


Abstract
We cloned cDNA encoding chicken cytoplasmic histone acetyltransferase-1, chHAT-1, comprising 408 amino acids including a putative initiation Met. It exhibits 80.4% identity to the human homolog and possesses a typical leucine zipper motif. The glutathione S-transferase (GST) pull-down assay, involving truncated and missense mutants of the chicken chromatin assembly factor-1 (chCAF-1)p48, revealed not only that a region (comprising amino acids 376–405 of chCAF-1p48 and containing the seventh WD dipeptide motif) binds to chHAT-1 in vitro, but also that mutation of the motif has no influence on the in vitro interaction. The GST pull-down assay, involving truncated and missense chHAT-1 mutants, established that a region, comprising amino acids 380–408 of chHAT-1 and containing the leucine zipper motif, is required for its in vitro interaction with chCAF-1p48. In addition, mutation of each of four Leu residues in the leucine zipper motif prevents the in vitro interaction. The yeast two-hybrid assay revealed that all four Leu residues within the leucine zipper motif of chHAT-1 are necessary for its in vivo interaction with chCAF-1p48. These results indicate not only that the proper leucine zipper motif of chHAT-1 is essential for its interaction with chCAF-1p48, but also that the propeller structure of chCAF-1p48 expected to act as a platform for protein–protein interactions may not be necessary for this interaction of chHAT-1.
INTRODUCTION
Alterations of the chromatin structure have been thought to be of fundamental importance in numerous DNA-utilizing processes in eukaryotes. In recent years, for example, knowledge concerning the participation of the acetylation of core histones in transcription regulation through alterations of the chromatin structure has rapidly accumulated (1–5). The acetylation induces an open chromatin conformation that allows the transcription machinery access to promoter regions. The acetylation and deacetylation of core histones are catalyzed by histone acetyltransferase(s) (HATs) and deacetylase(s) (HDACs), respectively. HATs have been identified as homologs of yeast Gcn5p, which is known to be a general controlled non-repressed protein (6). Then, the p300/CBP, P/CAF and TAFII230/250 complexes, respectively, were found to exhibit the ability to acetylate core histones (7–9). On the other hand, HDACs have been identified as homologs of yeast Rpd3, which negatively regulates the global expression of genes (10). In addition, it has been reported that HDACs exist in multiple forms, i.e. six to eight HDACs in mouse, man and chicken (10–14, and our unpublished data). These findings led us to expect that each member of the HDAC family plays an individual, particular role, and then participates, in combination with another member and/or HAT family members, in the acetylation of core histones, which is related to transcriptional activity.
Using the conventional gene targeting technique, by which many different genomic loci were easily disrupted (13–16), we have generated two homozygous DT40 mutants, ΔchHDAC-1 and -2, respectively, that are devoid of two alleles of chHDAC-1 and -2 (13). Systematic analysis of ΔchHDAC-2 revealed that chHDAC-2 dually controls the amount of the secreted form of the IgM H-chain at the steps of both the transcription of its gene and the switch from the membrane-bound to secreted form of mRNA. Recently, we generated the conditional, homozygous DT40 mutant lacking the two endogenous genes encoding chHDAC-3, and then established that the N- and C-terminal regions, proper nuclear export signal and deacetylation activity of the enzyme are necessary for the viability of DT40 cells (14).
Moreover, interestingly, transcription repressor complexes contain the p48 subunit, which is also present in the chromatin assembly factor-1 (CAF-1) involved in chromatin assembly in eukaryotes, together with two other subunits, p60 and p150 (17–23). The p48 subunit, CAF-1p48, with seven WD repeat motifs, is a member of the WD protein family (24–27). The chicken p48 subunit, chCAF-1p48, is essential for the viability of DT40 cells (and probably most vertebrate cells) (Y.Takami, T.Fukagawa, A.Ahyar, T.Ikemura and T.Nakayama, manuscript in preparation), although the depletion of CAF-1 is not lethal for yeast (28). These results together suggest that the interaction of CAF-1p48 with HDACs (and probably other proteins) should be involved in many aspects of DNA-utilizing processes. On the other hand, it has been reported that there are multiple forms of HATs, such as nuclear Gcn5p and P/CAF, and cytoplasmic HAT-1, similar to those of HDACs in eukaryotes (29). Furthermore, the heterodimer of HAT-1 and the p46 polypeptide, a p48 homolog, have been reported to participate in the acetylation of newly synthesized core histones. These findings suggest that the interaction of HATs with the p48 subunit (and/or the p46 polypeptide) is involved in numerous DNA-utilizing processes.
In this study, therefore, we cloned cDNA encoding the chicken HAT-1, chHAT-1, and demonstrated that it binds to chCAF-1p48 in vivo and in vitro. Through the glutathione S-transferase (GST) pull-down affinity assay, involving truncated and missense mutant proteins, we demonstrate the in vitro interaction of chHAT-1 with a region of chCAF-1p48 comprising amino acids 376–405, indicating that none of the seven WD dipeptide motifs is necessary for this interaction. We also demonstrate that this interaction requires a region of chHAT-1 comprising amino acids 380–408 and containing a leucine zipper motif. These results were confirmed in vivo, using the yeast two-hybrid assay system.
MATERIALS AND METHODS
Materials
In this study, the XL1-Blue MRF’ Escherichia coli strain, E.coli SORL strain (Stratagene), E.coli BL-21 strain (Pharmacia Biotech Inc.) and Saccharomyces cerevisiae MaV203 strain (Gibco BRL) were used. pBluescriptII SK(–) and pCite4a(–) were purchased from Promega. The pGEX-2TK gene fusion vector and glutathione–Sepharose beads were from Amersham Pharmacia Biotech Inc. The pDBLeu and pPC86 Gal4 fusion vectors were from Gibco BRL.
Cloning and sequencing of cDNA encoding chHAT-1
Based on the amino acid sequences (RPRVSQM and KINKQHA) of human HAT-1 (30), sense and antisense degenerate oligonucleotide primers containing sequences 5′-MGNCCNMGNGTITSICARATG-3′ and 5′-NGCRTGYTTRTTNATYTT-3′, respectively, were constructed. A PCR product of 279 bp, which corresponds to a part of the coding region of human HAT-1, was prepared from the chicken DT40 cDNA using degenerate primers. To obtain the full-length chHAT-1 cDNA, using the resultant PCR product as a probe, we screened the DT40 lambda ZAP II cDNA library using poly(A) mRNA prepared from DT40 cells, as described (13,31,32). The entire nucleotide sequences of both strands of the largest cDNA insert were determined by the dye terminator method (Applied Biosystems Division, Perkin Elmer).
Plasmid construction
The pGEX-2TKchCAF-1p48 plasmid, carrying the full-length chCAF-1p48 cDNA, was obtained as described (13,31). We first constructed the GAL4 yeast two-hybrid plasmid as follows. The parental pB(II)SKp48 plasmid was digested with NcoI, followed by treatment with T4 polymerase. From the resultant blunt-ended material, the NcoI blunt-end/ScaI fragment containing the full-length chCAF-1p48 cDNA was excised and subcloned into the SmaI site of the pDBLeu and pPC86 vectors (Gibco BRL) to yield pDBLeup48 and pPC86p48, respectively. The pGEX-2TKchHAT-1 plasmid was obtained as follows. First, we constructed a sense primer containing BamHI (5′) with sequence 5′-AGGATCCATGGCGGGACTTAC-3′ and an antisense primer containing EcoRI (3′) with sequence 5′-GAATTCTCATGCTTGCGCAAGTCGTTCTAGG-3′. Next, we prepared the DNA fragment encoding the full-length chHAT-1 cDNA by PCR using the parental chimeric plasmid [pB(II)SKchHAT-1], carrying the full-length chHAT-1 cDNA, as a template with generate primers, followed by digestion with BamHI plus EcoRI. We inserted the BamHI/EcoRI fragment between the BamHI and EcoRI sites of the pGEX-2TKH4 plasmid (Y.Takami et al., manuscript in preparation).
The pCiteHAp48 plasmid, carrying both the full-length chCAF-1p48 cDNA and the HA fragment, and various deletion mutants of it, were obtained as described (13,31). In addition, to newly generate pCiteHAp48-(360–425), we first constructed or used a sense primer containing NcoI (5′) with sequence 5′-ATTTTCCATGGAGGATGGTCCCCCAGAGC-3′ or an antisense 3′-Cite primer containing sequence 5′-CGATCAATAACGGTCGCCTGA-3′ (Novagen). Next, we prepared the DNA fragment encoding amino acids 360–425 by PCR using pCiteHAHAT-1 as a template with the generate primers, followed by digestion with NcoI plus XhoI. The NcoI/XhoI stick-ended fragment was introduced between the NcoI and XhoI sites of the pCiteHAp48 plasmid. To generate pCiteHAp48-(360–405), we used or constructed a sense 5′-Cite primer containing sequence 5′-GGGGACGTGGTTTTCCTTTGA-3′ (Novagen) or an antisense primer containing XhoI (3′) with sequence 5′-AATGCTCGAGCTATGCCATTTGCCAGACTTG-3′. Next, we prepared the DNA fragment lacking the regions that encode amino acids 1–359 and 406–425, respectively, by PCR using pCiteHAp48-(360–425) as a template with these generate primers, followed by digestion with NcoI plus XhoI. The NcoI/XhoI stick-ended fragment was introduced between the NcoI and XhoI sites of the pCiteHAp48 plasmid.
We constructed the pCiteHAHAT-1 plasmid as follows. The pCiteFlagHAT-1 plasmid was digested with XhoI, followed by partial digestion with NcoI, and then the 1.2 kb fragment carrying the full-length chHAT-1 cDNA was subcloned between the NcoI and XhoI sites of the pCiteHAHDAC-3 plasmid (Y.Takami et al., manuscript in preparation).
Deletion mutants of chHAT-1 were constructed as follows. The AlfII/XbaI fragment encoding amino acids 367–408 of chHAT-1 was deleted by digestion of pCiteHAHAT-1 with the corresponding enzymes, and then religated after being blunt-ended with T4 polymerase to generate pCiteHAHAT-1-(1–366). To generate pCiteHAHAT-1-(1–379), we first used or constructed a sense 5′-Cite primer containing sequence 5′-GGGGACGTGGTTTTCCTTTGA-3′ (Novagen) and an antisense primer containing XhoI (3′) with sequence 5′-GCAACAAGCTCGAGCTAGTCTATTTGGTTCAACTG-3′. Next, we prepared the DNA fragment lacking the fragment encoding amino acids 380–408 by PCR using pCiteHAHAT-1 as a template with these generate primers, followed by digestion with NdeI plus XhoI. The NdeI/XhoI stick-ended fragment was introduced between the NdeI and XhoI sites of the pCiteHAHAT-1 plasmid. pCiteHAHAT-1-(71–408) was generated by excision of the NdeI/BstPI fragment encoding amino acids 1–70 by digestion with NdeI plus BstPI, followed by religation after blunt-ending with T4 polymerase. To generate pCiteHAHAT-1-(334–408) and pCiteHAHAT-1-(366–408), we first constructed or used a sense primer containing NcoI (5′) with sequence 5′-GTTTCCATGGATGCAGAACAATCCAGAA-3′ and 5′-TTCTCCATGGGCTTAAGACCAGAGGAGT-3′ or an antisense 3′-Cite primer containing sequence 5′-CGATCAATAACGGTCGCCTGA-3′ (Novagen). Next, we prepared DNA fragments encoding amino acids 334–408 and 366–408 by PCR using pCiteHAHAT-1 as a template with these generate primers, followed by digestion with NcoI plus ClaI. The NcoI/ClaI stick-ended fragments were introduced between the NcoI and ClaI sites of the pCiteHAHAT-1 plasmid.
Alanine substitution mutagenesis plasmids, pCiteHAHAT-1-(L380A), pCiteHAHAT-1-(L387A), pCiteHAHAT-1-(L394A) and pCiteHAHAT-1-(L402A), were generated from pCiteHAHAT-1 as a template as follows. The series of mutagenic primer pairs in Table 1 was used to generate single leucine to alanine substitutions at the indicated positions by PCR.
Table 1. Oligonucleotide primer use for the alanine substitution PCR-mutagenesis of chHAT-1.
| Namea |
Sequenceb |
Mutationc |
| HAT-1/L380A-5′ |
GTTGAACCAAATAGACgcaAACATGCAACATGAAC |
L380A |
| HAT-1/L380A-3′ |
GTTCATGTTGCATGTTtgcGTCTATTTGGTTCAAC |
|
| HAT-1/L387A-5′ |
CATGCAACATGAACAGgcaGAAGAGAGCTTCCAAC |
L387A |
| HAT-1/L387A-3′ |
GTTGGAAGCTCTCTTCtgcCTGTTCATGTTGCATG |
|
| HAT-1/L394A-5′ |
GAGAGCTTCCAACAAgcaGTTTCAGATTACCAGAG |
L394A |
| HAT-1/L394A-3′ |
CTTCGGTAATCTGAAACtgcTTGTTGGAAGCTCTC |
|
| HAT-1/L402A-5′ |
GATTACCGAAGAGTCgcaGAACGACTTGCACAAGC |
L402A |
| HAT-1/L402A-3′ | GCTTGCGCAAGTCGTTCtgcGACTCTTCGGTAATC |
aTwo partially complementary primers (5′ and 3′) were used in each PCR mutagenesis for the substitution of alanine codons for the leucine codon.
bCapital letters indicate that the sequence is exactly the same as the wild-type sequence of chHAT-1 cDNA.
cDesignated amino acid substitution resulting from the use of each mutagenesis primer pair.
We constructed the GAL4 yeast two-hybrid plasmids as follows. pCiteHAHAT-1 was digested with NdeI, followed by treatment with T4 polymerase. From the resultant blunt-ended material, the NdeI blunt-end/SmaI fragment containing the full-length chHAT-1 cDNA was excised and subcloned into the SmaI site of the pDBLeu or pPC86 vector to yield pDBLeuHAT-1 or pPC86HAT-1. Similarly, we also constructed pDBLeuHAT-1-(L380A), pDBLeuHAT-1-(L387A), pDBLeuHAT-1-(L394A), pDBLeuHAT-1-(L402A), pPC86HAT-1-(L380A), pPC86HAT-1-(L387A), pPC86HAT-1-(L394A) or pPC86HAT-1-(L402A) from pCiteHAHAT-1-(L380A), pCiteHAHAT-1-(L387A), pCiteHAHAT-1-(L394A) or pCiteHAHAT-1(L402A). Each end point of the deletions and point mutation plasmids was determined by sequence analysis by the dye terminator method.
Expression and purification of the chHAT-1 and chCAF-1p48 GST fusion proteins
Escherichia coli BL-21 cells were transformed with pGEX-2TKchCAF-1p48 and pGEX-2TKchHAT-1, harboring the full-length chCAF-1p48 and chHAT-1 cDNAs, respectively, and grown as described (31). After induction by the addition of 50 µM isopropyl β-d-thiogalactopyranoside (IPTG) overnight at 20°C, the cells were collected by centrifugation, and then both the chHAT-1 and chCAF-1p48 GST fusion proteins were purified essentially as described (31).
HAT activity assay
The HAT liquid assay was carried out essentially as described (33). The concentrations of histones derived from DT40 cells (34) and recombinant GST–chHAT-1 fusion proteins in the assay were 0.6 and 0.4 mg/ml, respectively, and [14C]acetyl coenzyme A (57 µCi/mmol) was added to a final concentration of 1 mM. The reaction mixture was incubated for 20 min at 30°C. The reaction was stopped by the addition of 1.2 ml of ice-cold 20% trichloroacetic acid (TCA). The precipitated proteins were then washed with 5% TCA twice and with acetone once, dried, resolved by 15% SDS–PAGE, fixed, and then stained with Coomassie brilliant blue, followed by fluorography.
GST pull-down affinity assay and immunoprecipitation experiment
To produce [35S]Met-labeled full-length chCAF-1p48, chHAT-1, a set of truncated mutants and a double missense mutation (WN7AA) of Flag-tagged chCAF-1p48, and a set of truncated mutants and single missense mutations (alanine substitutions) of chHAT-1, the TNT-coupled transcription–translation system (Promega) was used. The in vitro binding pull-down assay was performed with 5 µl of [35S]Met-labeled protein fraction and 4 µg of the GST fusion proteins or 6 µg of GST essentially as described (31).
The in vitro immunoprecipitation experiment was performed using 5 µl fraction of [35S]Met-labeled full-length Flag-tagged chCAF-1p48 and 5 µl fraction of [35S]Met-labeled missense mutants of the HA-tagged chHAT-1 proteins essentially as described (31). The resultant immunoprecipitated proteins were analyzed by 12% SDS–PAGE, and then the gels were washed with a fluorographic reagent (Amersham Pharmacia Biotech Inc.), dried and subjected to fluorography.
Yeast two-hybrid assay
Yeast two-hybrid studies were carried out as recommended by the supplier (Gibco BRL) using the GAL4-based system and yeast S.cerevisiae. Qualitative lacZ assays were performed on filters placed on the YPD medium. Quantitative assays were performed as follows, with the O-nitrophenyl-β-d-galactopyranoside (ONPG) as a substrate. Overnight cultures of independent yeast colonies were grown in SD drop-out liquid medium lacking Leu and Trp. Prior to the assay, overnight cultures were diluted 1:2 with fresh YPD medium and then grown for 4 h at 30°C. For OD600 measurements, 500 µl aliquots of cultures were diluted with 500 µl of YPD medium. Aliquots of 1 ml of cultures were pelleted and washed with Z buffer (Gibco BRL). After repelleting, the samples were frozen in liquid N2, thawed at room temperature and then resuspended in 150 µl of Z buffer containing 0.27% β-mercaptoethanol, 50 µl of chloroform and 20 µl of 0.1% SDS. The cell suspension was vortexed vigorously for 1 min. The cell lysate was added to 700 µl of 1 mg/ml ONPG substrate in Z buffer containing 0.27% β-mercaptoethanol, and the reaction was then allowed to proceed at 30°C. The reactions were stopped by the addition of 0.5 ml of 1 M Na2CO3, cellular debris was pelleted by centrifugation and then the reaction product was measured at OD420. The number of β-galactosidase units, 1 U of β-galactosidase being defined as the amount that hydrolyzed 1 µmol of ONPG to o-nitrophenol and d-galactose per min (35), was calculated using the following formula: 1000 × OD420/(t × V × OD600), where t is the reaction time in minutes and V the volume of culture used in the assay in milliliters. The reactions were performed in triplicate using independent colonies for co-transfection assays.
RESULTS
Cloning of cDNA encoding chHAT-1
To identify chHAT-1 as a novel enzyme, we cloned and sequenced the cDNA encoding it. Based on conserved amino acid sequences in Caenorhabditis elegans and human HAT-1s (29,30), we prepared the 279 bp PCR fragment, which corresponds to a part of the cDNA encoding the human HAT-1, by PCR using cDNAs from DT40 cells with degenerate primers, i.e. a sense primer and an antisense primer. Our screening, with the resultant PCR product, of a DT40 lambda ZAP II cDNA library yielded six positive cDNA clones. Sequence analysis of the largest cDNA insert (1639 bp) among them revealed that the clone contained both an initiation codon and a 3′ poly(A) tail, and thus appeared to contain the full-length sequence of the chicken HAT-1 cDNA. The amino acid sequence deduced from the nucleotide sequence comprises 408 residues including a putative initiation Met. This chicken protein exhibits 80.4% identity in amino acid sequence to the human HAT-1. Therefore, it is the chicken homolog of HAT-1s (accession number AF257739). A typical leucine zipper motif is located at positions 366–408 in chHAT-1.
In vitro HAT activity and substrate specificity of recombinant chHAT-1
First, to construct pGEX-2TKchHAT-1, expressing the GST–chHAT-1 fusion protein, chHAT-1 cDNA was subcloned into the pGEX-2TK plasmid in frame. GST fusion proteins were synthesized in E.coli, extracted and then purified as described (31). As shown in Figure 1A, the electrophoretic patterns on SDS–PAGE of whole cell lysates before and after induction with IPTG revealed that GST–chHAT-1 fusion proteins of ~68 kDa were dramatically accumulated in E.coli BL-21 cells containing the pGEX-2TKchHAT-1 plasmid. In addition, the GST–chHAT-1 fusion proteins were purified to >95% homogeneity using glutathione–Sepharose beads (see lane 4).
Figure 1.
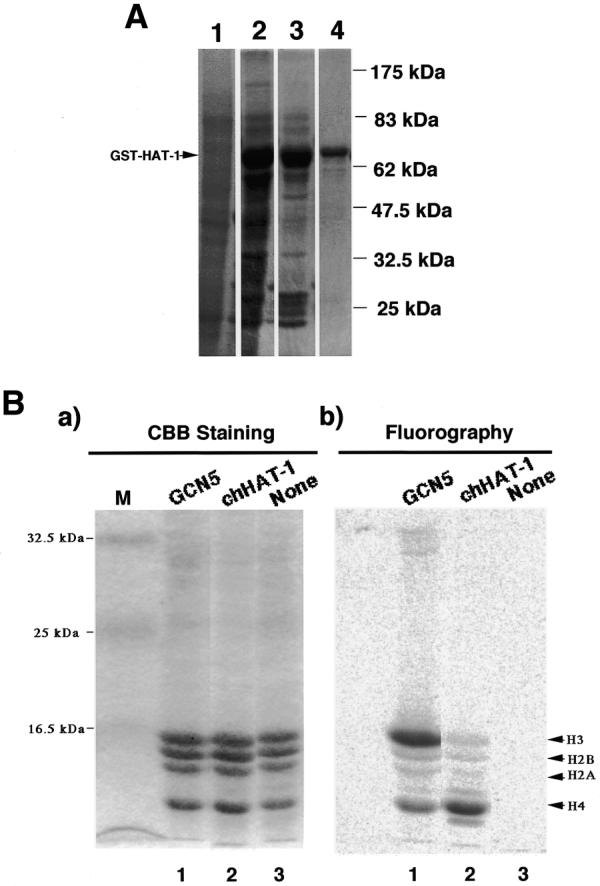
SDS–gel electrophoretic patterns of GST–chHAT-1 fusion protein-containing fractions at different purification steps, and substrate specificity of the recombinant chHAT-1. (A) Protein samples prepared were subjected to 10% SDS–PAGE, followed by Coomassie brilliant blue staining. Lane 1, whole cell lysate of BL-21 cells containing the pGEX-2TKchHAT-1 plasmid without induction by IPTG; lane 2, lysate of BL-21 cells containing the pGEX-2TKchHAT-1 plasmid with induction by 50 µM IPTG; lane 3, complex beads containing chHAT-1; lane 4, chHAT-1 fraction purified with glutathione–Sepharose beads. The standard molecular weights are indicated. GST–HAT-1, GST–chHAT-1 fusion protein. (B) The in vitro acetylation reaction mixture (described below) was subjected to 15% SDS–PAGE, fixed, stained with Coomassie brilliant blue (a) and then exposed to an imaging plate (b). Lane 1, recombinant chicken GCN5 (our unpublished data); lane 2, recombinant chHAT-1; lane 3, without the enzyme as a negative control, using chicken histones prepared from DT40 cells as substrates.
To determine both the HAT activity and substrate specificity of the recombinant GST–chHAT-1 fusion protein in vitro, we measured [14C] acetylated histones by means of the activity gel assay with chicken core histones as substrates (Fig. 1B). The recombinant GST chicken Gcn5 fusion protein, the cDNA of which was recently cloned by us (our unpublished data), as with yeast Gcn5, preferentially acetylated histone H3 over histone H4, but weakly acetylated histones H2A and H2B (lane 1). On the other hand, the recombinant GST–chHAT-1 fusion protein preferentially acetylated histone H4, but weakly acetylated the other core histones, i.e. H2A, H2B and H3 (lane 2). This substrate specificity was virtually the same as that of yeast HAT-1, as described (36).
Approximately 30 amino acids in the C-terminal region of chCAF-1p48 are essential for in vitro interaction with chHAT-1
We have reported that not only does chCAF-1p48, a member of the WD protein family, possess seven copies of the WD repeat motif, but that it also binds tightly to chHDACs (31). To determine whether or not the p48 subunit bound to chHAT-1, the GST pull-down assay was carried out. chCAF-1p48 was translated in vitro in the presence of [35S]Met, and then its ability to interact with chHAT-1 was assayed. Samples were separated by 12% SDS–PAGE and proteins were stained with Coomassie brilliant blue, followed by fluorography. The GST–chHAT-1 fusion protein bound to chCAF-1p48 (Fig. 2A and B). This finding was confirmed in vivo with both the yeast two-hybrid assay system (see later) and the immunoprecipitation experiment (data not shown), suggesting the existence of a region(s) in chHAT-1 to which chCAF-1p48 binds.
Figure 2.
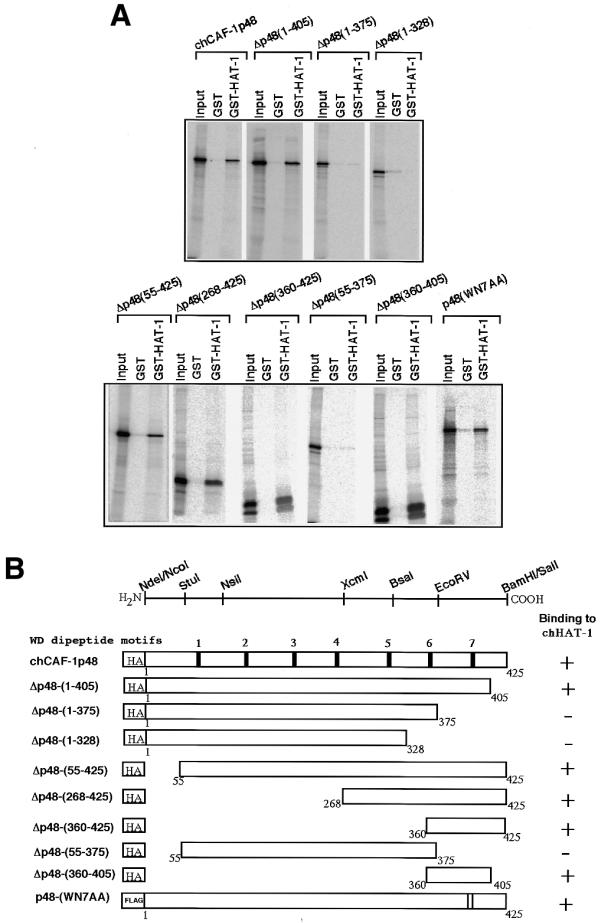
In vitro interaction of truncated and double missense chCAF-1p48 mutant proteins with the GST–chHAT-1 fusion protein. (A) A series of mutant proteins devoid of the N- and/or C-terminal regions of HA-tagged chCAF-1p48, and a double missense mutant of it were constructed, and then their binding activities, as compared to the GST–chHAT-1 fusion protein, were examined as described in Materials and Methods. The mutant proteins used are denoted by appropriate abbreviations: for example, Δp48(1–405) represents ΔchCAF-1p48-(1–405) and p48(WN7AA) represents chCAF-1p48-(WN7AA). GST–HAT-1, GST–chHAT-1 fusion protein. (B) The results obtained for the in vitro interaction of HA-tagged chCAF-1p48, and truncated and missense mutants of it with chHAT-1 are presented schematically.
To clarify the putative binding region(s) of chCAF-1p48 as compared to chHAT-1, we first constructed a series of C-terminal truncated mutants of chCAF-1p48, and then studied their in vitro interaction with the GST–chHAT-1 fusion protein (Fig. 2A and B). Two truncated proteins, ΔchCAF-1p48-(1–375) and ΔchCAF-1p48-(1–328), exhibited no binding activity, although the other protein [ΔchCAF-1p48-(1–405)] exhibited similar binding activity to that of the parental chCAF-1p48 protein. These findings suggested that the C-terminal region of chCAF-1p48, comprising amino acids 376–405, is necessary for its binding activity towards chHAT-1. We next constructed a series of N-terminal truncated mutants of HA-tagged chCAF-1p48, and then studied their binding activity as compared to chHAT-1. The activities of all the truncated mutant proteins, ΔchCAF-1p48-(55–425), ΔchCAF-1p48-(268–425) and ΔchCAF-1p48-(360–425), were the same as that of the parental chCAF-1p48 protein. These results suggested that not only does the region of chCAF-1p48 comprising amino acids 360–425 (in fact, 360–405) exhibit binding activity similar to chHAT-1, but also that six of the seven WD dipeptide motifs (first to sixth) are not necessary for binding activity.
Next, we constructed two mutants with simultaneous deletion of both the N- and C-terminal regions of chCAF-1p48, and then assayed their binding activity. As shown in Figure 2A and B, as expected, one mutant, ΔchCAF-1p48-(55–375), exhibited no binding activity. On the other hand, the remaining mutant, ΔchCAF-1p48-(360–405), exhibited similar binding activity to the parental chCAF-1p48 protein, indicating that the region comprising amino acids 360–405 (more specifically 376–405) and carrying the seventh WD dipeptide motif is enough for its in vitro interaction with chHAT-1.
To determine whether or not the seventh WD (WN) dipeptide motif participated in the binding activity of chCAF-1p48 towards chHAT-1, we constructed a double missense mutant protein carrying alanine substitutions in the dipeptide motif, chCAF-1p48-(WN7AA), and assayed its binding activity as compared to chHAT-1. The binding activity of chCAF-1p48-(WN7AA) was the same as that of the parental protein (Fig. 2A and B). These results strongly indicate that the seventh WN dipeptide motif within amino acids 376–405 of chCAF-1p48 is not involved in its binding activity as compared to chHAT-1.
Leucine zipper motif in the C-terminal region of chHAT-1 required for in vitro interaction with chCAF-1p48
To determine the putative binding region(s) of chHAT-1 as compared to chCAF-1p48, we constructed two C-terminal truncated mutants of HA-tagged chHAT-1, labeled them with [35S]Met, and then studied their in vitro interaction with the GST–chCAF-1p48 fusion protein. Two deletion mutant proteins, ΔchHAT-1-(1–379) and ΔchHAT-1-(1–366), exhibited no binding ability (Fig. 3A and B). These findings suggested that deletion of the C-terminal region of chHAT-1, comprising amino acids 380–408, caused the loss of the binding ability, as with chCAF-1p48. Next we constructed three N-terminal truncated mutants of HA-tagged chHAT-1, and then studied their binding ability as compared to chCAF-1p48. The abilities of the three truncated mutant proteins, ΔchHAT-1-(71–408), ΔchHAT-1-(344–408) and ΔchHAT-1-(366–408), were the same as that of the parental protein (Fig. 3A and B). Taken together, these results strongly indicate that the C-terminal region of chHAT-1, comprising amino acids 366–408 (more specifically 380–408), is necessary and sufficient for its in vitro interaction with chCAF-1p48.
Figure 3.
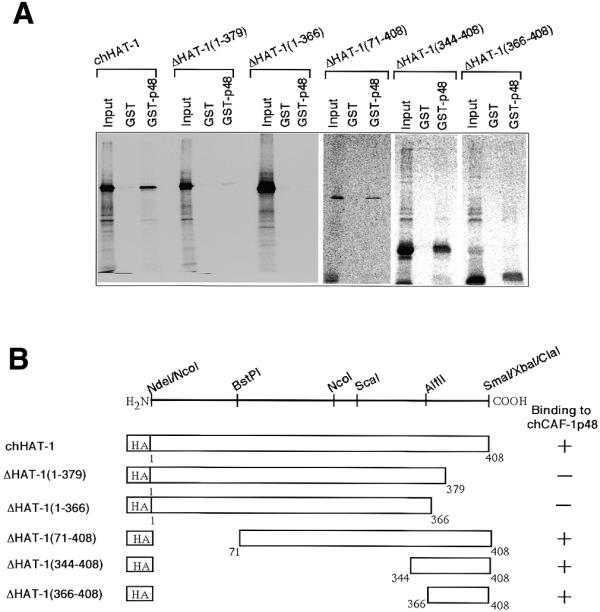
In vitro interaction of truncated mutant proteins of chHAT-1 with the GST–chCAF-1p48 fusion protein. (A) A series of mutant proteins devoid of the N- plus C-terminal regions of HA-tagged chHAT-1 were constructed and their binding activities, as compared to the GST–chCAF-1p48 fusion protein, were examined as in Figure 2. The mutant proteins used are denoted by appropriate abbreviations: for example, ΔHAT-1(1–379) represents ΔchHAT-1-(1–379). GST–p48, GST–chCAF-1p48 fusion protein. (B) The results obtained for the in vitro interaction of HA-tagged chHAT-1 and truncated mutants of it with chCAF-1p48 are presented schematically.
The C-terminal region of chHAT-1 comprising amino acids 366–408, in particular the presumably minimal active region (comprising amino acids 380–408), was thought to be a typical leucine zipper motif containing four Leu residues at positions 380, 387, 394 and 402. To determine whether or not this leucine zipper motif really does facilitate the interaction of chHAT-1 with chCAF-1p48, we constructed single missense mutants with Ala substitutions of the four Leu residues, chHAT-1-(L380A), chHAT-1-(L387A), chHAT-1-(L394A) and chHAT-1-(L402A), and then assayed their binding ability as compared to the GST–chCAF-1p48 fusion protein. As shown in Figure 4A, the GST pull-down assay, involving these missense mutants, revealed that all the Ala substitutions of the four Leu residues resulted in loss of binding ability. These findings were confirmed by the immunoprecipitation experiment involving anti-Flag M2 beads, i.e. these four mutant proteins lost their binding ability as compared to in vitro translated Flag-tagged chCAF-1p48, whereas the wild-type chHAT-1 could interact with it (Fig. 4B). Taken together, these results indicate that the leucine zipper motif within the C-terminal region of chHAT-1, comprising amino acids 380–408, is essential for its interaction with chCAF-1p48.
Figure 4.
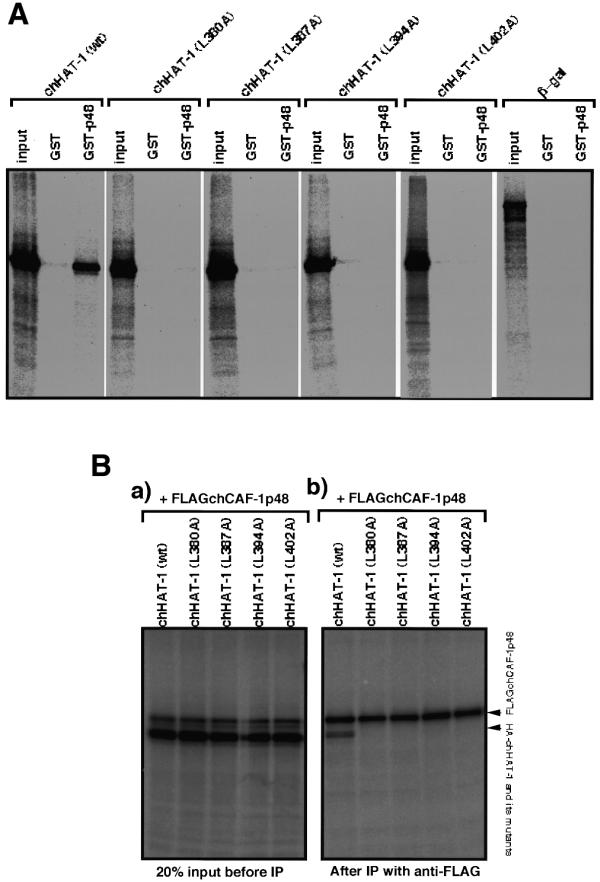
In vitro interaction of single missense mutant proteins as compared to the leucine zipper motif of chHAT-1 with the GST–chCAF-1p48 fusion protein. (A) A series of single missense mutant proteins, as compared to the leucine zipper motif of chHAT-1, were constructed and then their binding activities, as compared to the GST–chCAF-1p48 fusion protein, were examined. The mutant proteins used are denoted by appropriate abbreviations. (B) An in vitro immunoprecipitation experiment on the interaction of the single missense mutant proteins of chHAT-1 with the Flag-tagged chCAF-1p48 protein was carried out using [35S]Met-labeled Flag-tagged chCAF-1p48 and [35S]Met-labeled chHAT-1, or missense mutants of it, followed by the addition of anti-Flag M2 beads. Aliquots of eluates from the affinity beads were analyzed by 12% SDS–PAGE and the gels were then subjected to fluorography (b). A portion (20%) of each reaction mixture was removed before the immunoprecipitation as an input sample (a).
Leucine zipper motif in the C-terminal region of chHAT-1 is required for in vivo interaction with chCAF-1p48
We examined both the in vivo interaction of chHAT-1 with chCAF-1p48 and the necessity of the leucine zipper motif of the former for the in vivo interaction, using the GAL4-based yeast two-hybrid system, which is applicable to studies on in vivo protein–protein interactions. We first subcloned the full-length chCAF-1p48 and chHAT-1 cDNAs, respectively, into the pDBLeu and pPC86 vectors (Gibco BRL). Yeast S.cerevisiae strain MaV203 was co-transfected with pairs of the GAL4–chCAF-1p48 and chHAT-1 fusion protein-encoding plasmids, in combination with available vectors, or negative control plasmids lacking inserts and the pCL1 plasmid (Clontech) as a positive control. The yeast colonies containing paired plasmids were all grown on SD drop-out plates lacking Leu and Trp (Fig. 5A, a). When the yeast colonies were replica-cleaned on SD drop-out plates lacking Leu, Trp and His, but with the addition of 10 mM 3-AT (Fig. 5A, b), noticeable alterations were observed as follows. In the case of the GAL4 DNA binding domain and GAL4 activation domain containing chCAF-1p48 and chHAT-1, respectively, yeast colonies grew well, as in the case of yeast containing both the pCL1 and pDBTrp plasmids as positive controls. In addition, yeast colonies grew weakly when the GAL4 activation domain was fused to chCAF-1p48 and the GAL4 binding domain was fused to chHAT-1, although all other combinations failed to activate the histidine reporter gene.
Figure 5.
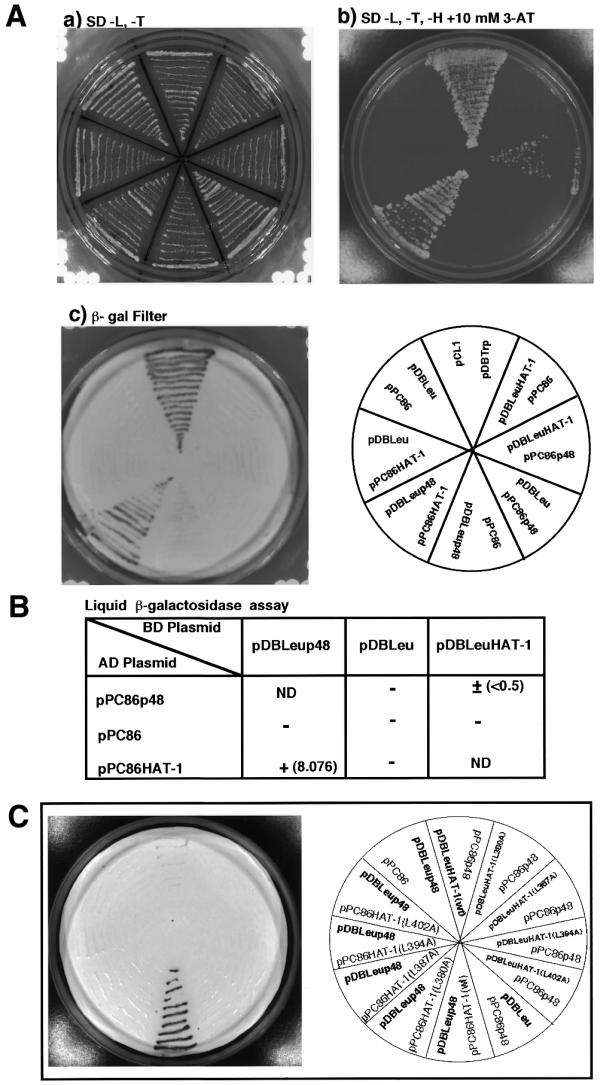
Yeast two-hybrid assay for the in vivo interaction of chHAT-1 or single missense mutants of it as compared to the leucine zipper motif with chCAF-1p48. The GAL4-based system and yeast S.cerevisiae were used for the two-hybrid study. (A) The in vivo interaction of chHAT-1 with chCAF-1p48 was performed on an SD drop-out plate lacking Leu and Trp (a) and an SD drop-out plate lacking Leu, Trp and His, but with the addition of 10 mM 3-AT (b). Qualitative lacZ assays for the in vivo interaction of the two proteins were performed on filters placed on YPD medium (c). (B) Quantitative assays for the in vivo interaction of the two proteins were performed with ONPG as a substrate. (C) Qualitative lacZ assays for the in vivo interaction of single missense mutants of chHAT-1 with chCAF-1p48 were performed on filters placed on YPD medium. The expression of proteins in combination with available vectors was verified by western blotting using anti-GAL4 antiserum as the primary antibodies (data not shown).
To confirm these findings, the lacZ assay with X-gal as the substrate was performed on filters placed on the YPD medium (Fig. 5A, c). As expected, the interaction of chCAF-1p48 with chHAT-1 provided a robust two-hybrid interaction only when the GAL4 activation domain was fused to chHAT-1 and the GAL4 binding domain was fused to chCAF-1p48. In addition, as shown in Figure 5B, these findings were confirmed by the liquid β-galactosidase assay, with OPNG as the substrate. Taken together, these results indicated that chHAT-1 binds tightly to chCAF-1p48 in vivo.
Next, using the GAL4-based yeast two-hybrid system, we examined whether or not the leucine zipper motif of chHAT-1 was really necessary for its in vivo interaction with chCAF-1p48. The full-length chCAF-1p48 cDNA and the four missense mutants of chHAT-1 cDNA: chHAT-1-(L380A), chHAT-1-(L387A), chHAT-1-(L394A) and chHAT-1-(L402A), were subcloned into the pDBLeu and pPC86 vectors, respectively. Yeast strain MaV203 was co-transfected with pairs of the GAL4–chCAF-1p48 and mutant chHAT-1 fusion protein-encoding plasmids, in combination with available vectors, or negative control plasmids lacking inserts. The lacZ assay was performed on filters placed on the YPD medium. As shown in Figure 5C, the interaction of chCAF-1p48 with chHAT-1 provided a robust two-hybrid interaction when the GAL4 activation domain was fused to the wild-type chHAT-1 protein and the GAL4 binding domain was fused to chCAF-1p48. Similarly, the yeast lacZ reporter gene was weakly expressed when the GAL4 activation domain was fused to chCAF-1p48 and the GAL4 binding domain was fused to the wild-type of chHAT-1, although all other combinations failed to activate the lacZ reporter gene. On the other hand, interestingly, in the two distinct experiments, all other combinations involving Ala substitution mutations of Leu at four positions failed to activate the reporter gene. Taken together, these results indicate that the leucine zipper motif of chHAT-1 is essential for its in vivo interaction with chCAF-1p48.
DISCUSSION
The p48 subunit has been reported to be a component of CAF-1, which is involved in chromatin assembly in eukaryotes (17–23). In addition, the p48 subunit, together with the p46 polypeptide (a p48 homolog), exists in repressor complexes with HDAC-1 and -2, and mSin3, Rb or Mi2 plus MeCP2, to repress transcription (37). On the other hand, the p46 polypeptide associates with HAT-1, and this complex possesses the ability to acetylate particular Lys residues, i.e. Lys-5 and Lys-12, of histone H4. Moreover, p48 and p46 themselves have been reported to differentially bind to core histones (23,30,38,39). Our recent study, involving the gene targeting technique and the chicken DT40 B cell line, revealed that chCAF-1p48 is essential for cell viability (Y.Takami et al., manuscript in preparation).
Taken together, these results indicate that chCAF-1p48 is separately involved in numerous DNA-utilizing processes, probably through different protein–protein interactions. An initial step for assessing such fundamental participation of the p48 subunit in important cell functions was to clarify how it interacts with other proteins. Previously, we established that two N-terminal, two C-terminal or one N-terminal and one C-terminal WD repeat motifs of chCAF-1p48 are required for its in vitro interaction with chHDAC-2 (31). In line with this view, we first cloned and sequenced the cDNA encoding chHAT-1, which exhibits 80.4% identity to its human homolog. The in vitro substrate specificity of the recombinant chHAT-1, i.e. the preferential acetylation of histone H4 relative to other core histones, H2A, H2B and H3 (Fig. 1B), is virtually the same as of the yeast and human HAT-1s (36).
The HIS6 pull-down assay revealed the in vitro interaction of HAT-1 with yeast HAT2, a yeast p48 homolog (CAC3 or MS1) (36). On the other hand, the monoclonal antibody that reacts equally with both p46 and p48 recognized an ~50 kDa polypeptide in the purified human HAT-1 holoenzyme preparation, but specific anti-p48 antiserum did not detect any polypeptide in the purified HAT-1 preparation (30). These findings indicate that the human p46 polypeptide could be co-purified with HAT-1 but that the p48 subunit could not be present in the HAT-1 holoenzyme complex.
We then studied the molecular basis of the interaction of chCAF-1p48 with chHAT-1, because the former is essential for the viability of the DT40 cell line and is present in a large amount relative to p46 (Y.Takami et al., manuscript in preparation). The GST pull-down assay revealed the obvious in vitro interaction of chHAT-1 with chCAF-1p48 (Fig. 2). The C-terminal region of chCAF-1p48, comprising amino acids 376–405, wherein six WD dipeptide motifs (first to sixth motifs) are deleted, i.e. only the seventh WD (WN) dipeptide motif is present, is enough for this in vitro interaction of the two proteins (Fig. 2A and B). In addition, the double missense mutation of this dipeptide motif, WN7AA, had no influence on this in vitro protein–protein interaction, indicating that the propeller structure of chCAF-1p48 is not necessary for its in vitro interaction with chHAT-1. These results obtained with the in vitro assay system most likely reflect what happens in vivo, because chCAF-1p48 also interacted with chHAT-1 in the yeast two-hybrid assay system (Fig. 5).
The GST pull-down assay, involving truncated mutants, revealed that the C-terminal region of chHAT-1, comprising amino acids 366–408 (probably more specifically 380–408), wherein the typical leucine zipper motif is included, is sufficient for its in vitro interaction with chCAF-1p48 (Fig. 3A and B). Interestingly, as indicated by both the GST pull-down assay and the immunoprecipitation experiment involving single missense mutants, all the missense mutations, as with the four Leu residues: L380A, L387A, L394A and L402A, caused the complete loss of the in vitro protein–protein interaction (Fig. 4A and B). Moreover, experiments involving the yeast two-hybrid assay established that these missense mutations within the leucine zipper motif all resulted in the loss of the in vivo interaction of chHAT-1 with chCAF-1p48 (Fig. 5). Taken together, these results indicated that the proper structure of the leucine zipper motif is essential for the in vitro and in vivo interaction of chHAT-1 with chCAF-1p48. As described previously (30), the human HAT-1 could not associate with the human p48 subunit in vivo. The reason for the discrepancy between our results regarding the interaction of chHAT-1 with chCAF-p48 and those for the interaction of human HAT-1 with the p48 subunit is probably due to the fact that two Leu residues at positions 380 and 402 within the leucine zipper motif of chHAT-1 are substituted by Ile residues in the corresponding region of the human HAT-1.
Based on the results obtained for series of both truncated and missense chCAF-1p48 and chHAT-1 mutant proteins, we propose a model for the interaction of these two proteins. Most protein–protein interactions are probably attributable to the proper propeller structures of WD protein family members and should be involved in numerous biological processes, such as gene expression, RNA processing, cell division, signal transduction and vesicular trafficking (24). Conversely, the interaction of chCAF-1p48 with chHAT-1 should require only a restricted narrow region surrounding the seventh WD dipeptide motif of the former protein, comprising amino acids 376–405, i.e. it should not require its proper propeller structure as a common platform. On the other hand, the interaction of these two proteins should require the leucine zipper motif of chHAT-1, comprising amino acids 380–408. In particular, the four Leu residues at positions 380, 387, 394 and 402 within the motif are essential for the interaction of chHAT-1 with chCAF-1p48, probably because the bulky hydrophobic side chains of Leu residues should be the major hydrophobic force allowing the leucine zipper motif to facilitate their interaction. This manner of interaction of chCAF-1p48 with chHAT-1 is obviously distinct from that of the interaction of chCAF-1p48 with chHDAC-2, for which the propeller structure of the former protein should be maintained properly (31). In addition to these two distinct types of protein–protein interactions, chCAF-1p48, as well as the p48 subunits of most vertebrate cells, is expected to participate in other protein–protein interactions that depend on variable regions (5–62 amino acids) preceding the conserved cores of 32 amino acids of the WD repeat motifs. Thus, these chCAF-1p48-participating interactions (or chCAF-1p48-carrying complexes) should be involved directly or indirectly in a number of fundamental events necessary for cell viability.
Acknowledgments
ACKNOWLEDGEMENT
This work was supported in part by a Grant-in-Aid from the Ministry of Education, Science, Sports and Culture of Japan.
DDBJ/EMBL/GenBank accession no. AF257739
References
- 1.Wolffe A.P. (1996) Histone deacetylase: A regulator of transcription. Science, 272, 371–372. [DOI] [PubMed] [Google Scholar]
- 2.Wolffe A.P. and Pruss,D. (1996) Targeting chromatin disruption: transcription regulators that acetylate histones. Cell, 84, 817–819. [DOI] [PubMed] [Google Scholar]
- 3.Pazin M.J. and Kadonaga,J.T. (1997) What’s up and down with histone deacetylation and transcription? Cell, 89, 325–328. [DOI] [PubMed] [Google Scholar]
- 4.Pennisi E. (1997) Opening the way to gene activity. Science, 275, 155–157. [DOI] [PubMed] [Google Scholar]
- 5.Wolffe A.P., Wong,J. and Pruss,D. (1997) Activators and repressors: making use of chromatin to regulate transcription. Genes Cells, 2, 291–302. [DOI] [PubMed] [Google Scholar]
- 6.Brownell J.E., Zhou,J., Ranalli,T., Kobayashi,R., Edmondson,D.G., Roth,S.Y. and Allis,C.D. (1996) Tetrahymena histone acetyltransferase A: a homolog of yeast Gcn5p linking histone acetylation to gene activation. Cell, 84, 843–851. [DOI] [PubMed] [Google Scholar]
- 7.Mizzen C.A., Yang,X.J., Kokubo,T., Brownell,J.E., Bannister,A.J., Owen-Hughes,T., Workman,J., Wang,L., Berger,S.L., Kouzarides,T., Nakatani,Y. and Allis,D. (1996) The TAFII250 subunit of TFIID has histone acetyltransferase activity. Cell, 87, 1261–1270. [DOI] [PubMed] [Google Scholar]
- 8.Ogryzko V.V., Schiltz,R.L., Russanova,V., Howard,B.H. and Nakatani,Y. (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell, 87, 953–959. [DOI] [PubMed] [Google Scholar]
- 9.Yang X.J., Ogryzko,V.V., Nishikawa,J.I., Howard,B. and Nakatani,Y. (1996) A p300/CBP-associated factor that competes with the adenoviral E1A oncoprotein. Nature, 382, 319–324. [DOI] [PubMed] [Google Scholar]
- 10.Taunton J., Hassig,C.A. and Schreiber,S.L. (1996) A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science, 272, 408–411. [DOI] [PubMed] [Google Scholar]
- 11.Yang W.M., Inoue,C., Zeng,Y., Bearss,D. and Seto,E. (1996) Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl Acad. Sci. USA, 93, 12845–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang W.M., Yao,Y.L., Sun,J.M., Davie,J.R. and Seto,E. (1997) Isolation and characterization of cDNA corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem., 272, 28001–28007. [DOI] [PubMed] [Google Scholar]
- 13.Takami Y., Kikuchi,H. and Nakayama,T. (1999) Chicken histone deacetylase-2 controls the amount of the IgM H-chain at the steps of both transcription of its gene and alternative processing of its pre-mRNA in the DT40 cell line. J. Biol. Chem., 274, 23977–23990. [DOI] [PubMed] [Google Scholar]
- 14.Takami Y. and Nakayama,T. (2000) N-Terminal region, C-terminal region, nuclear export signal and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J. Biol. Chem., 275, 16191–16201. [DOI] [PubMed] [Google Scholar]
- 15.Buerstedde J.-M. and Takeda,S. (1991) Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell, 67, 179–188. [DOI] [PubMed] [Google Scholar]
- 16.Takami Y., Takeda,S. and Nakayama,T. (1995) Targeted disruption of H2B-V encoding a particular H2B histone variant causes changes in protein patterns on two-dimensional polyacrylamide gel electrophoresis in the DT40 chicken B cell line. J. Biol. Chem., 270, 30664–30670. [DOI] [PubMed] [Google Scholar]
- 17.Stillman B. (1986) Chromatin assembly during SV40 DNA replication in vitro. Cell, 45, 555–565. [DOI] [PubMed] [Google Scholar]
- 18.Smith S. and Stillman,B. (1989) Purification and characterization of CAF-1, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell, 58, 15–25. [DOI] [PubMed] [Google Scholar]
- 19.Smith S. and Stillman,B. (1991) Stepwise assembly of chromatin during DNA replication in vitro. EMBO J., 10, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krude T. (1995) Chromatin assembly factor 1 (CAF-1) colocalizes with replication foci in Hela cell nuclei. Exp. Cell Res., 220, 304–311. [DOI] [PubMed] [Google Scholar]
- 21.Kaufman P.D., Kobayashi,R., Kessler,N. and Stillman,B. (1995) The p150 and p60 subunits of chromatin assembly factor-1: A molecular link between newly synthesized histones and DNA replication. Cell, 81, 1105–1114. [DOI] [PubMed] [Google Scholar]
- 22.Ito T., Bulger,M., Kobayashi,R. and Kadonaga,J.T. (1996) Drosophila NAP-1 is a core histone chaperone that functions in ATP-facilitated assembly of regularly-spaced nucleosomal arrays. Mol. Cell. Biol., 16, 3112–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verreault A., Kaufman,P.D., Kobayashi,R. and Stillman,B. (1996) Nucleosome assembly by a complex of CAF-1 and acetylated histones H3/H4. Cell, 87, 95–104. [DOI] [PubMed] [Google Scholar]
- 24.Neer E.J., Schmidt,C.J., Nambudripad,R. and Smith,T.F. (1994) The ancient regulatory-protein family of WD-repeat proteins. Nature, 371, 297–300. [DOI] [PubMed] [Google Scholar]
- 25.Lambright D.G., Sondek,J., Bohm,A., Skiba,N.P., Hamm,H.E. and Sigler,P.B. (1996) The 2.0 Å crystal structure of a heterotrimeric G protein. Nature, 379, 311–319. [DOI] [PubMed] [Google Scholar]
- 26.Neer E.J. and Smith,T.F. (1996) G protein heterodimers: new structures propel new questions. Cell, 84, 175–178. [DOI] [PubMed] [Google Scholar]
- 27.Sondek J., Bohm,A., Lambright,D.G., Hamm,H.E. and Sigler,P.B. (1996) Crystal structure of a GA protein βγ dimer at 2.1 A resolution. Nature, 379, 369–374. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman P.D., Kobayashi,R. and Stillman,B. (1997) Ultraviolet radiation sensitivity and reduction of telomeric silencing in Saccharomyces cerevisiae cells lacking chromatin assembly factor-1. Genes Dev., 11, 345–357. [DOI] [PubMed] [Google Scholar]
- 29.Dutnall R.N., Tafrov,S.T., Sternglanz,R. and Ramakrishnan,V. (1998) Structure of the histone acetyltransferase Hat1: A paradigm for the GCN5-related N-acetyltransferase superfamily. Cell, 94, 427–438. [DOI] [PubMed] [Google Scholar]
- 30.Verreault A., Kaufman,P.D., Kobayashi,R. and Stillman,B. (1998) Nucleosomal DNA regulates the core-histone binding subunit of the human hat1 acetyltransferase. Curr. Biol., 8, 96–108. [DOI] [PubMed] [Google Scholar]
- 31.Ahmad A., Takami,Y. and Nakayama,T. (1999) WD repeats of the p48 subunit of chicken chromatin assembly factor-1 required for in vitro interaction with chicken histone deacetylase-2. J. Biol. Chem., 274, 16646–16653. [DOI] [PubMed] [Google Scholar]
- 32.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 33.Brownell J.E. and Allis,C.D. (1995) An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei. Proc. Natl Acad. Sci. USA, 92, 6364–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takami Y., Takeda,S. and Nakayama,T. (1997) An approximately half set of histone genes is enough for cell proliferation and a lack of several histone variants causes protein pattern changes in the DT40 chicken B cell line. J. Mol. Biol., 265, 394–408. [DOI] [PubMed] [Google Scholar]
- 35.Miller J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 36.Parthun M.R., Widom,J. and Gottschling,D.E. (1996) The major cytoplasmic histone acetyltransferase in yeast: links to chromatin replication and histone metabolism. Cell, 87, 85–94. [DOI] [PubMed] [Google Scholar]
- 37.Nagy L., Kao,H.-Y., Chakravarti,D., Lin,R.J., Hassig,C.A., Ayer,D.E., Schreiber,S.L. and Evans,R.M. (1997) Nuclear receptor repression mediated by a complex containing SMRT, mSin3A and histone deacetylase. Cell, 89, 373–380. [DOI] [PubMed] [Google Scholar]
- 38.Vermaak D., Wada,P.A., Jones,P.L., Shi,Y.-B. and Wolffe,A.P. (1999) Functional analysis of the SIN3-histone deacetylase RPD3-RbAp48-histone H4 connection in the Xenopus oocyte. Mol. Cell. Biol., 19, 5847–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmad A., Takami,Y. and Nakayama,T. (2000) Distinct regions of the chicken p46 polypeptide are required for its in vitro interaction with histones H2B and H4 and histone acetyltransferase-1. Biochem. Biophys. Res. Commun., 279, 95–102. [DOI] [PubMed] [Google Scholar]