061 (Boehringer Ingelheim, Ingelheim, Germany), which was halted due to cardiac toxicity in an animal model;41 however, two other protease inhibitors have progressed to Phase III trials.
Figure 1.

The HCV genome and gene products – potential STAT-C targets. The 9.6-kb HCV RNA genome encoding a single open-reading frame (ORF) flanked by highly structured 5′ and 3′ untranslated regions (UTRs) is depicted. The internal ribosome entry site (IRES) located in the 5′ UTR-mediated translation of a polyprotein precursor that is cotranslationally cleaved (arrows) by host (SPP = maroon and SP = blue) and viral (NS2/3 = black and NS3/4a = orange) proteases into mature structural (green boxes) and nonstructural (purple boxes) viral proteins. Descriptive titles and known activities are listed for each viral protein.
Telapravir (TVR; VX-950; Vertex Cambridge, MA) is a peptidomimetic NS3/4a protease inhibitor that covalently but reversibly binds NS3/4a. It is dosed orally every 8 hours. In Phase I, TVR monotherapy achieved a 4.4-log mean reduction in viral load over 14 days, but rapid selection of viral resistance at four different sites (T54, V36, R155, and A156) was observed.42,43 To minimize viral breakthrough, TVR has since been studied in combination with PegIFN/RBV and three Phase II PROVE studies indicate that addition of TVR to SOC allows for shorter therapy duration in GT1 patients while increasing SVR ~20%–30% in treatment-naive patients and ~30% in previous nonresponders (Tables 1–3).
Table 1.
Phase II clinical data from protease inhibitors in ongoing Phase III development: TVR PROVE 1
| Arms | n | RVR (%) | SVR (%) | Relapse (%) | Halted (%) | |
|---|---|---|---|---|---|---|
| 260 Noncirrhotic treatment-naive GT1 (USA) | TVR/PR 12 wk | 17 | 59 | 35 | 33 | 18 |
| TVR/PR 12 wk + PR 12 wk (24 wk total) | 79 | 81 | 61 | 2 | ||
| TVR/PR 12 wk + PR 36 wk (48 wk total) | 79 | 81 | 67 | 6 | ||
| SOC 48 wk | 85 | 11 | 41 | 23 | 4 |
Notes: P = PegIFN; R = RBV; SOC = PegIFN/RBV; “/” = in combination with; “+” = followed by.
Table 3.
Phase II clinical data from protease inhibitors in ongoing Phase III development: TVR PROVE 3
| Arms | SVR (%)
|
||||
|---|---|---|---|---|---|
| Total | Prior nonresponder | Prior relapser | Prior viral resistance | ||
| 453 Treatment failure GT1 | TVR/PR 12 wk + 12 wk PR | 51 | 39 | 69 | 57 |
| TVR/PR 24 wk + 24 wk PR | 53 | 38 | 76 | 63 | |
| TVR/P 24 wk | 24 | 11 | 42 | 36 | |
| SOC 48 wk | 14 | 9 | 20 | 40 | |
Notes: P = PegIFN; R = RBV; SOC = PegIFN/RBV; “/” = in combination with; “+” = followed by.
PROVE 1 randomized treatment-naive GT1 patients in the United States into three arms of 12 weeks TVR plus varying durations of PegIFN/RBV (12, 24, or 48 weeks) compared with 48 weeks SOC (Table 1).44 Inferior SVR in the 12-week TVR/IFN/RBV arm showed the need for continued PegIFN/RBV treatment after the initial 12 weeks of triple therapy; however, TVR in combination with 24 or 48 weeks PegIFN/RBV improved SVR by 20% or more. Viral breakthrough occurred in 12/175 of patients receiving TVR who did not achieve viral suppression with breakthrough rates higher for GT1a than GT1b because the most common TVR resistance mutation (R155T/I) requires one nucleotide change in GT1a compared with two nucleotide changes in GT1b.45 Rash, gastrointestinal toxicity, and anemia were the main adverse effects associated with TVR requiring discountinuation of treatment in 18% of TVR subjects, most frequently due to severe rash (7%).45
PROVE 2 studied treatment-naive GT1 patients receiving 12 weeks of TVR in combination with 12 weeks of PegIFN alone or PegIFN/RBV for 12 or 24 weeks (Table 2).45 Again, 12 weeks triple therapy followed by an additional 12 weeks of PegIFN/RBV resulted in more than a 20% increase in SVR compared to SOC (69% vs 46%). Overall discontinuation due to side effects was 11% in the TVR arms and 7% for SOC, again with severe rash in 5% of TVR patients. Notably, 24% viral breakthrough in patients taking TVR/PegIFN without RBV revealed the importance of RBV in the treatment regimen.
PROVE 3 evaluated GT1 patients who previously failed SOC therapy (Table 3).46 Importantly, the addition of TVR to SOC increased SVR in these treatment experience patients. Again, viral breakthrough was highest in the group that did not receive RBV (21% vs 3%–1 1%) and severe rash occurred in 3%–5% of TVR patients.
Table 2.
Phase II clinical data from protease inhibitors in ongoing Phase III development: TVR PROVE 2
| Arms | N | RVR (%) | SVR (%) | Relapse (%) | Halted (%) | |
|---|---|---|---|---|---|---|
| 334 Treatment-naive GT1 (Europe) | TVR/P 12 wk | 78 | 51 | 36 | 48 | 10 |
| TVR/PR 12 wk | 82 | 80 | 60 | 30 | 11 | |
| TVR/PR 12 wk + PR 12 wk | 81 | 69 | 69 | 14 | 14 | |
| SOC 48 wk | 93 | 13 | 46 | 22 | 7 |
Notes: P = PegIFN; R = RBV; SOC = PegIFN/RBV; “/” = in combination with; “+” = followed by.
Three Phase III trials (ADVANCE, ILLUMINATE, and REALIZE) with TVR in combination with various lengths of PegIFN/RBV therapy are in progress.
Boceprevir (BVR) (formerly SCH503034; Schering, Kenilworth, NJ) is another NS3/4a protease inhibitor administered orally every 8 hours that has entered Phase III clinical trials. In a Phase Ib trial with GT1 nonresponders, 400 mg BVR 3 times per day (TID) monotherapy resulted in a 1.61-log HCV reduction at day 14,47 but analogous to TVR, BVR monotherapy led to rapid selection of drug-resistant mutations including V36M/A, T54A/S, V55A, R155K/T, A156S, and V170A.48 BVR has since been studied in combination with PegIFN/RBV and has been found to improve SVR in GT1 treatment-naive patients, but might require 48 weeks of triple therapy for optimal results.
The Phase II SPRINT-1 study assessed 800 mg TID BVR in combination with PegIFN/RBV in treatment-naive GT1 patients.49 Initial treatment arms were used to assess the use of a 4-week PegIFN/RBV lead-in for reducing viral load and hence resistance during subsequent triple BVR/PegIFN/RBV (Table 4). Although the 4-week PegIFN/RBV lead-in made no difference in SVR of patients in the 28-week therapy arms (56% and 55%), SVR was slightly higher in the 48-week therapy arm that received the PegIFN/RBV lead-in (74% vs 66%). More notably however, the lead-in arms showed higher RVR (64% vs 37%–40%) and fewer virological breakthroughs (4%-5% vs 7%–12%). In a subsequent branch of the study, low-dose RBV was associated with higher viral breakthrough and lower SVR rates (36%), demonstrating again the importance of RBV
In another Phase II trial in GT1 nonresponders, BVR/PegIFN/RBV or BVR/PegIFN alone yielded SVR of 7%–14% vs 2% for SOC; however, adjustments where made during the trial such as increasing BVR dosing and adding RBV to all groups, making it difficult to interpret the results (reviewed in Thompson and McHutchison).50
Table 4.
Phase 2 clinical data from protease inhibitors in ongoing Phase III development: BVR SPRINT 1
| Arms | RVR (%) | SVR (%) | Breakthrough (%) | Halted (%) | |
|---|---|---|---|---|---|
| 520 Treatment-naive GT1 | 4 wk PR + BVR/PR 24 wk (28 wk total) | 64 | 56 | 4 | 26 |
| 4 wk PR + BVR/PR 44 wk (48 wk total) | 64 | 74 | 5 | 26 | |
| BVR/PR 28 wk | 37 | 55 | 7 | 28 | |
| BVR/PR 48 wk | 40 | 66 | 11 | 38 | |
| SOC 48 wk | 8 | 38 | 0 | 15 |
Notes: P = PegIFN; R = RBV; SOC = PegIFN/RBV; “/” = in combination with; “+” = followed by.
Two Phase III trials, SPRINT-2 and RESPOND-2, are underway in treatment-naive and nonresponders, respectively.
Many other HCV NS3/4a inhibitors are currently in preclinical and clinical development. Some inhibitors that have reached Phase II (Table 5) include the following:
Table 5.
Phase 2 clinical data from NS3/4a inhibitors
| NS3/4a inhibitors in phase 2 | Arms | Log HCV reduction | RVR (%) | |
|---|---|---|---|---|
| MK7009 (Merck) | Phase I; (29 GT 1 treatment-naive and experienced) | 25 mg BID MK7009 8 d | 1.9 | – |
| 75 mg BID MK7009 8 d | 2.5 | – | ||
| 250 mg BID MK7009 8 d | 2.8 | – | ||
| 500 mg BID MK7009 8 d | 3.3 | – | ||
| 700 mg BID MK7009 8 d | 4.6 | – | ||
| 125 mg/d MK7009 8 d | 1.8 | – | ||
| 600 mg/d MK7009 8 d | 2.4 | – | ||
| Phase IIb; (85 GT1 treatment-naive) | 300 mg BID MK7009/PR 4 wk | 75 | ||
| 600 mg BID MK7009/PR 4 wk | 79 | |||
| 600 mg/d MK7009/PR 4 wk | 69 | |||
| 800 mg/d MK7009/PR 4 wk | 82 | |||
| SOC 4 wk | 5.6 | |||
| TMC435 (Medivir/Tibotec) | Phase I; (6 GT1 experienced) | 200 mg/d TMC 6 d | 3.9 | |
| Phase IIb; (GT1 treatment-naive) | 25 mg/d TMC/PR 4 wk + SOC 24/48 wk | 4.7 | ||
| 75 mg/d TMC/PR 4 wk + SOC 24/48 wk | 5.5 | |||
| 200 mg/d TMC/PR 4 wk + SOC 24/48 wk | 5.4 | |||
| Phase IIb; (GT1 experienced) | 75 mg/d TMC/PR 4 wk + SOC 24/48 wk | 4.3 | ||
| 150 mg/d TMC/PR 4 wk + SOC 24/48 wk | 5.5 | |||
| 200 mg/d TMC/PR 4 wk + SOC 24/48 wk | 5.3 | |||
| SOC 24/48 wk | 1.5 | |||
| R7227 (intermune/Roche) | Phase I; (GT1 naive) | 600 mg/d R7227 14 d | 3.8 | – – |
| Phase I; (GT1 experienced) | 600 mg/d R7227 14 d | 2.5 | – – |
|
| Phase I; (57 GT1 treatment-naive) | 100 mg R7227/PR 14 d | 4.7 | – | |
| 600 mg R7227/PR 14 d | 5.7 | |||
| SOC 14 d | 2.0 | |||
| BI201335 (Boehringer ingelheim) | Phase I; (34 GT1 treatment-naive) | 20 mg/d BI 14 d | 3.0 | – |
| 40 mg/d BI 14 d | 3.6 | – | ||
| 120 mg/d BI 14 d | 3.7 | – | ||
| 240 mg/d BI 14 d | 4.2 | – | ||
| Phase I; (19 GT1 experienced) | 48 mg/d BI/PR 4 wk | 5.0 | – | |
| 120 mg/d BI/PR 4 wk | 5.2 | – | ||
| 240 mg/d BI/PR 4 wk | 5.3 | – | ||
| Phase Ib; (13 GT1 nonresponders) | 240 mg/d BI/PR 4 wk | 4.8 | – | |
| 240 mg Bid BI/PR 4 wk | 5.4 | – | ||
| Phase II; (GT1 treatment-naive)(ongoing) | 240 mg/d BI/PR 28 wk | 92 | ||
| 240 mg BI 3 d + 240 mg/d BI/PR 28 wk | 84 | |||
| 120 mg BI 3 d + 240 mg/d BI/PR 28 wk | 90 | |||
| SOC 28 wk | 16 | |||
Notes: P = PegIFN; R = RBV; “/” = in combination with; “+” = followed by.
MK7009 (Merck, Whitehouse Station, NJ) is a noncovalent competitive inhibitor of the HCV NS3/4a protease that was shown to have antiviral activity when administered for 8 days as monotherapy51 In Phase II trials combining various doses of MK7009 with PegIFN/RBV for 28 days, higher RVR was achieved in patients receiving MK7009 when compared to those in the SOC control group (70%–83% vs 5.6%), and all patients receiving 600 mg BID MK7009 were HCV RNA undetectable at day 42 of treatment.51 Virological failure in the 300 mg BID and 800 mg/d dosing groups were associated with HCV resistance mutations R155K and D168V No serious adverse events were noted.
TMC435 (TMC435350-C201; Medivir/Tibotec, Stockholm, Sweden) monotherapy led to a 3.9-log mean reduction in previous treatment failures in Phase I trials.52 In the ongoing Phase II OPERA-1 trials of GT1 treatment-naive and treatment-experienced patients, arms include varying doses of TMC435 with PegIFN/RBV for 4 weeks followed by 24 or 48 weeks of PegIFN/RBV53–55 At treatment week 4, TMC435/PegIFN/RBV arms have achieved mean HCV RNA reductions of 4.3–5.5 logs compared with 1.5 logs in the placebo/PegIFN/RBV arm with no increase in adverse reactions.
R7227 (formerly ITMN191; Intermune/Roche, Brisbane, CA) in multiple 14-day Phase I studies decreased HCV RNA levels 2.5–3.8 logs as monotherapy and 5.5 logs when administered 600 mg/d with PegIFN/RBV compared a 2-log reduction in the SOC only. In the combination study, no viral rebound occurred in any of the R7227 arms, and 13%–57% of R7227/PegIFN/RBV recipients had undetectable HCV RNA vs none with SOC.56 Adverse events were similar to SOC, except hyperbilirubinemia developed in two patients.
BI201335 (Boehringer Ingelheim) in a Phase I trial led to ~5 log reductions in viral load in treatment-experienced patients after 28 days of combination treatment of BI201335 (240 mg/d or BID) with PegIFN/RBV57,58 Phase II studies are ongoing in treatment-naive patients and cirrhotic nonresponders.
HCV polymerase inhibitors
The HCV RNA-dependant RNA polymerase, NS5B, synthesizes the viral RNA and thus plays a key role in viral replication. Although final triple therapy SVR results are still pending, current data indicate that HCV polymerase inhibitors tend to display lower potency during monotherapy than NS3/4a protease inhibitors (Table 6 vs Table 5), but show broader effectiveness against multiple GTs and can exhibit a higher barrier to viral resistance. Both nucleoside analog inhibitors (NIs) and nonnucleoside inhibitors (NNIs) are in development (Table 6). NIs target the highly conserved active site of NS5B by mimicking the polymerase substrate and hence tend to be more potent and have lower resistance rates than NNIs. NNIs interact with the NS5B polymerase outside of the catalytic site, causing allosteric changes that compromise NS5B complex formation and function. Five binding sights have been identified; however, rapid resistance can limit these targets.
Table 6.
Phase 2 clinical data from NS5B inhibitors
| NS5B inhibitors in phase 2 | Arms | Log HCV reduction | RVR (%) | |
|---|---|---|---|---|
|
R7128 (Roche/Pharmasset) (NI) |
Phase I | 1500 mg BID R7128 2 wk | 2.7 | − |
| Phase Ib; (81 GT1 treatment-naive) | 500 mg BID R7128/PR 4 wk | 3.3 | 30 | |
| 1000 mg BID R7128/PR 4 wk | 5 | 88 | ||
| 1500 mg BID R7128/PR 4 wk | 5 | 85 | ||
| SOC 4 wk | 2 | 19 | ||
| Phase Ib; (25 GT2/3 treatment-experienced) | 1500 mg BID R7128/PR 4 wk | 5 | 90 | |
| SOC 4 wk | 3.7 | 60 | ||
| IDX184 (idenix) (Ni) | Phase I; (12 GT1 treatment-naive) | 25 mg/d IDX184 14 d | 0.47 | − |
| 50 mg/d IDX184 14 d | 0.69 | − | ||
| 75 mg/d IDX184 14 d | 0.70 | − | ||
| 100 mg/d IDX184 14 d | 0.74 | − | ||
| Phase IIa; (GT1 treatment-naive) | 50 mg/d IDX184/PR 14 d + PR 14 d | 2.7 (14 d) | − | |
| 50 mg bid IDX184/PR 14 d + PR 14 d | 4.0 (14 d) | − | ||
| 100 mg/d IDX184/PR 14 d + PR 14 d | 4.2 (14 d) | − | ||
| SOC 28 d | 1.2 (14 d) | − | ||
| Filibuvir (Pfizer) (NNi) | Phase I; (32 GT1 treatment-naive) | 100 mg BID FVR 8 d | 0.68 | − |
| 300 mg BID FVR 8 d | 1.26 | − | ||
| 450 mg BID FVR 8 d | 1.21 | − | ||
| 300 mg TID FVR 8 d | 1.95 | − | ||
| Phase Ib; (35 GT1 treatment-naive) | 200 mg FVR/PR × 28 d + 44 wk PR | 3.6 | 60 | |
| 300 mg FVR/PR × 28 d + 44 wk PR | 4.5 | 75 | ||
| 500 mg FVR/PR × 28 d + 44 wk PR | 4.7 | 63 | ||
| SOC 48 wk | 2.1 | 0 | ||
| ANA 598 (Anadys) (NNi) | Phase I; (10 GT1 treatment-naive) | 200 mg BID ANA 3 d | 2.4 | − |
| 400 mg BID ANA 3 d | 2.3 | − | ||
| 800 mg BID ANA 3 d | 2.9 | − | ||
| Phase II; (GT1 treatment-naive) | 200 mg Bid ANA/PR 12 wk + PR 12/36 wk | − | 56 | |
| 400 mg BID ANA/PR 12 wk + PR 12/36 wk | − | 42 | ||
| SOC 24 wk or 48 wk | − | 13 | ||
| VCH 759 (vertex) (NNI) | Phase I; (GT1 treatment-naive) | 400 mg TID VCH 759 10 d | 1.0 | − |
| 800 mg BID VCH 759 10 d | 1.0 | − | ||
| 800 mg TID VCH 759 10 d | 2.5 | − | ||
| GS 9190 (Gilead) (NNI) | Phase I; (31 GT1 treatment-naive) | 40 mg GS9190 8 d | 1.4 | − |
| 120 mg GS9190 8 d | 1.7 | − | ||
| BI 207127 (Boerhinger ingelheim) (NNI) | Phase I; (60 GT1 treatment-naive) | 100 mg TID BI207127 5 d | 0.4 | − |
| 200 mg TID BI207127 5 d | 0.8 | − | ||
| 400 mg TID BI207127 5 d | 1.3 | − | ||
| 800 mg TID BI207127 5 d | 3.8 | − | ||
| 1200 mg TID BI207127 5 d | 3.2 | − | ||
Notes: P = PegIFN; R = RBV; “/” = in combination with; “+” = followed by
Two oral NIs in Phase II development are R7128 (Roche/Pharmasset, Basel, Switzerland) and IDX184 (Idenix, Cambridge, MA). While a 2-week 1,500 mg BID R7128 monotherapy trial showed a 2.7-log HCV RNA reduction,59 a 4-week combination trial of R7128 (1,000 and 1,500 mg BID) with PegIFN/RBV in treatment-naive GT1 patients gave a 5-log mean reduction in HCV RNA vs a 2-log decrease in the SOC arm, resulting in higher RVR (88% and 85%) than SOC alone (19%).60 Likewise, in a Phase Ib study in GT2/3 relapsers and nonresponders, addition of R7128 was found to increase RVR (90%) compared with SOC alone (60%).61 No resistance was identified at 4 weeks. Phase II R7128 trials are currently enrolling patients. In a Phase I IDX184 study, treatment-naive GT1 patients receiving 25–100 mg/d IDX184 had mean HCV RNA reductions ranging from 0.47–0.74 log at 2 weeks, with no serious adverse events (Table 6).62 However, IDX184 is synergistic with PegIFN/RBV in vitro,63 and results from an ongoing Phase IIa trial indicate a 4-log mean HCV RNA reduction at day 14 after the combination therapy using IDX184 (50 mg BID or 100 mg/d) with PegIFN/RBV.64
Several NNIs have also begun Phase II development (Table 6). In Phase I trials, ANA596 (Anadys Pharmaceuticals, San Diego, CA) at 200, 400, or 800 mg BID for 3 days achieved log reductions of 2.4, 2.3, and 2.9, respectively, without viral rebound or serious adverse events. In an ongoing Phase II study, patients are receiving ANA569 (200 or 400 mg BID) with PegIFN/RBV for 12 weeks followed by 12 or 36 weeks of SOC. RVR rates of 42%–56% for the ANA596 arms compared with only 13% with SOC have been reported.65 Filibuvir (FBV; formerly PF868554; Pfizer, NewYork, NY) is another NNI. When given as monotherapy for 8 days (100–450 mg BID or 300 mg TID), FBV demonstrated dose-dependent inhibition of HCV in treatment-naive GT1 patients, with mean HCV RNA reductions of 0.97–2.13 log.65 A second Phase I study in which FBV (200, 300, or 500 mg BID) was given in combination with PegIFN/RBV for 4 weeks showed enhanced mean reductions of 3.62, 4.46, and 4.67 log, respectively, compared with 2.10 logs in the SOC-alone group. The Phase II FITNESS trial is currently enrolling patients to receive FBV/PegIFN/RBV for 24 weeks with or without an additional 24 weeks of PegIFN/RBV.
Protease and polymerase inhibitor combination therapy
Although data thus far indicate that IFN and RBV help prevent the emergence of viral resistance when used in combination with protease inhibitors, dual treatment of the protease inhibitor R7227 and the polymerase inhibitor R7128 was also found to suppress the emergence of R7227 resistance in the replicon system.67 Using this strategy, the INFORM-1 trial evaluated escalating doses of R7227 and R7128 in GT1 treatment-naive, treatment-experienced, and IFN-null responders.68 Overall, patients who received R7227/R7128 therapy had a mean 4.9-log HCV RNA reduction at day 14 without resistance or serious adverse effects. SVR data are pending. In the meantime, Phase II INFORM-2 trials are enrolling patients to receive 4 weeks of R7227/R7128 alone or in combination with PegIFN, RBV, or PegIFN/RBV Additional studies testing other compound combinations are also being conducted (eg, VX-950 + VCH222, ClinicalTrials.gov Identifier: NCT00958152; BMS790052 + BMS650032, ClinicalTrials.gov Identifier: NCT00904059; NCT01012895).
Elucidation of the viral life cycle and identification of novel drug targets
Each step of the viral life cycle represents a potential drug target; however, progress in the development of specifically targeted HCV inhibitors has been hindered because the experimental cell culture and small animal models needed to elucidate the HCV life cycle and host–virus interactions that determine infection outcome have not been available. Notably however, the development of HCV replicons,69–72 HCV pseudotyped particles (pp)73 and most recently the infectious HCV cell culture system74–76 have advanced our understanding of the viral life cycle (Figure 2) resulting in the identification of unprecedented number of putative inhibitors directed against both virus and host targets (Table 7). Of course, with these tools now available, each step of the HCV life cycle is being further studied to identify and test potentially more effective drug targets.
Figure 2.
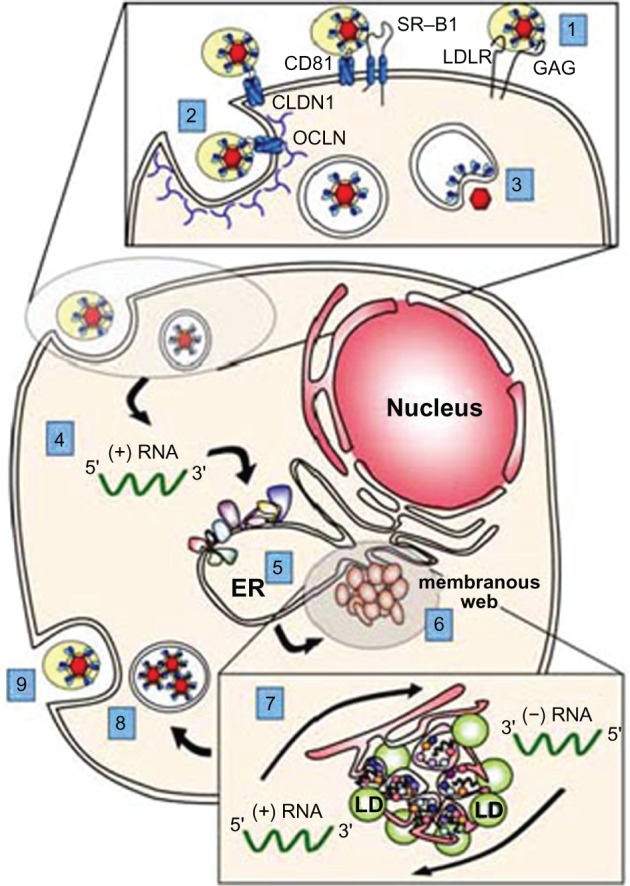
The HCV lifecycle and potential antiviral drug targets. (1) Binding of the virus to cell surface receptors; (2) HCV entry into the cell via endocytosis; (3) release of the viral genome into cytoplasm (ie, fusion and uncoating); (4) IRES-mediated polyprotein translation; (5) polyprotein processing; (6) formation of viral replication complexes (ie, the membranous web); (7) viral RNA replication; (8) packaging and assembly of progeny virions; (9) virion maturation and release via the cellular secretory system. Although depicted separately, many of these viral functions likely occur concurrently and within close proximity. in particular, we show here lipid droplets (LDs), which are thought to be the site of HCV particle assembly, closely associated with the membranous web; however, the spatial relationship of these two viral activities remains to be determined.
Table 7.
Specifically targeted HCV inhibitors (April 2010)
| Stage in viral life cycle | Viral/host target | Inhibitor class | Agent | Phase |
|---|---|---|---|---|
| Entry | E1/E2 | Neutralizing antibody | Civacir | 2 |
| SR-B1 | SR-B1 inhibitor | ITX5061 | 2 | |
| Protein synthesis | NS3/4a | Protease inhibitors | Telepravir | 3 |
| Boceprevir | 3 | |||
| TM 435 | 2 | |||
| MK 7009 | 2 | |||
| BI 201335 | 2 | |||
| R 7227 (iTMN-191) | 2 | |||
| SCH 900518 | 2 | |||
| BMS-650032 | 2 | |||
| VX 813 | 1 | |||
| VX 985 | 1 | |||
| ABT 450 | 1 | |||
| VX 500 | 1 | |||
| PHX 1766 | 1 | |||
| ACH 1625 | 1 | |||
| Genome replication | NS5B | Nucleoside analog | R 7128 | 2 |
| IDX 184 | 2 | |||
| PSI-7977 | 2 | |||
| Nonnucleoside analog | ANA 598 | 2 | ||
| PF 868554 (Filibuvir) | 2 | |||
| GS 9190 | 2 | |||
| BI 207127 | 2 | |||
| A 837093 | 1 | |||
| VCH 759 | 1 | |||
| ABT 333 | 1 | |||
| VCH 916 | 1 | |||
| VX 222 (VCH222) | 1 | |||
| R05024048 | 1 | |||
| IDX 375 | 1 | |||
| ABT-072 | 1 | |||
| NS5A | BS 790052 | 2 | ||
| A-832 | 2 | |||
| A-689 | 1 | |||
| PPI-461 | Pre | |||
| NS4B | Clemizole | 1 | ||
| miR-122 | SPC3649 | 1 | ||
| Cyclophilins | Debio025 | 2 | ||
| NIM811 | 2 | |||
| SYC635 | 1 | |||
| HMGCO-A | Statins | 2 | ||
| Assembly/secretion | Glucosidases | Celgosivir | 2 | |
| LDL pathway | Naringenin | Pre |
HCV entry
HCV infection begins with binding of the virus to cell surface receptors. Four cellular receptors – the tetraspanin protein CD81,73,76–79 the scavenger receptor class B member I (SR-B1),73,80–83 and the tight junction proteins claudin-184 and occludin85–87 – have all been shown to be required for HCV entry (Figure 2, step 1). In addition, the low-density lipoprotein receptor,79,88–90 asialoglycoprotein receptor,91 and glycosaminoglycans (heparin sulfate) have been implicated but their exact roles have not been determined. Following this multistep binding process, HCV enters the cell via clathrin-mediated endocytosis92,93 (Figure 2, step 2). Fusion between the viral envelope and endosomal membrane occurs in the acidified endosomal compartment77,94–98 via E1/E2-mediated class II fusion,99,100 ultimately resulting in the release of the viral genomic RNA into the cytoplasm (Figure 2, step 3). With the recent identification of several new HCV entry factors and progress identifying antibodies that neutralize the virus,101–104 we will likely see a wave of novel HCV entry inhibitors in clinical evaluation soon. Entry inhibitors in clinical trials now include the following:
Civacir (Nabi Biopharmacueticals, Rockville, MD) is a pool of fractionated and concentrated immunoglobulin from HCV antibody-positive human plasma that was shown to neutralize HCV in preclinical development. In a Phase I trial, 18 HCV-positive transplant recipients received a total of 17 infusions of 75 mg/kg, 200 mg/kg, or no treatment at the time of transplantation and for the following 14 weeks. The drug was well tolerated and associated with reduced aminotransferase (ALT) levels, but did not suppress hepatitis C viremia.105 The use of higher doses is being studied.
ITX5061 (iTherX Pharmaceuticals, San Diego, CA) is an SR-B1 inhibitor106 that blocks HCV entry in vitro and has demonstrated both preclinical and clinical safety107 Phase Ib and IIa proof-of-concept studies in HCV patients have been initiated.
Viral protein synthesis and processing
After the ~9.6 kb viral RNA genome is released into the host cell cytoplasm, it serves as template for the translation of the viral proteins which are encoded in a single open-reading frame flanked by highly structured 5′ and 3′ untranslated regions (UTRs) (Figure 1). The 5′ UTR contains an internal ribosome entry site (IRES) for cap-independent translation of the ~3,010-amino acid viral polyprotein (Figure 2, step 4), which is cleaved by cellular and viral proteases into individual structural and nonstructural (NS) proteins (Figure 2, step 5). Although past efforts targeting HCV IRES translation with antisense oligonucleotides, ribozyme-directed cleavage, and siRNAs have been halted due to the lack of response and/or toxicity, the positive clinical performance of NS3/4a protease inhibitors highlights viral protein production as an effective drug target.
Viral replication
Once expressed, the viral NS proteins (NS2-NS5B) assemble on and remodel endoplasmic reticulum membranes to form viral replication complexes (known as the membranous web; Figure 2, step 6).108 HCV RNA replication occurs within these membranous structures in a two-step process in which the viral NS5B polymerase uses the input genome as template to synthesize a negative-strand RNA intermediate, which then provides the template for ~ 10-fold amplification of positive-strand genomic RNA (Figure 2, step 7). In addition to NS5B polymerase inhibitors, there is a growing list of compounds that block viral replication by inhibiting one of the other HCV NS proteins or by targeting a host cellular protein shown to be essential for viral replication. Some of those currently in clinical development include the following:
NS5A inhibitors: The viral NS5A protein is essential for HCV replication, virus assembly, and also plays a role in IFN resistance. As such, it represents a promising virus-specific drug target (Table 1). In a phase 1 study, a single dose of the NS5A inhibitor BMS-790052 (Bristol-Myers Squibb, New York, NY) was found to be well tolerated in HCV GT1 patients at 1, 10, and 100 mg doses with an average mean log reduction of 1.8 logs, 3.2 logs, and 3.3 logs, respectively, at 24 hours.109 Phase II studies comparing SOC ± BMS-790052 are underway.
NS4B inhibitors: NS4B plays a major role in replication by inducing the membrane alteration required for formation of the HCV replication complexes. One NS4B inhibitor, Clemizole (Eiger BioPharmaceuticals, Palo Alto, CA), has recently entered Phase I trials.
HMG Co A reductase inhibitors: In vitro studies indicate that HCV replication is dependent on interaction between FBL2 and NS5A.110 FBL2 is a host protein that requires anchoring by geranylgeranyl pyrophospahate (GGP), an intermediate in cholesterol synthesis. Because statins inhibit HMG CoA reductase lowering GGP levels, statins have anti-HCV activity in vitro.111,112 Although some in vivo studies have shown no antiviral effect,113 a recent post hoc analysis of the IDEAL study revealed a correlation between elevated cholesterol as well as statin use and SVR to PegIFN/RBV114 Hence, targeting lipid metabolism as an additive treatment remains an area of interest.
Cyclophilin inhibitors: Several laboratories have identified a number of cyclophilins to be involved in HCV replication (reviewed in Gallay115 and Gaither et al116), and cyclophilin inhibitors have been shown to have anti-HCV activity in vitro.117–120 A Phase II trial evaluated the cyclophilin inhibitor DeBIO-025 (DebioPharm, Lausanne, Switzerland) alone or in combination with PegIFN/RBV compared with SOC for 29 days. There was a 4.8-log reduction in HCV RNA with triple therapy (vs 2.5 logs in the SOC group, and 2.2 logs in the DeBIO-025 monotherapy group).121 DeBIO-025 was associated with reversible conjugated hyperbilirubinemia and hypertension. Phase II trials using combination therapy with PegIFN/RBV in treatment-naive GT1 patients are underway. Other cyclophilin inhibitors in clinical development are SCY-635 (ScyNexis, Research Triangle Park, NC) and NIM 811 (Novartis, Basel, Switzerland).
miR-122 inhibitors: Multiple binding sites for the liver-specific microRNA miR-122 have been identified in the HCV genome, and sequestering miR-122 using antisense oligonucleotides results in the inhibition of HCV infection in cell culture.122–124 Importantly, preclinical analysis of an miR-122 inhibitor, LNA-antimiR/SPC3649 (Santaris Pharma, Copenhagen, Denmark), in HCV infected chimpanzees was well tolerated and HCV RNA levels were reduced 1.3–2.6 logs depending on the dose (1 and 5 mg/kg, respectively) without rebound (which occurred ~10 weeks after treatment conclusion).125,126
Progeny virus assembly and release
After positive-strand HCV RNA has been replicated in the cell, this RNA can serve as a template for additional translation and negative-strand synthesis or be packaged into progeny virus127 (Figure 2, step 8). Although the molecular details are not well defined the viral capsid is believed to become enveloped when it buds into the endoplasmic reticulum and then exits the cell via the lipoprotein secretory pathway (Figure 2, step 9).128,129 With the availability of the cell culture HCV infection system, the viral proteins and virus-host interactions that mediate capsid assembly, envelopment, and virion maturation are now being elucidated providing a plethora of promising drug targets (eg, viral proteins p7,130 NS2,131,132 and HCV core dimerization,133 as well as host targets related to lipoprotein secretion).
Lipoprotein synthesis inhibitors: The body of evidence linking the HCV life cycle to lipid metabolism continues to grow, whether it is the need for HCV core to associate with lipid droplets, the dependence of particle morphogenesis on vLDL pathway components, or the presence of lipoproteins on the viral particle being involved in particle entry (reviewed in Popescu and Dubuisson128). Along these lines, it has been shown in cell culture that the secretion of infectious HCV particles is blocked by the vLDL secretion inhibitor, naringenin, found in grapefruit.134
α-Glucosidase inhibitors: α -Glucosidases I and II are host enzymes involved in glycoprotein processing, but viruses are more sensitive to decreases in the activity of these enzymes resulting in misfolding of the viral glycoproteins proteins and inhibition of viral assembly and release. Celgosivir (Mignex, Vancouver, Canada) is a prodrug of castanospermine that has been shown to inhibit α-glucosidase I (reviewed in Durantel135). A Phase II trial in GT1 patients showed that 200 and 400 mg celgosivir once daily, and 200 mg BID were tolerable, but of the 35 patients who completed the 12-week treatment, only two had peak reductions of ≥1 log. Phase II combination trials of celgosivir + PegIFN/RBV are underway.
Identification of new drug targets
To identify new drug targets on a more global scale, high-throughput screening (HTS) approaches using the infectious HCV cell culture system are rapidly being developed. Yu et al136 recently developed a simple mix-and-measure fluorescence resonance energy transfer (FRET)-based in vitro HCV infection HTS capable of identifying inhibitors that act at any point in the viral life cycle. In addition, high-throughput assays are being used to screen siRNA libraries to functionally identify host factors involved in HCV infection.137–140 For example, Li et al140 reported a two-step screening approach in which imaging for HCV core protein in the initially inoculated culture was performed to identify siRNA-silenced genes that act early in infection, whereas imaging for HCV core protein in secondary cell cultures exposed to supernatant from the initial infection was used to identify genes that act late during infection. With these efforts ongoing, the list of targeted inhibitors is expected to grow.
Therapeutics to boost HCV immune response and limit viral pathology
In addition to elucidating the details of the viral life cycle, efforts to better understand the immunological basis of spontaneous HCV clearance and the mechanism of action of IFN and RBV are underway to provide insight into the host immune mechanisms that may be used to clear the virus. Other studies are focused on understanding the mechanistic basis of HCV-associated liver disease. Hence, alternative immunomodulators and antifibrotics are emerging as possible HCV therapeutics (Table 8).
Table 8.
Other HCV inhibitors under evaluation
| Drug class | Type | Name | Phase |
|---|---|---|---|
| Immunomodulators | Alternative interferons | Omega IFN | 2 |
| PegIFN lambda | 1 | ||
| TLR agonist and immunostimulatory peptides | SCV 07 | 2 | |
| ANA 773 | 1 | ||
| SD-101 | 1 | ||
| IMO-2125 | 1 | ||
| Ogulfanide | 1 | ||
| Therapeutic vaccines | GI 5005 (Tarmogen) | 2 | |
| IC41 | 2 | ||
| CT 1011 | 1 | ||
| Chronvac C (DNA) | 1 | ||
| TG4040 (MvA-HCV) | 1 | ||
| PeviPROTM | 1 | ||
| CSL123 | 1 | ||
| MBL-HCV1 | 1 | ||
| HuMax-HepC | 1 | ||
| PKR activator | Nitazoxanide | 2 | |
| NOV 205 | 2 | ||
| Bavituximab (Tarvacin) | 1 | ||
| CYT 107 | 1 | ||
| Antifibrotic agents | Caspase inhibitors | GS 9450 | 2 |
| PF03491390 (iDN-6556) | 2 | ||
| Antioxidant | Mitoquinone | 2 | |
| Silybinin | 2 | ||
| Matrix metalloproteinase | CTS 1027 | 2 | |
| PPAR γ agonist | G 1262570 (Farglitazar) | 2 |
Immunomodulators
Although specifically targeted HCV therapeutics are currently getting a great deal of attention, concerns about viral escape and past success with IFN are still driving the development of additional immunomodulators as potential therapeutics for HCV infection.
Alternative IFNs
Many different IFNs have been identified in humans and several are being considered as possible HCV therapeutics. Omega IFN is a type 1 IFN that shares 62% amino acid identity with IFNα-2141 and has been shown to have anti-HCV activity in vitro.142 In a Phase II study of GT1 treatment-naive patients, daily omega-IFN plus RBV led to a SVR of 37%.143 The type III IFN, IFN-λ (IL-29), has demonstrated inhibitory activity against HCV in vitro,144,145 and during a 4-week Phase I trial PegIFN-λ plus RBV achieved a 2-log HCV RNA reduction in GT1 treatment-naive patients and previous relapsers, most notably without the flu-like symptoms or hematological alterations associated with IFN-α.146–148 Phase II trials have begun in treatment-naive patients.
Toll-like receptor agonists
Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs) are pattern recognition receptors that recognize structurally conserved molecules shared by pathogens that distinguish them from host molecules. Binding to TLRs and RLRs activate host cytokine signaling defenses, thus similar to IFN, TLR and RLR agonists stimulate host immune defenses. Although some TLR agonists have been halted due to side effects, other compounds in this class are under clinical evaluation. ANA 773 (Anadys Pharmacueticals, San Diego, CA) induces IFN via the TLR7 pathway and in a Phase I trial ANA773 every other day (QOD) reduced HCV RNA 1.3 logs compared with a 0.3-log decline with placebo (P = 0.037)149 with no serious adverse events. IMO2125 (Idera Pharmaceuticals) is a TLR9 ligand that has shown anti-HCV activity in HCV replicon studies and is now entering Phase I studies.150 Two other immunomodulatory dipeptides being tested are Oglufanide (formally IM862; Implicit Bioscience, Brisbane, Australia) and SCV07 (SciClone Pharmaceuticals, San Francisco, CA).
Therapeutic vaccines
Because chronic HCV infection results from a failure of the host immune response to clear infection, it is logical to hypothesize that an effective immune stimulant may allow for viral clearance. As such, therapeutic vaccine strategies are being pursed. One example is GI-5005 (Globeimmune, Louisville, CO), which uses modified HCV NS3 and core protein expressed by yeast to stimulate an anti-HCV T-cell response (reviewed in Habersetzer et al151). Phase I trials showed that GI 5005 was tolerated and reduced HCV RNA levels.152 A Phase II trial evaluated GI-5005 + PegIFN/RBV compared with SOC alone in GT1 treatment-naive patients and previous nonresponders. GI-5005 was given for a 12-week lead-in period followed by 48 weeks of GI-5005 + PegIFN/RBV. In treatment-naive patients, the GI-5005 arm had a significantly higher end-of-treatment response (74% vs 59% with SOC) and a higher ALT normalization rate (55% vs 31%).153 Previous IFN nonresponders showed equivalent treatment response regardless of GI-5005 administration (31%); however, more ALT normalization was observed in patients receiving GI-5005 when compared with those receiving SOC alone (33% vs 20%). Several other therapeutic vaccines are in Phase I development including DNA-based vaccines, T-cell vaccines, and dendritic cell immunotherapy (Table 8).
PKR inducers
Protein kinase R (PKR) is part of the cellular antiviral defense system. Once activated, PKR induces phosphorylation of translation initiation factor 2α, resulting in the inhibition of host protein synthesis. Nitazoxanide (NTZ) is an approved antihelminthic agent, which was noted to reduce ALT levels in HCV/HIV coinfected patients being treated for cryptosporidium. Subsequent in vitro HCV replicon studies confirmed the anti-HCV activity of NTZ.154 The mechanism of action is not completely understood; however, one study provides evidence that NTZ induces the PKR antiviral pathway155 The phase 2 STEALTH-C1 study evaluated HCV GT4 patients in Egypt.156 A 12-week NTZ lead-in followed by 36 weeks of NTZ + PegIFN/RBV or NTZ + PegIFN was compared with 48 weeks of SOC, and the SVR in patients receiving NTZ + PegIFN/RBV was higher than those receiving SOC alone (79% vs 50%). Adverse events were similar across the groups, with the exception of increased anemia in patients receiving RBV. The STEALTH C-2 study is exaluating lead-in NTZ + PegIFN/RBV for GT1 cirrhotic patients who were previous nonresponders, and preliminary results indicate an EVR of 38% vs 25% in patients receiving SOC. A STEALTH C-3 (ERAIS-C) trial is also underway in which treatment-naive GT1 subjects are receiving NTZ for 2 weeks, followed by NTZ + RBV for 2 weeks and then 12 weeks of NTZ/PegIFN/RBV157,158 Initial results show a 12% RVR in the NTZ group vs 19% in the SOC group with EVRs of 80% vs 68%.157,158
Antifibrotic therapy
Although SVR has been associated with HCV-associated fibrosis regression,159 there is interest in developing therapies to directly prevent or reverse fibrosis independent of SVR, and a variety of anti-inflammatory and antifibrotic drugs are in clinical development.
Caspase inhibitors
HCV-infected livers have higher than normal numbers of apoptotic cells, which promote inflammation and fibrogenesis.160,161 GS 9450 and PF-03491390 are caspase inhibitors, which block apoptosis. Although one concern is that antiapoptotic drugs might increase the risk of malignancy, GS-9450 was not associated with adverse effects with short-term administration in healthy adults and is now being studied in HCV patients. PF-03491390 (formerly IDN-6556; Pfizer) is a irreversible caspase inhibitor.162,163 Doses from 5 to 400 mg (given daily to TID) for 14 days in 80 HCV patients resulted in the reduction of ALT levels in all but the lowest dosing group; however, HCV RNA levels did not change significantly.164
Matrix metallopreinase inhibitors
Matrix metallopreinases (MMPs) are proteolytic enzymes that digest the scaffolding and architecture of the liver. Phase II studies of the MMP inhibitor CTS-1027 (Conatus, San Diego, CA) are underway.
Antioxidants
HCV infection is associated with oxidative stress, which leads to hepatic stellate cell activation, increasing collagen production and fibrogenesis. As such, antioxidants may help reduce HCV-induced fibrosis.
Mitoquinone (Antipodean Pharmaceuticals, Auckland New Zealand) is an antioxidant that covalently binds to Co-Q10 (known as ubiquinone), causing it to accumulate more than 100-fold in the mitochondria, and thus decrease oxidative stress.165 In a Phase II study, mitoquinone led to 26.4% (40 mg dose group) and 28% (80 mg dose group) reductions in ALT level in HCV patients, with no significant safety issues reported.
Silibinin is the main component of milk thistle (silymarin) and has strong antioxidant and antifibrotic properties. A Phase I trial to assess the safety and efficacy of high oral doses of silymarin in previous nonresponders showed no adverse events and no antiviral efficacy166 However, an intravenous (IV) administration of silbinin in nonresponders showed a dose-dependant decline in HCV RNA level.167 Specifically, patients received IV silbinin for 7 days before and 7 day after the initiation of PegIFN/RBV before being switched to oral silymarin. Silbinin monotherapyied of 5, 10, 15 or 20 mg\kg for 7 days led to viral load decreases ranging from 0.55 to 3.02 logs. The addition of PegIFN/RBV at day 8 resulted in a further decline in viremia (giving a peak decline of 4.85 logs in the 20 mg/kg group), but some patients had slight rebound viremia after IV silbinin treatment was discontinued despite continued SOC.
Future research challenges
Although the future of HCV research and drug development looks promising, there are still many research challenges to overcome.
Robust in vitro infection with other HCV GTs
Except for the JFH-1 GT2a clone, robust infection of other HCV clones has not been achieved in cell culture. Attempts to propagate GT1a168 and GT1b169 virus in cell culture have achieved detectable infection, but low de novo virus production still impedes the use of these systems as experimental tools. Meanwhile, although the obstacles preventing the propagation of other HCV GTs in vitro are not completely understood, the 3′ end of the JFH-1 clone can confer replication permissiveness, and thus chimeric HCV genomes expressing the necessary parts of the JFH-1 clone recombined with the structural proteins p7 and parts of the NS2 protein from GTs 1–6 have been developed as alternative systems.170–174
More physiological hepatocyte culture systems
To date, robust HCV infection has only been reported in Huh7-derived human hepatoma cells.175 Huh7 cells allow for the study of the viral life cycle, but they are transformed and only minimally mimic the state of hepatocytes in vivo176,177 limiting the ability to study how HCV alters hepatocyte function and induces HCV-associated liver disease. One approach to achieve more in vivo-like cell cultures has been to alter culture conditions to coax Huh7 cells to upregulate hepatocyte-specific genes, become competent for Phase I and Phase II drug metabolism, and increase expression and specific localization of tight junction, cell adhesion, and polarity markers.178–180 Although these systems may prove useful for elucidating how HCV interacts with polarized cells or disrupts specific aspects of hepatocyte physiology, Huh7 cells by nature remain transformed. Hence, numerous laboratories are trying to develop methods for maintaining primary human hepatocytes in culture without loss of differentiation. Recent publications reporting infection of primary human hepatocytes with patient serum containing HCV GTs 1, 2, 3, and 4 may represent a step forward in the use of primary human hepatocyte in HCV research.181–183
Development of small animal models
Efforts to develop small animal HCV infection models have included the attempts to transmit HCV to tree shrews,184,185 marmosets/tamarins,186–188 and other primates189 however, only the chimpanzee has proven to be a reliable HCV animal model. Although xenorepopulation approaches in which mice are repopulated with human hepatocytes have proven to be acceptable for studying HCV infection in vivo, serious technical challenges, such as hepatocyte availability, surgical technical expertise, variability in the degree of repopulation among mice, and the fragile nature of the repopulated mice themselves, all limit the widespread use of this humanized mouse model.190–192 To study the effects of HCV proteins in vivo, several noninfectious, nonreplicating transgenic mouse lineages expressing one or more HCV proteins have been created.193–198 Despite variations in transgene expression levels and in resulting pathology, these mice have indicated that some HCV proteins can induce disturbances in lipid metabolism and possibly contribute to the development of HCC.
To create a more authentic HCV mouse model that includes active viral replication, which may be responsible for some of the clinically observed HCV-associated pathology, one goal is the development of a mouse permissive for HCV replication and/or infection. To this end, we and others have achieved HCV replication in cultured mouse cells, with JFH-1 GT2a RNA replicons replicating efficiently after transfection into mouse cells.199,200 Additionally, Ploss et al86 determined the species-specific determinants for HCV psuedoparticle (HCVpp) entry into mouse cells to be human occludin and human CD81. Hence, advancements toward HCV infection mouse models are being made.
The future of patient care
It is expected that personalized therapy, based on more efficacious treatment options and possibly more sophisticated viral and host genetic analysis, will reshape the future of HCV patient care.
Personalized therapy
In the past, treatment strategies were based on a one-size-fits-all approach; however, simple algorithms do not take into account complex viral host interactions and patient-specific factors. As such, personalized HCV therapy is emerging as the preferred management approach, whereby the duration, dose, and type of treatment are tailored to an individual. As the field of HCV mathematical modeling continues to grow,201–206 we will undoubtedly adopt more of these modeling approaches into our SOC not only in terms of predicting patient response, but also to monitor viral response, particularly the emergence of drug-resistant mutant, which may necessitate strategic changes in treatment.
Expanded treatment options
Likewise, as STAT-C and other novel therapeutics become available, the approach to HCV treatment will change. The ideal would be to have available a variety of drugs with distinct mechanisms of action that are effective across a range of GTs with shorter treatment duration and improved tolerability. However, even a moderate increase in treatment options (e.g. SOC plus 1 OR 2 STAT-C drugs) would likely have a great impact on SVR rates. Although we are rapidly approaching the STAT-C era, it will still be some time until these therapies are available for all patients and have been optimized to control viral drug resistance.
Viral and host genetic analysis
Within the realm of individualized patient care is the potential analysis of viral and host biomarkers as tools for predicting HCV patient response to therapy. In particular, specific host genetic polymorphisms in IL28 have been identified as determinants of response to PegIFN/RBV,207,208 and it is possible that ongoing research will reveal other genetic factors that could be analyzed. Likewise, analysis of viral factors, such as sequence variation, have proven efficacious in correlating HCV quasispecies complexity and treatment outcome.209–211 As such, the analysis of both viral and host markers together might significantly improve virological response predictions and aid in clinical treatment decisions.208,212
Conclusion and Discussion
Despite substantial progress optimizing the current HCV SOC, there are still many patients who do not successfully respond to treatment and/or remain untreated due to the severe adverse effects associated with this IFN/RBV-based therapy. Improved versions of IFN and RBV hold promise of less frequent, more convenient dosing and decreased toxicity; however, the largest improvements in treatment efficacy in the near future will likely be achieved by increasing the complexity of the anti-HCV regimen to include more compounds, particularly compounds that are directly targeted against the virus and critical virus–host interactions. With the great diversity of HCV and its ability to rapidly mutate to escape targeted therapeutics, strategies to design the proper combination of inhibitors will be critical; however, the development the HCV experimental systems needed to elucidate the molecular mechanisms that mediate HCV infection have greatly advanced our understanding of the viral life cycle, resulting in the identification of unprecedented number of putative HCV inhibitors. With many of these HCV antiviral compounds already in clinical development, it is clear that we are about to witness a great change in HCV patient care. In the immediate future, this is expected to include viral protease and/or polymerase inhibitors in combination with PegIFN and RBV, but in the long term, new therapeutics will ideally include a choice of multiple agents that block different aspects of infection while controlling the emergence of viral resistance ultimately allowing for efficacy across HCV GTs and in more patient populations, with decreased duration of treatment and improved tolerability.
Acknowledgments
The authors thank Danyelle Martin for graphic illustrations. Drs. Uprichard and Sainz were supported by National Institutes of Health grants AI070827 and CA33266 and American Cancer Society grant RSG-09-076-01. Drs. Uprichard and Cotler were supported by the UIC Walter Payton Center GUILD. Dr. TenCate was supported by an Institutional Ruth L. Kirchstein National Research Service Award (DK-007788-07) from the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Myers RP, Regimbeau C, Thevenot T, et al. Interferon for interferon naive patients with chronic hepatitis C. Cochrane Database Syst Rev. 2002;2:CD000370. doi: 10.1002/14651858.CD000370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fried MW, Shiffman ML, Reddy KR, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 3.Manns MP, McHutchison JG, Gordon SC, et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet. 2001;358(9286):958–965. doi: 10.1016/s0140-6736(01)06102-5. [DOI] [PubMed] [Google Scholar]
- 4.Glue P, Fang JW, Rouzier-Panis R, et al. Pegylated interferon-alpha2b: pharmacokinetics, pharmacodynamics, safety, and preliminary efficacy data. Hepatitis C Intervention Therapy Group. Clin Pharmacol Ther. 2000;68(5):556–567. doi: 10.1067/mcp.2000.110973. [DOI] [PubMed] [Google Scholar]
- 5.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436(7053):967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 6.Poordad F, Reddy KR, Martin P. Rapid virologic response: a new milestone in the management of chronic hepatitis C. Clin Infect Dis. 2008;46(1):78–84. doi: 10.1086/523585. [DOI] [PubMed] [Google Scholar]
- 7.Ferenci P, Fried MW, Shiffman ML, et al. Predicting sustained virological responses in chronic hepatitis C patients treated with peginterferon alfa-2a (40 KD)/ribavirin. J Hepatol. 2005;43(3):425–433. doi: 10.1016/j.jhep.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 8.Dienstag JL, McHutchison JG. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology. 2006;130(1):231–264. doi: 10.1053/j.gastro.2005.11.010. ; quiz 214–237. [DOI] [PubMed] [Google Scholar]
- 9.National Institutes of Health Consensus Development Conference Statement: Management of hepatitis C. 2002 Jun 10–12. Gastroenterology. 2002;123(6):2082–2099. doi: 10.1053/gast.2002.1232082. [DOI] [PubMed] [Google Scholar]
- 10.El-Serag HB. Hepatocellular carcinoma: recent trends in the United States. Gastroenterology. 2004;127(5 Suppl 1):S27–S34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 11.Pradat P, Tillmann HL, Sauleda S, et al. Long-term follow-up of the hepatitis C HENCORE cohort: response to therapy and occurrence of liver-related complications. J Viral Hepat. 2007;14(8):556–563. doi: 10.1111/j.1365-2893.2006.00829.x. [DOI] [PubMed] [Google Scholar]
- 12.Pradat P, Voirin N, Tillmann HL, Chevallier M, Trepo C. Progression to cirrhosis in hepatitis C patients: an age-dependent process. Liver Int. 2007;27(3):335–339. doi: 10.1111/j.1478-3231.2006.01430.x. [DOI] [PubMed] [Google Scholar]
- 13.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348(17):1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 14.Chen CL, Yang HI, Yang WS, et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology. 2008;135(1):111–121. doi: 10.1053/j.gastro.2008.03.073. [DOI] [PubMed] [Google Scholar]
- 15.Bruno S, Silini E, Crosignani A, et al. Hepatitis C virus genotypes and risk of hepatocellular carcinoma in cirrhosis: a prospective study. Hepatology. 1997;25(3):754–758. doi: 10.1002/hep.510250344. [DOI] [PubMed] [Google Scholar]
- 16.Degos F, Christidis C, Ganne-Carrie N, et al. Hepatitis C virus related cirrhosis: time to occurrence of hepatocellular carcinoma and death. Gut. 2000;47(1):131–136. doi: 10.1136/gut.47.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poustchi H, Negro F, Hui J, et al. Insulin resistance and response to therapy in patients infected with chronic hepatitis C virus genotypes 2 and 3. J Hepatol. 2008;48(1):28–34. doi: 10.1016/j.jhep.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 18.Nasta P, Gatti F, Puoti M, et al. Insulin resistance impairs rapid virologic response in HIV/hepatitis C virus coinfected patients on peginterferon-alfa-2a. AIDS. 2008;22(7):857–861. doi: 10.1097/QAD.0b013e3282fbd1c4. [DOI] [PubMed] [Google Scholar]
- 19.Grasso A, Malfatti F, de Leo P, et al. Insulin resistance predicts rapid virological response in non-diabetic, non-cirrhotic genotype 1 HCV patients treated with peginterferon alpha-2b plus ribavirin. J Hepatol. 2009;51(6):984–990. doi: 10.1016/j.jhep.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Kumar D, Farrell GC, Kench J, George J. Hepatic steatosis and the risk of hepatocellular carcinoma in chronic hepatitis C. J Gastroenterol Hepatol. 2005;20(9):1395–1400. doi: 10.1111/j.1440-1746.2005.04007.x. [DOI] [PubMed] [Google Scholar]
- 21.Ohata K, Hamasaki K, Toriyama K, et al. Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer. 2003;97(12):3036–3043. doi: 10.1002/cncr.11427. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Torres M, Jeffers LJ, Sheikh MY, et al. Peginterferon alfa-2a and ribavirin in Latino and non-Latino whites with hepatitis C. N Engl J Med. 2009;360(3):257–267. doi: 10.1056/NEJMoa0805062. [DOI] [PubMed] [Google Scholar]
- 23.McHutchison JG, Manns M, Patel K, et al. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology. 2002;123(4):1061–1069. doi: 10.1053/gast.2002.35950. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian GM, Fiscella M, Lamouse-Smith A, Zeuzem S, McHutchison JG. Albinterferon alpha-2b: a genetic fusion protein for the treatment of chronic hepatitis C. Nat Biotechnol. 2007;25(12):1411–1419. doi: 10.1038/nbt1364. [DOI] [PubMed] [Google Scholar]
- 25.Zeuzem S, Yoshida EM, Benhamou Y, et al. Albinterferon alfa-2b dosed every two or four weeks in interferon-naive patients with genotype 1 chronic hepatitis C. Hepatology. 2008;48(2):407–417. doi: 10.1002/hep.22403. [DOI] [PubMed] [Google Scholar]
- 26.Sulkowski MS, Zeuzem S, Lawitz E, et al. Efficacy and Safety of albinterferon alfa – 2b in Combination with ribavirin in Treatment Naive Patients with chronic hepatitis C genotype 1; The Liver Meeting – The American Association for the Study of Liver Diseases; 2009; Boston, MA. [Google Scholar]
- 27.Sulkowski M, Zeuzem S, Lawitz E, et al. Efficacy and Safety of albinterferon alfa – 2b in Combination with ribavirin in Treatment Naive Patients with chronic hepatitis C genotype 1. Hepatology; 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 333A. [Google Scholar]
- 28.Benhamou Y, Nelson D, Chuang W, et al. Efficacy and Safety Results of albinterferon alfa – 2b in Combination with ribavirin in Treatment Naive Subjects with Chronic Hepatitis C Genotype 2 or 3. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 1026A. [Google Scholar]
- 29.de Leede LG, Humphries JE, Bechet AC, et al. Novel controlled-release Lemna-derived IFN-alpha2b (Locteron): pharmacokinetics, pharmacodynamics, and tolerability in a phase I clinical trial. J Interferon Cytokine Res. 2008;28(2):113–122. doi: 10.1089/jir.2007.0073. [DOI] [PubMed] [Google Scholar]
- 30.Dzyublyk I, Yegorova T, Moroz L, et al. Phase 2a study to evaluate the safety and tolerability and anti-viral of 4 doses of a novel, controlled-release interferon alfa-2b (Locteron) given every 2 weeks for 12 weeks in treatment-naive patients with chronic hepatitis C (genotype 1). Hepatology; The 58th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2007. p. S8. [Google Scholar]
- 31.Thompson AJ, McHutchison JG. Review article: investigational agents for chronic hepatitis C. Aliment Pharmacol Ther. 2009;29(7):689–705. doi: 10.1111/j.1365-2036.2009.03927.x. [DOI] [PubMed] [Google Scholar]
- 32.Bacon BR, Shiffman ML, Mendes F, et al. Retreating chronic hepatitis C with daily interferon alfacon-1/ribavirin after nonresponse to pegylated interferon/ribavirin: DIRECT results. Hepatology. 2009;49(6):1838–1846. doi: 10.1002/hep.22871. [DOI] [PubMed] [Google Scholar]
- 33.Benhamou Y, Afdhal NH, Nelson DR, et al. A phase III study of the safety and efficacy of viramidine versus ribavirin in treatment-naive patients with chronic hepatitis C: ViSER1 results. Hepatology. 2009;50(3):717–726. doi: 10.1002/hep.23073. [DOI] [PubMed] [Google Scholar]
- 34.Marcellin P, Gish RG, Gitlin N, et al. Safety and efficacy of viramidine versus ribavirin in ViSER2: Randomized, double-blind study in therapy-naive hepatitis C patients. J Hepatol. 2009;52(1):32–38. doi: 10.1016/j.jhep.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 35.Poordad F, Lawitz E, Hassanein T, et al. Sustained Virologic Response (SVR) Results for Weight-Based-Taribavirin Versus Weight-Based-Ribavirin, in Naive Chronic – Hepatitis C, Genotype 1 Patients. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 334A. [Google Scholar]
- 36.Sulkowski MS. Specific targeted antiviral therapy for hepatitis C. Curr Gastroenterol Rep. 2007;9(1):5–13. doi: 10.1007/s11894-008-0015-x. [DOI] [PubMed] [Google Scholar]
- 37.Ferreon JC, Ferreon AC, Li K, Lemon SM. Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J Biol Chem. 2005;280(21):20483–20492. doi: 10.1074/jbc.M500422200. [DOI] [PubMed] [Google Scholar]
- 38.Foy E, Li K, Wang C, et al. Regulation of interferon regulatory factor-3 by the hepatitis C virus serine protease. Science. 2003;300(5622):1145–1148. doi: 10.1126/science.1082604. [DOI] [PubMed] [Google Scholar]
- 39.Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A. 2005;102(8):2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A. 2005;102(49):17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lamarre D, Anderson PC, Bailey M, et al. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426(6963):186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 42.Sarrazin C, Kieffer TL, Bartels D, et al. Dynamic hepatitis C virus genotypic and phenotypic changes in patients treated with the protease inhibitor telaprevir. Gastroenterology. 2007;132(5):1767–1777. doi: 10.1053/j.gastro.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 43.Reesink HW, Zeuzem S, Weegink CJ, et al. Rapid decline of viral RNA in hepatitis C patients treated with VX-950: a phase Ib, placebo-controlled, randomized study. Gastroenterolog y. 2006;131(4):997–1002. doi: 10.1053/j.gastro.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 44.McHutchison JG, Everson GT, Gordon SC, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360(18):1827–1838. doi: 10.1056/NEJMoa0806104. [DOI] [PubMed] [Google Scholar]
- 45.Hezode C, Forestier N, Dusheiko G, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360(18):1839–1850. doi: 10.1056/NEJMoa0807650. [DOI] [PubMed] [Google Scholar]
- 46.McHutchison J, Manns M, Muir A, et al. PROVE3 Final Results and 1 – Year Durability of SVR with Telaprevir – Based Regimen in Hepatitis C Genotype 1 – Infected Patients with Prior Non-response, Viral Breakthrough or Relapse to peginterferon – Alfa – 2a/b and Ribavirin Therapy. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 334A. [Google Scholar]
- 47.Sarrazin C, Rouzier R, Wagner F, et al. SCH 503034, a novel hepatitis C virus protease inhibitor, plus pegylated interferon alpha-2b for genotype 1 nonresponders. Gastroenterology. 2007;132(4):1270–1278. doi: 10.1053/j.gastro.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 48.Susser S, Welsch C, Wang Y, et al. Characterization of resistance to the protease inhibitor boceprevir in hepatitis C virus-infected patients. Hepatology. 2009;50(6):1709–1718. doi: 10.1002/hep.23192. [DOI] [PubMed] [Google Scholar]
- 49.Kwo P, Lawitz E, McCone J, et al. Boceprevir plus Peginterferon alfa-2b/Ribavirin for Treatment of Genotype 1 Chronic Hepatitis C in Previously Untreated Patients. Hepatology; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; San Francisco, CA. 2008. p. 1027A. [Google Scholar]
- 50.Thompson AJ, McHutchison JG. Antiviral resistance and specifically targeted therapy for HCV (STAT-C) J Viral Hepat. 2009;16(6):377–387. doi: 10.1111/j.1365-2893.2009.01124.x. [DOI] [PubMed] [Google Scholar]
- 51.Manns M, Gane E, Rodriguez-Torres M, et al. Early Viral Response (EVR) Rates in Treatment - na ve Patients with Chronic Hepatitis C (CHC) Genotype 1 Infection Treated with MK - 7009, a Novel NS3/4a Protease Inhibitor, in Combination with Pegylated Interferon Alfa - 2a and Ribavirin for 2. Hepatotology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 332A. [Google Scholar]
- 52.van’t Klooster G, Vanwelkenhuysen I, Verloes R, et al. Pharmacokinetics of once-daily regimens of the novel HCV NS3/4A-protease inhibitor TMC435350, with and without pegIFN and ribavirin, in HCV-infected individuals; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. Abstract 1895. [Google Scholar]
- 53.Manns M, Reesink H, Moreno C, et al. Safety and antiviral activity of TMC435350 in treatment-naïve genotype 1 HCV-infected patients. Hepatology; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; San Francisco, CA. 2008. p. LB8. [Google Scholar]
- 54.Manns MP, Reesink HW, Moreno C, et al. Safety and antiviral activity of TMC435350 in treatment-naïve genotype 1 HCV-infected patients; 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. [Google Scholar]
- 55.Tsantrizos YS. TMC-435, an NS3/4A protease inhibitor for the treatment of HCV infection. Curr Opin Investig Drugs. 2009;10(8):871–881. [PubMed] [Google Scholar]
- 56.Forestier N, Larrey D, Marcellin P, et al. Antiviral activity and safety of ITMN-191 in combination with Peginterferon alfs-2a and ribavirin in patients with chronic hepatitis C virus EASL 44th Annual Meeting; 2009; Copenhagen, Denmark. Abstract 17. [Google Scholar]
- 57.Manns M, Bourliere M, Benhamou Y, et al. Safety and antiviral activity of BI201335, a new HCV NS3 protease inhibitor, in combination therapy with Peginterferon alfa 2a (P) and Ribavirin (R) for 28 days in P + R treatment-experienced patients with chronic hepatitis C genotype-1 infection; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. Abstract 1882. [Google Scholar]
- 58.Pol S, Berg T, Bonacini M, et al. Virological Response and Safety of BI 201335 protease inhibitor, Peginterferon alfa 2a and Ribavirin treatment of HCV genotype - 1 patients with compensated liver cirrhosis and non - response to previous peginterferon/ribavirin; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; 2009; Boston, MA. Abstract LB16. [Google Scholar]
- 59.Reddy R, Rodriguez-Torres M, Gane E, et al. Antiviral activity, pharmacokinetics, safety, and tolerability of R7128, a novel nucleoside HCV RNA polymerase inhibitor, following multiple, ascending, oral doses in patients with HCV genotype 1 infection who have failed prior interferon therapy; The 58th Annual Meeting of the American Association for the Study of Liver Diseases; 2007; Boston, MA. Abstract LB9. [Google Scholar]
- 60.Rodriguez-Torres M, Lalezari J, Gane E, et al. Potent antiviral response to the HCV Nucleoside Polymerase Inhibitor R7128 For 2 With Peg-Ifn and Ribavirin: Subanalysis by Race/Ethnicity, Weight and HCV Genotype; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. Abstract 1899. [Google Scholar]
- 61.Gane E, Rodriguez-Torres M, Nelson D, et al. Antiviral activity of the HCV nucleoside polymerase inhibitor R7128 in HCV genotype 2 and 3 prior non-responder; results of R 7128 1500 mg BID with PEG-IFN and Ribavirin for 28 days. Hepatology; 59th Annual Meeting of the American Association for the Study of Liver Diseases; San Francisco, CA. 2008. p. 1024A. [Google Scholar]
- 62.Lalezari J, Asmuth D, Casir A, et al. Antiviral Activity Safety and Pharmacokinetics of IDX184 A, Liver – Targeted Nucleotide HCV Polymerase Inhibitor, in Patients with Chronic Hepatitis C; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; 2009; Boston, MA. Abstract LB18. [Google Scholar]
- 63.Lallos L, LaColla M, Serra I, et al. Combination of IDX184, a Nucleotide Prodrug Polymerase Inhibitor, With Other Classes of HCV Inhibitors is Additive to Synergistic in the HCV Replicon In vitro. Hepatology; 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 333A. [Google Scholar]
- 64.Lalezari J, Poordad F, Mehra P, et al. Antiviral Activity Pharmacokinetics and Safety of IDX184 in Combination with Pegylated Interferon (pegIFN) and Ribavirin (RBV) in Treatment-Naive HCV Genotype 1-infected Subjects; European Association for the Study of the Liver; 2010; Vienna, Austria. Poster 2013. [Google Scholar]
- 65.Lawitz E, Rodriquez-Torres M, Rustgi V, et al. Safety and Anti viral Activity of ANA598 in Combination with Pegylated Interferon alpha-2A Plus Ribavirin in Treatment-Naive Genotype 1 Chronic HCV Patients; The 45th Annual Meeting of the European Association for the Study of the Liver; 2010; Vienna, Austria. [Google Scholar]
- 66.Hammond J, Purohit V, Fang J, et al. Safety Tolerability Pharmacokinetics of the HCV Polymerase Inhibitor PF-00868554 Following Multiple Dose Administration in Healthy Volunteers. Hepatology; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; San Francisco, CA. 2008. p. LB11. [Google Scholar]
- 67.Tan H, Rajyaguru S, Wu T, et al. Combination of the NS3/4A Protease Inhibitor ITMN-191 (R7227) with the Active Moiety of the NS5B Inhibitors R1626 or R7128 Enhances Replicon Clearance and Reduces the Emergence of Drug Resistant Variants; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. Abstract 1885. [Google Scholar]
- 68.Gane E, Roberts S, Stedman C, et al. Combination therapy with a nucleoside polymerase (R7128) and protease (R7227/ITMN–191) inhibitor in HCV: Safety, pharmacokinetics, and virologic results from INFORM – 1. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 394A. [Google Scholar]
- 69.Lohmann V, Korner F, Koch J, et al. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285(5424):110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 70.Blight KJ, McKeating JA, Marcotrigiano J, Rice CM. Efficient Replication of Hepatitis C Virus Genotype 1a RNAs in Cell Culture. J Virol. 2003;77(5):3181–3190. doi: 10.1128/JVI.77.5.3181-3190.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290(5498):1972–1975. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- 72.Ikeda M, Yi M, Li K, Lemon SM. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J Virol. 2002;76(6):2997–3006. doi: 10.1128/JVI.76.6.2997-3006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bartosch B, Dubuisson J, Cosset FL. Infectious hepatitis C virus pseudo-particles containing functional E1–E2 envelope protein complexes. J Exp Med. 2003;197(5):633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309(5734):623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 75.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11(7):791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhong J, Gastaminza P, Cheng G, et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102(26):9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hsu M, Zhang J, Flint M, et al. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A. 2003;100(12):7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pileri P, Uematsu Y, Campagnoli S, et al. Binding of hepatitis C virus to CD81. Science. 1998;282(5390):938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 79.Wunschmann S, Medh JD, Klinzmann D, Schmidt WN, Stapleton JT. Characterization of hepatitis C virus (HCV) and HCV E2 interactions with CD81 and the low-density lipoprotein receptor. J Virol. 2000;74(21):10055–10062. doi: 10.1128/jvi.74.21.10055-10062.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grove J, Huby T, Stamataki Z, et al. Scavenger receptor BI and BII expression levels modulate hepatitis C virus infectivity. J Virol. 2007;81(7):3162–3169. doi: 10.1128/JVI.02356-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kapadia SB, Barth H, Baumert T, McKeating JA, Chisari FV. Initiation of hepatitis C virus infection is dependent on cholesterol and cooperativity between CD81 and scavenger receptor B type I. J Virol. 2007;81(1):374–383. doi: 10.1128/JVI.01134-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scarselli E, Ansuini H, Cerino R, et al. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. Embo J. 2002;21(19):5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zeisel MB, Koutsoudakis G, Schnober EK, et al. Scavenger receptor class B type I is a key host factor for hepatitis C virus infection required for an entry step closely linked to CD81. Hepatology. 2007;46(6):1722–1731. doi: 10.1002/hep.21994. [DOI] [PubMed] [Google Scholar]
- 84.Evans MJ, von Hahn T, Tscherne DM, et al. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature. 2007;446(7137):801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 85.Liu S, Yang W, Shen L, et al. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol. 2009;83(4):2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ploss A, Evans MJ, Gaysinskaya VA, et al. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature. 2009;457(7231):882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benedicto I, Molina-Jimenez F, Bartosch B, et al. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J Virol. 2009;83(16):8012–8020. doi: 10.1128/JVI.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A. 1999;96(22):12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Monazahian M, Bohme I, Bonk S, et al. Low density lipoprotein receptor as a candidate receptor for hepatitis C virus. J Med Virol. 1999;57(3):223–229. doi: 10.1002/(sici)1096-9071(199903)57:3<223::aid-jmv2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 90.Molina S, Castet V, Fournier-Wirth C, et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol. 2007;46(3):411–419. doi: 10.1016/j.jhep.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 91.Saunier B, Triyatni M, Ulianich L, et al. Role of the asialoglycoprotein receptor in binding and entry of hepatitis C virus structural proteins in cultured human hepatocytes. J Virol. 2003;77(1):546–559. doi: 10.1128/JVI.77.1.546-559.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Blanchard E, Belouzard S, Goueslain L, et al. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol. 2006;80(14):6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Meertens L, Bertaux C, Dragic T. Hepatitis C virus entry requires a critical postinternalization step and delivery to early endosomes via clathrin-coated vesicles. J Virol. 2006;80(23):11571–11578. doi: 10.1128/JVI.01717-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bartosch B, Vitelli A, Granier C, et al. Cell entry of hepatitis C virus requires a set of co-receptors that include the CD81 tetraspanin and the SR-B1 scavenger receptor. J Biol Chem. 2003;278(43):41624–41630. doi: 10.1074/jbc.M305289200. [DOI] [PubMed] [Google Scholar]
- 95.Lavillette D, Bartosch B, Nourrisson D, et al. Hepatitis C virus glycoproteins mediate low pH-dependent membrane fusion with liposomes. J Biol Chem. 2006;281(7):3909–3917. doi: 10.1074/jbc.M509747200. [DOI] [PubMed] [Google Scholar]
- 96.Tscherne DM, Jones CT, Evans MJ, et al. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol. 2006;80(4):1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koutsoudakis G, Kaul A, Steinmann E, et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol. 2006;80(11):5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haid S, Pietschmann T, Pecheur EI. Low pH-dependent Hepatitis C Virus Membrane Fusion Depends on E2 Integrity, Target Lipid Composition, Density of Virus Particles. J Biol Chem. 2009;284(26):17657–17667. doi: 10.1074/jbc.M109.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Garry RF, Dash S. Proteomics computational analyses suggest that hepatitis C virus E1 and pestivirus E2 envelope glycoproteins are truncated class II fusion proteins. Virology. 2003;307(2):255–265. doi: 10.1016/s0042-6822(02)00065-x. [DOI] [PubMed] [Google Scholar]
- 100.Lavillette D, Pecheur EI, Donot P, et al. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J Virol. 2007;81(16):8752–8765. doi: 10.1128/JVI.02642-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Broering TJ, Garrity KA, Boatright NK, et al. Identification and characterization of broadly neutralizing human monoclonal antibodies directed against the E2 envelope glycoprotein of hepatitis C virus. J Virol. 2009;83(23):12473–12482. doi: 10.1128/JVI.01138-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Burioni R, Perotti M, Mancini N, Clementi M. Perspectives for the utilization of neutralizing human monoclonal antibodies as anti-HCV drugs. J Hepatol. 2008;49(2):299–300. doi: 10.1016/j.jhep.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 103.Law M, Maruyama T, Lewis J, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med. 2008;14(1):25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 104.Meunier JC, Russell RS, Goossens V, et al. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J Virol. 2008;82(2):966–973. doi: 10.1128/JVI.01872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Davis GL, Nelson DR, Terrault N, et al. A randomized, open-label study to evaluate the safety and pharmacokinetics of human hepatitis C immune globulin (Civacir) in liver transplant recipients. Liver Transpl. 2005;11(8):941–949. doi: 10.1002/lt.20405. [DOI] [PubMed] [Google Scholar]
- 106.Masson D, Koseki M, Ishibashi M, et al. Increased HDL cholesterol and apoA-I in humans and mice treated with a novel SR-BI inhibitor. Arterioscler Thromb Vasc Biol. 2009;29(12):2054–2060. doi: 10.1161/ATVBAHA.109.191320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McKelvy J, Lee H, Syder D, et al. ITX5061, an orally bioavailable, clinical stage compound, targets the scavanger receptor B1 to inhibit HCV entry; 16th International Symposium on Hepatitis C Virus and Related Viruses; 2009; Nice, France. [Google Scholar]
- 108.Gosert R, Egger D, Lohmann V, et al. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol. 2003;77(9):5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nettles R, Chien C, Chung E, et al. BMS-790052 is a First-in-class Potent Hepatitis C Virus (HCV) NS5A Inhibitor for Patients with Chronic HCV Infection: Results from a Proof-of-concept Study; 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. [Google Scholar]
- 110.Wang C, Gale M, Jr, Keller BC, et al. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol Cell. 2005;18(4):425–434. doi: 10.1016/j.molcel.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 111.Ikeda M, Abe K, Yamada M, et al. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology. 2006;44(1):117–125. doi: 10.1002/hep.21232. [DOI] [PubMed] [Google Scholar]
- 112.Ye J, Wang C, Sumpter R, Jr, et al. Disruption of hepatitis C virus RNA replication through inhibition of host protein geranylgeranylation. Proc Natl Acad Sci U S A. 2003;100(26):15865–15870. doi: 10.1073/pnas.2237238100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.O’Leary JG, Chan JL, McMahon CM, Chung RT. Atorvastatin does not exhibit antiviral activity against HCV at conventional doses: a pilot clinical trial. Hepatology. 2007;45(4):895–898. doi: 10.1002/hep.21554. [DOI] [PubMed] [Google Scholar]
- 114.Harrison S, Rossaro L, Hu K, et al. Relationship of the Use of Statins and Elevated Low – Density Lipoprotein or Total Cholesterol to Virologic Response in Patients Treated for Hepatitis C Virus in the IDEAL Study. 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 85A. [Google Scholar]
- 115.Gallay PA. Cyclophilin inhibitors. Clin Liver Dis. 2009;13(3):403–417. doi: 10.1016/j.cld.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 116.Gaither LA, Borawski J, Anderson LJ, et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology. 2010;397(1):43–55. doi: 10.1016/j.virol.2009.10.043. [DOI] [PubMed] [Google Scholar]
- 117.Ma S, Boerner JE, TiongYip C, et al. NIM811, a cyclophilin inhibitor, exhibits potent in vitro activity against hepatitis C virus alone or in combination with alpha interferon. Antimicrob Agents Chemother. 2006;50(9):2976–2982. doi: 10.1128/AAC.00310-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Nakagawa M, Sakamoto N, Tanabe Y, et al. Suppression of hepatitis C virus replication by cyclosporin a is mediated by blockade of cyclophilins. Gastroenterology. 2005;129(3):1031–1041. doi: 10.1053/j.gastro.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 119.Paeshuyse J, Kaul A, de Clercq E, et al. The non-immuno-suppressive cyclosporin DEBIO-025 is a potent inhibitor of hepatitis C virus replication in vitro. Hepatology. 2006;43(4):761–770. doi: 10.1002/hep.21102. [DOI] [PubMed] [Google Scholar]
- 120.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology. 2003;38(5):1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 121.Flisiak R, Feinman SV, Jablkowski M, et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNalpha2a significantly reduces viral load in treatment-naive hepatitis C patients. Hepatology. 2009;49(5):1460–1468. doi: 10.1002/hep.22835. [DOI] [PubMed] [Google Scholar]
- 122.Jopling CL. Regulation of hepatitis C virus by microRNA-122. Biochem Soc Trans. 2008;36(Pt 6):1220–1223. doi: 10.1042/BST0361220. [DOI] [PubMed] [Google Scholar]
- 123.Jopling CL, Schutz S, Sarnow P. Position-dependent function for a tandem microRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe. 2008;4(1):77–85. doi: 10.1016/j.chom.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science. 2005;309(5740):1577–1581. doi: 10.1126/science.1113329. [DOI] [PubMed] [Google Scholar]
- 125.Roberts AP, Jopling CL. Targeting viral infection by microRNA inhibition. Genome Biol. 2010;11(1):201. doi: 10.1186/gb-2010-11-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lanford RE, Hildebrant-Eriksen ES, Petri A, et al. LNA-Modified Antisense oligonucleotide (SPC3649) suppresses HCV RNA anundance by inhibition of microRNA-122 in chronically infected chimpanzees. 16th International Symposium on Hepatitis C Vius and related Viruses; 2009; Nice, France. [Google Scholar]
- 127.Lindenbach BD, Rice CM. Unravelling hepatitis C virus replication from genome to function. Nature. 2005;436(7053):933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 128.Popescu CI, Dubuisson J. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol Cell. 2010;102(1):63–74. doi: 10.1042/BC20090125. [DOI] [PubMed] [Google Scholar]
- 129.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21(1):33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Griffin S. Inhibition of HCV p7 as a therapeutic target. Curr Opin Investig Drugs. 2010;11(2):175–181. [PubMed] [Google Scholar]
- 131.Jirasko V, Montserret R, Appel N, et al. Structural and functional characterization of nonstructural protein 2 for its role in hepatitis C virus assembly. J Biol Chem. 2008;283(42):28546–28562. doi: 10.1074/jbc.M803981200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jones CT, Murray CL, Eastman DK, Tassello J, Rice CM. Hepatitis C virus p7 and NS2 proteins are essential for production of infectious virus. J Virol. 2007;81(16):8374–8383. doi: 10.1128/JVI.00690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kota S, Scampavia L, Spicer T, et al. A Time-Resolved Fluorescence-Resonance Energy Transfer Assay for Identifying Inhibitors of Hepatitis C Virus Core Dimerization. Assay Drug Dev Technol. 2009;8(1):96–105. doi: 10.1089/adt.2009.0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nahmias Y, Goldwasser J, Casali M, et al. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology. 2008;47(5):1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Durantel D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr Opin Investig Drugs. 2009;10(8):860–870. [PubMed] [Google Scholar]
- 136.Yu X, Sainz B, Jr, Uprichard SL. Development of a cell-based hepatitis C virus infection fluorescent resonance energy transfer assay for high-throughput antiviral compound screening. Antimicrob Agents Chemother. 2009;53(10):4311–4319. doi: 10.1128/AAC.00495-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tai AW, Benita Y, Peng LF, et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5(3):298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Randall G, Panis M, Cooper JD, et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc Natl Acad Sci U S A. 2007;104(31):12884–12889. doi: 10.1073/pnas.0704894104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ng TI, Mo H, Pilot-Matias T, et al. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology. 2007;45(6):1413–1421. doi: 10.1002/hep.21608. [DOI] [PubMed] [Google Scholar]
- 140.Li Q, Brass AL, Ng A, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci U S A. 2009;106(38):16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Adolf GR. Human interferon omega – a review. Mult Scler. 1995;1(Suppl 1):S44–S47. [PubMed] [Google Scholar]
- 142.Buckwold VE, Wei J, Huang Z, et al. Antiviral activity of CHO-SS cell-derived human omega interferon and other human interferons against HCV RNA replicons and related viruses. Antiviral Res. 2007;73(2):118–125. doi: 10.1016/j.antiviral.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 143.Novozhenov V, Zakharova NEV, et al. Phase 2 study of omega interferon alone or in combination with ribavirin in subjects with chronic hepatitis C genotype-1 infection. J Hepatology; 58th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2007. p. S8. [Google Scholar]
- 144.Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79(6):3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pagliaccetti NE, Eduardo R, Kleinstein SH, et al. Interleukin-29 functions cooperatively with interferon to induce antiviral gene expression and inhibit hepatitis C virus replication. J Biol Chem. 2008;283(44):30079–30089. doi: 10.1074/jbc.M804296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lawitz E, Zaman A, Muir A, et al. Interim results from a phase 1b dose-escalation study of 4 weeks of peg-interferon lambda (PEG-RIL-29) treatment in subjects with hepatitis C virus (HCV) genotype 1 with prior virologic response and relapse to peginterferon alfa and ribavirin; The 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. Abstract 170. [Google Scholar]
- 147.Shiffman E, Lawitz A, Zaman J, et al. Peg-IFN-[lambda]: antiviral activity and safety profile in a 4-week phase 1b study in relapsed genotype 1 hepatitis C infection, 2009. Hepatology. 50(Suppl 1):S327. [Google Scholar]
- 148.Lawitz EJ, Zaman A, Muir AJ, et al. Interim results from a phase 1b dose-escalation study of 4 weeks of peg-interferon lambda (PEG-RIL-29) treatment in subjects with hepatitis C virus (HCV) genotype 1 with prior virologic response and relapse to peginterferon alfa and ribavirin; 59th Annual Meeting of the American Association for the Study of Liver Diseases; 2008; San Francisco, CA. [Google Scholar]
- 149.Janssen H, de Bruijne J, Bergmann J, et al. ANA773, an Oral Inducer of Endogenous Interferons that Acts Via TLR7, Reduced Serum Viral Load in Patients Chronically Infected with HCV. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 1022A. [Google Scholar]
- 150.Bhagat L, Yu D, Trombino A, et al. IMO-2125, a TLR9 agonist, induces Th1-type cytokines and interferons with potent anti-HCV activity in human peripheral blood mononuclear cells and plasmacytoid dendritic cells; 60th Annual Meeting of the American Association for the Study of Liver Diseases; 2009; Boston, MA. Abstract 1593. [Google Scholar]
- 151.Habersetzer F, Baumert TF, Stoll-Keller F. GI-5005, a yeast vector vaccine expressing an NS3-core fusion protein for chronic HCV infection. Curr Opin Mol Ther. 2009;11(4):456–462. [PubMed] [Google Scholar]
- 152.Schiff ER, Everson GT, Tsai N, et al. HCV-specific cellular immunity, RNA reductions, and normalization of ALT in chronic HCV subjects after treatment with GI-5005, a yeast-based immunotherapy targeting NS3 and core: a randomized, doubleblind, placebo controlled phase 1B study; 58th Annual Meeting of the American Association for the Study of Liver Diseases; 2007; Boston, MA. [Google Scholar]
- 153.McHutchison J, Jacobson I, Boyer T, et al. GI – 5005 Therapeutic Vaccine Plus Peg – IFN/Ribavirin Improves End of Treatment Response at 4s Versus Peg – IFN/Ribavirin in Naive Genotype 1 Chronic HCV Patients; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; 2009; Boston, MA. Abstract LB15. [Google Scholar]
- 154.Korba BE, Montero AB, Farrar K, et al. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 2008;77(1):56–63. doi: 10.1016/j.antiviral.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 155.Elazar M, Liu M, McKenna SA, et al. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2alpha via protein kinase activated by double-stranded RNA activation. Gastroenterology. 2009;137(5):1827–1835. doi: 10.1053/j.gastro.2009.07.056. [DOI] [PubMed] [Google Scholar]
- 156.Rossignol JF, Kabil SM, El-Gohary Y, Elfert A, Keeffe EB. Clinical trial: randomized, double-blind, placebo-controlled study of nitazoxanide monotherapy for the treatment of patients with chronic hepatitis C genotype 4. Aliment Pharmacol Ther. 2008;28(5):574–580. doi: 10.1111/j.1365-2036.2008.03781.x. [DOI] [PubMed] [Google Scholar]
- 157.Basu P, Rayapudi K, Shah N, Pacana T, Brown R. Effects of High Dose Ribavirin (RBV), Alinia (Nitazoxanide) and Pegylated Interferon (PEG) alfa – 2a in Attaining Sustained Viral Response (SVR) in Treatment of Chronic Hepatitis C (ERAIS – C trial) – Interim Results in Naive Genotype 1 Patients. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 730A. [Google Scholar]
- 158.Yoffe B, Gasitashvili K, Khaoustov V. Pilot Study of Lead – In Nitazoanide Plus Pegylated Aplha – 2A Interferon and Ribavirin in HCV – Genotype 1 Nonresponders with cirrhosis: Interim Results. Hepatology; The 60th Annual Meeting of the American Association for the Study of Liver Diseases; Boston, MA. 2009. p. 1034A. [Google Scholar]
- 159.Arif A, Levine RA, Sanderson SO, et al. Regression of fibrosis in chronic hepatitis C after therapy with interferon and ribavirin. Dig Dis Sci. 2003;48(7):1425–1430. doi: 10.1023/a:1024196201684. [DOI] [PubMed] [Google Scholar]
- 160.Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54(7):1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hoglen NC, Chen LS, Fisher CD, et al. Characterization of IDN-6556 (3-[2-(2-tert-butyl-phenylaminooxalyl)-amino]-propionylamino]-4-oxo-5-(2,3,5,6-tetrafuoro-phenoxy)-pentanoic acid): a liver-targeted caspase inhibitor. J Pharmacol Exp Ther. 2004;309(2):634–640. doi: 10.1124/jpet.103.062034. [DOI] [PubMed] [Google Scholar]
- 163.Canbay A, Feldstein A, Baskin-Bey E, Bronk SF, Gores GJ. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther. 2004;308(3):1191–1196. doi: 10.1124/jpet.103.060129. [DOI] [PubMed] [Google Scholar]
- 164.Pockros PJ, Schiff ER, Shiffman ML, et al. Oral IDN-6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology. 2007;46(2):324–329. doi: 10.1002/hep.21664. [DOI] [PubMed] [Google Scholar]
- 165.Gane E, Orr D, Weilert F, et al. Phase II study of the mitochondrial antioxidant mitoquinone for hepatitis C. 43rd Annual Meeting of the European Association for the Study of the Liver 2008. Journal of Hepatology. 48(Suppl 2):S318. [Google Scholar]
- 166.Hawke RL, Schrieber SJ, Soule TA, et al. Silymarin Ascending Multiple Oral Dosing Phase I Study in Noncirrhotic Patients With Chronic Hepatitis C. J Clin Pharmacol. 2009;50(4):434–449. doi: 10.1177/0091270009347475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ferenci P, Scherzer TM, Kerschner H, et al. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology. 2008;135(5):1561–1567. doi: 10.1053/j.gastro.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 168.Yi M, Villanueva RA, Thomas DL, Wakita T, Lemon SM. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc Natl Acad Sci U S A. 2006;103(7):2310–2315. doi: 10.1073/pnas.0510727103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pietschmann T, Zayas M, Meuleman P, et al. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 2009;5(6):e1000475. doi: 10.1371/journal.ppat.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gottwein JM, Scheel TK, Hoegh AM, et al. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology. 2007;133(5):1614–1626. doi: 10.1053/j.gastro.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 171.Jensen TB, Gottwein JM, Scheel TK, et al. Highly effcient JFH1-based cell-culture system for hepatitis C virus genotype 5a: failure of homologous neutralizing-antibody treatment to control infection. J Infect Dis. 2008;198(12):1756–1765. doi: 10.1086/593021. [DOI] [PubMed] [Google Scholar]
- 172.Scheel TK, Gottwein JM, Jensen TB, et al. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A. 2008;105(3):997–1002. doi: 10.1073/pnas.0711044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gottwein JM, Scheel TK, Jensen TB, et al. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology. 2009;49(2):364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 174.Pietschmann T, Kaul A, Koutsoudakis G, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A. 2006;103(19):7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J. Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res. 1982;42(9):3858–3863. [PubMed] [Google Scholar]
- 176.Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 177.Suzuki T, Aizaki H, Murakami K, Shoji I, Wakita T. Molecular biology of hepatitis C virus. J Gastroenterol. 2007;42(6):411–423. doi: 10.1007/s00535-007-2030-3. [DOI] [PubMed] [Google Scholar]
- 178.Choi S, Sainz B, Jr, Corcoran P, Uprichard SL, Jeong H. Characterization of increased drug metabolism activity in dimethyl sulfoxide (DMSO)-treated Huh7 hepatoma cells. Xenobiotica. 2009;39(3):205–217. doi: 10.1080/00498250802613620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sainz B, Jr, Chisari FV. Production of infectious hepatitis C virus by well-differentiated, growth-arrested human hepatoma-derived cells. J Virol. 2006;80(20):10253–10257. doi: 10.1128/JVI.01059-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sainz B, Jr, TenCate V, Uprichard SL. Three-dimensional Huh7 cell culture system for the study of Hepatitis C virus infection. Virol J. 2009;6:103. doi: 10.1186/1743-422X-6-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Aly HH, Shimotohno K, Hijikata M. 3D cultured immortalized human hepatocytes useful to develop drugs for blood-borne HCV. Biochem Biophys Res Commun. 2009;379(2):330–334. doi: 10.1016/j.bbrc.2008.12.054. [DOI] [PubMed] [Google Scholar]
- 182.Buck M. Direct infection and replication of naturally occurring hepatitis C virus genotypes 1, 2, 3 and 4 in normal human hepatocyte cultures. PLoS One. 2008;3(7):e2660. doi: 10.1371/journal.pone.0002660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhu H, Elyar J, Foss R, et al. Primary human hepatocyte culture for HCV study. Methods Mol Biol. 2009;510:373–382. doi: 10.1007/978-1-59745-394-3_28. [DOI] [PubMed] [Google Scholar]
- 184.Xie Z, Riezu-Boj J, Lasarte J, et al. Transmission of hepatitis C virus infection to tree shrews. Virology. 1998;244(2):513–520. doi: 10.1006/viro.1998.9127. [DOI] [PubMed] [Google Scholar]
- 185.Zhao X, Tang Z, Klumpp B, et al. Primary hepatocytes of Tupaia belangeri as a potential model for hepatitis C virus infection. J Clin Invest. 2002;109(2):221–232. doi: 10.1172/JCI13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Feinstone S, Alter H, Dienes H, et al. Non-A, non-B hepatitis in chimpanzees and marmosets. J Infect Dis. 1982;144(6):588–598. doi: 10.1093/infdis/144.6.588. [DOI] [PubMed] [Google Scholar]
- 187.Karayiannis P, Scheuer PJ, Bamber M, et al. Experimental infection of Tamarins with human non-A, non-B hepatitis virus. J Med Virol. 1983;11:251–256. doi: 10.1002/jmv.1890110308. [DOI] [PubMed] [Google Scholar]
- 188.Watanabe T, Katagiri J, Kojima H, et al. Studies on transmission of human non-A, non-B hepatitis to marmosets. J Med Virol. 1987;22:143–156. doi: 10.1002/jmv.1890220205. [DOI] [PubMed] [Google Scholar]
- 189.Abe K, Kurata T, Teramoto Y, Shiga J, Shikata T. Lack of susceptibility of various primates and woodchucks to hepatitis C virus. J Med Primatol. 1993;22(7–8):433–434. [PubMed] [Google Scholar]
- 190.Kneteman NM, Mercer DF. Mice with chimeric human livers: who says supermodels have to be tall? Hepatology. 2005;41(4):703–706. doi: 10.1002/hep.20681. [DOI] [PubMed] [Google Scholar]
- 191.Kneteman NM, Toso C. In vivo study of HCV in mice with chimeric human livers. Methods Mol Biol. 2009;510:383–399. doi: 10.1007/978-1-59745-394-3_29. [DOI] [PubMed] [Google Scholar]
- 192.Ploss A, Rice CM. Towards a small animal model for hepatitis C. EMBO Rep. 2009;10(11):1220–1227. doi: 10.1038/embor.2009.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kawamura T, Furusaka A, Koziel MJ, et al. Transgenic expression of hepatitis C virus structural proteins in the mouse. Hepatology. 1997;25(4):1014–1021. doi: 10.1002/hep.510250437. [DOI] [PubMed] [Google Scholar]
- 194.Pasquinelli C, Shoenberger JM, Chung J, et al. Hepatitis C virus core and E2 protein expression in transgenic mice. Hepatology. 1997;25(3):719–727. doi: 10.1002/hep.510250338. [DOI] [PubMed] [Google Scholar]
- 195.Honda A, Arai Y, Hirota N, et al. Hepatitis C virus structural proteins induce liver cell injury in transgenic mice. J Med Virol. 1999;59(3):281–289. doi: 10.1002/(sici)1096-9071(199911)59:3<281::aid-jmv4>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 196.Moriya K, Yotsuyanagi H, Shintani Y, et al. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J Gen Virol. 1997;78(Pt 7):1527–1531. doi: 10.1099/0022-1317-78-7-1527. [DOI] [PubMed] [Google Scholar]
- 197.Lerat H, Honda M, Beard MR, et al. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122(2):352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- 198.Koike K, Moriya K, Ishibashi K, et al. Expression of hepatitis C virus envelope proteins in transgenic mice. J Gen Virol. 1995;76:3031–3038. doi: 10.1099/0022-1317-76-12-3031. [DOI] [PubMed] [Google Scholar]
- 199.Chang KS, Cai Z, Zhang C, et al. Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: protein kinase R (PKR)-dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating interferon activities. J Virol. 2006;80(15):7364–7374. doi: 10.1128/JVI.00586-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Uprichard SL, Chung J, Chisari FV, Wakita T. Replication of a hepatitis C virus replicon clone in mouse cells. Virol J. 2006;3:89. doi: 10.1186/1743-422X-3-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Neumann AU, Lam NP, Dahari H, et al. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science. 1998;282(5386):103–107. doi: 10.1126/science.282.5386.103. [DOI] [PubMed] [Google Scholar]
- 202.Dahari H, Major M, Zhang X, et al. Mathematical modeling of primary hepatitis C infection: noncytolytic clearance and early blockage of virion production. Gastroenterology. 2005;128(4):1056–1066. doi: 10.1053/j.gastro.2005.01.049. [DOI] [PubMed] [Google Scholar]
- 203.Dahari H, Perelson AS. Hepatitis C virus RNA kinetics: Drug efficacy and the rate of HCV-infected cells loss. World J Gastroenterol. 2007;13(21):3020–3021. doi: 10.3748/wjg.v13.i21.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Dahari H, Perelson AS. Hepatitis C virus kinetics in chimeric mice during antiviral therapy. Hepatology. 2007;46(6):2048–2049. doi: 10.1002/hep.21798. [DOI] [PubMed] [Google Scholar]
- 205.Dahari H, Ribeiro RM, Perelson AS. Triphasic decline of hepatitis C virus RNA during antiviral therapy. Hepatology. 2007;46(1):16–21. doi: 10.1002/hep.21657. [DOI] [PubMed] [Google Scholar]
- 206.Dahari H, Sainz B, Jr, Perelson AS, Uprichard SL. Modeling subgenomic hepatitis C virus RNA kinetics during treatment with alpha interferon. J Virol. 2009;83(13):6383–6390. doi: 10.1128/JVI.02612-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Torres-Puente M, Cuevas JM, Jimenez-Hernandez N, et al. Genetic variability in hepatitis C virus and its role in antiviral treatment response. J Viral Hepat. 2008;15(3):188–199. doi: 10.1111/j.1365-2893.2007.00929.x. [DOI] [PubMed] [Google Scholar]
- 208.Persico M, Capasso M, Russo R, et al. Elevated expression and polymorphisms of SOCS3 influence patient response to antiviral therapy in chronic hepatitis C. Gut. 2008;57(4):507–515. doi: 10.1136/gut.2007.129478. [DOI] [PubMed] [Google Scholar]
- 209.Aurora R, Donlin MJ, Cannon NA, Tavis JE. Genome-wide hepatitis C virus amino acid covariance networks can predict response to antiviral therapy in humans. J Clin Invest. 2009;119(1):225–236. doi: 10.1172/JCI37085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Oh TS, Rice CM. Predicting response to hepatitis C therapy. J Clin Invest. 2009;119(1):5–7. doi: 10.1172/JCI38069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Moreau I, Levis J, Crosbie O, Kenny-Walsh E, Fanning LJ. Correlation between pre-treatment quasispecies complexity and treatment outcome in chronic HCV genotype 3a. Virol J. 2008;5:78. doi: 10.1186/1743-422X-5-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.El-Shamy A, Sasayama M, Nagano-Fujii M, et al. Prediction of efficient virological response to pegylated interferon/ribavirin combination therapy by NS5A sequences of hepatitis C virus and anti-NS5A antibodies in pre-treatment sera. Microbiol Immunol. 2007;51(4):471–482. doi: 10.1111/j.1348-0421.2007.tb03922.x. [DOI] [PubMed] [Google Scholar]