

Abstract
The goal was to assess regional patterns of metabolite abnormalities in mild cognitive impairment (MCI) and Alzheimer disease (AD) patients using proton magnetic resonance spectroscopy imaging at 1.5 Tesla. Fourteen MCI, 17 AD, and 16 healthy control (HC) subjects were studied. MCI was associated with higher myo-inositol (mIn) concentration in right parietal white matter compared with HC and lower mIn levels in frontal white matter compared with AD. AD was associated with higher mIn concentration in frontal and parietal white matter compared with HC. N-acetylaspartate (NAA) concentration of white matter was similar in all groups, whereas NAA concentration of gray matter showed a trend toward lower values in the right parietal lobe in AD compared with MCI and HC. A mIn increase in white matter in absence of significant NAA reduction suggests that mIn is a more robust and sensitive marker of white matter pathology in AD and MCI than NAA. Furthermore, the dissociation between mIn and NAA alterations in white matter could provide important information regarding the role of glial and neuronal damage in MCI and AD.
Keywords: mild cognitive impairment, H-MRS, Alzheimer disease
There is growing evidence in Alzheimer disease (AD) for a cerebral disconnection syndrome that involves both gray matter (GM) and white matter (WM)1 besides the well-documented pathology of neuronal cell bodies, which primarily include cortical and hippocampal GM locations.1
Evidence for a more prominent role of WM in AD pathology comes from histopathologic studies showing a differential myelin breakdown of connection fibers that resembles the progressive regional spread of the pathognomonic lesions of AD (neuritic plaques and neurofibrillary tangles).2 In addition, in vivo diffusion tensor imaging (DTI) studies on patients suggest progressive disintegration of WM fibers in AD, such as the cingulum bundle.3,4 Proton magnetic resonance spectroscopy (1H MRS) studies of WM integrity in AD reported abnormal metabolite concentrations, particularly decreased N-acetylaspartate (NAA), a putative marker of neuronal processes5 and increased myo-inositol (mIn), a proposed marker for gliosis.6 In addition to myelin breakdown, there is also accumulative evidence that inflammation expressed as reactive microglial activation, reactive astrocytosis, and presence of inflammatory mediators could be an important pathogenetic factor for AD.7 Neuroinflammatory species like activated microglia surrounding senile plaques and increased level of cytokines, chemokines, and free radical have been observed in AD.7 These processes might develop in response to amyloid deposition and the associated loss of cerebral tissue. Inflammation is also consistent with MRS findings of increased mIn8 and DTI findings of increased diffusivity9 in AD. A better understanding of WM abnormalities in AD and potential targets for intervention to prevent cognitive decline has therefore assumed greater urgency.
Clinical observations show that patients with mild cognitive impairment (MCI) have a higher risk of conversion to AD than cognitively normal elderly subjects of similar age,10 consistent with the view that MCI is a prodromal stage of AD.11 The support of the fact that MCI might be a transitional stage to AD also comes from neuroimaging studies including morphometric magnetic resonance imaging (MRI), magnetization transfer imaging, 1H MRS, and DTI.12–15 For example, increased mIn/creatinine (Cr) ratios in paratrigonal WM in MCI were reported in a 1H MRS study, similar to the biochemical pattern seen in AD.16 Furthermore, converters to AD exhibited lower NAA/Cr ratios in paratrigonal WM than those who remained clinically stable.17 WM abnormalities in MCI were also assessed using DTI3 and magnetization transfer imaging.13 Together, the studies suggest vulnerability of WM in MCI similar to AD.
However, the fast majority of previous MRS studies in MCI and AD, provided limited information about systematic regional alterations, because single voxel MRS was used, which facilitates measuring metabolites only from 1 or a few regions of interest at a time. To our knowledge, there has been only 1 report of regional mIn alterations in AD18 using magnetic resonance spectroscopic imaging (MRSI) technology, which can simultaneously measure metabolites from multiple regions (dozens to hundreds of voxels) across the brain, thus mapping the regional variation of metabolites in both GM and WM.19,20 However, the previous MRSI report did not include MCI. Furthermore, the extent to which vascular disease often present in MCI and AD are associated with the metabolite alterations has not thoroughly been investigated.
In this study, we used MRSI to determine the characteristic regional pattern of metabolite abnormalities, especially mIn, in MCI and AD. In addition, we co-registered 1H MRSI to tissue-segmented high-resolution MRI data and regressed metabolite variations against tissue compositions to obtain metabolite concentrations independent of partial volume effects of GM and WM lesions.20–22 The overall goal of this study was to determine difference in GM and WM: Therefore, our first hypothesis was:
MCI and AD are associated with increased mIn and decreased NAA in GM and normal appearing WM. As the parietal lobe is thought to be involved early in the course of AD, our second hypothesis was:
The most prominent metabolite abnormalities in MCI and AD occur in the parietal lobe. In addition, we determined the extent to which vascular pathology, expressed as WM hyperintensities (WMH) on MRI, can explain metabolite abnormalities.
METHODS
Patients
Fourteen subjects meeting the criteria for amnestic MCI (6 women, 8 men, mean age 77.1±6.3 y), according to Petersen et al’s criteria,10 were studied using MRI and MRSI and compared with 16 healthy elderly control subjects (11 women, 5 men, mean age 72.5±5.2 y) and 17 patients with a diagnosis of AD (7 women, 10 men, mean age 74.5±7.7 y). The MCI subjects were recruited from the Memory and Aging Clinic of the University of California at San Francisco (UCSF). The healthy controls were recruited from the community and received the same standard neurologic examinations than the MCI patients. The AD patients were also recruited from the Memory and Aging Clinic at UCSF and met criteria for probable or possible AD according to the National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRA) criteria. 23 The MCI subjects had on average a Mini-Mental State Examination score (MMSE)24 of 27.6±1.5, the AD patients had 21.4±5.4, and control subjects had 29.5±0.9. The AD patients presented different stages of the disease; mean disease duration was 3.9 years (range, 0 to 6.9 y), mean age at disease onset was 69.6 years (range, 56 to 80 y), and cognitive impairment ranged from 17 to 27 in MMSE score. All participants were free of a clinical history of psychiatric disease, diabetes, major heart disease, had trauma, or signs of major neuropathology, such as stroke, tumors, vascular malformations on MRI, which were read by a trained neuroradiologist, who was blinded to the diagnosis. In addition, subjects were excluded if poor data quality of MRI or MRSI prohibited postprocessing analyses.
Written informed consent was obtained from each subject or his/her legal guardian before participating in the study, which was approved by the committees for human research at UCSF and the San Francisco Veterans Affairs Medical Center.
Structural MRI and MRSI Acquisition
MRI and MRSI were obtained in 1 session on a 1.5 Tesla Siemens Vision System (Siemens Inc, Iselin, NJ), as described previously.21 In short, the structural MRI protocol included axially oblique dual spin echo (DSE) images with repetition time (TR)=2500 ms, echo times (TE1/TE2)=20/80 ms, 1.0×1.4mm2 in-plane resolution, and 3-mm slice thickness with no section gap. In addition, a volumetric magnetization-prepared rapid gradient echo sequence was acquired with TR/TE=9/4 ms timing, 300 ms inversion time (TI), 15-degree flip angle, 1.0×1.0mm2 in-plane resolution, and 1.5-mm thick coronal partitions, oriented approximately orthogonal to the long axis of the hippocampus. Multislice MRSI data (TR/TI/TE=1800/170/25 ms, 30 min total acquisition) were acquired from axially oblique, 15-mm thick slices, with a nominal in-plane resolution of 7.8×7.8mm2, parallel to the axial-oblique DSE images (Fig. 1). The top 2 slices covered primarily the frontal and parietal lobes of the brain and part of occipital lobe whereas the bottom slice covered subcortical structures and parts of the superior temporal lobe. Slice selective inversion recovery pulses with TI=170 ms were applied for nulling the lipid signal as described in detail earlier.21 Due to technical limitations, we were unable to reliably obtain 1H MR spectra from the lower parts of the brain including the hippocampal and entorhinal cortex region, which are major targets of AD pathology. Although measurement of mIn has previously been technically challenging using 1H MRSI, because of problems related to incomplete suppression of resonances from solvent water and lipids at short spin echo times, this laboratory recently reported progress in reliably obtaining short TE MRSI data, facilitating clinical studies with this technique.21,22 To prevent head movement during scanning, we used head pads to stabilize the head position of subjects. In addition, all data were retrospectively screened for movement artifacts and excluded if artifacts were visible.
FIGURE 1.
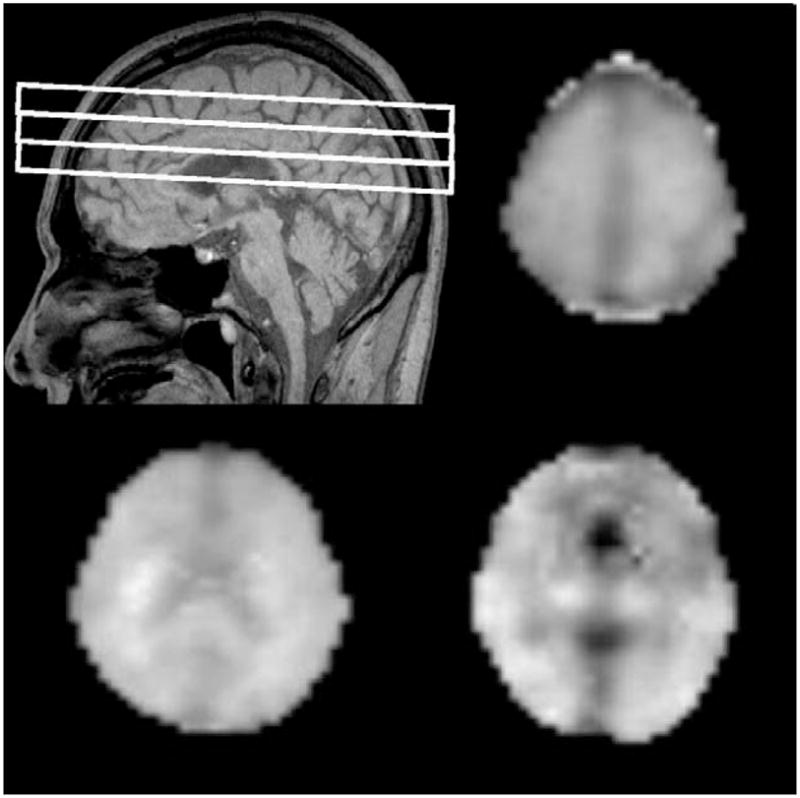
A, Sagittal scout indicating MRSI slice position. B to D, Myo-inositol image of each slice.
MRI and MRSI Processing
The proton density and T2-weighted images from DSE and T1-weighted magnetization-prepared rapid gradient echo images were used together for brain tissue segmentation into GM, WM, cerebrospinal fluid, and WMH. Image segmentation was based on SPM99 (Wellcome Department of Imaging Neuroscience, University College, London, UK) and EMS (Expectation-Maximization Segmentation, Medical Imaging Computing, University Hospital Gasthuisberg, Leuven, Belgium) software.
The fully automated processing of 1H MRSI data has been described before20 In brief, the MRSI data were zero-padded from 32×32 to 64×64 points in the spatial domain and from 1024 to 2048 in the spectral domain. Furthermore, to reduce voxel ‘bleeding’ of the intense signal from extracranial lipids, an iterative method for k-space extrapolation of the lipid signal was applied.25 After Fourier reconstruction of MRSI, metabolite peaks were fitted using a fully automated spectral fitting algorithm.26,27
Quality control of the fits was accomplished by including only spectra with a line width between 2 and 11 Hz. To reduce variations due to instrumental instability, the metabolite intensities were corrected for receiver gain, coin loading, and normalized to the intensity of cerebrospinal fluid on each subject’s proton density MRI. To estimate metabolite concentrations separately of ‘pure’ GM and WM in the left and right frontal and parietal lobes, metabolite intensities were regressed against the tissue composition within each MRSI voxel, as previously described.20 Tissue composition of the MRSI voxels was obtained by aligning the tissue-segmented MRI to the MRSI data and blurring MRI to the MRSI resolution. Because T1 and T2 relaxation times of the metabolites could not be measured due to prohibitively long acquisition times, metabolite concentrations are expressed in arbitrary units rather than in mmol/L.
Statistics
Differences in demographic data were tested using Student t test or χ2 test, as appropriate. Metabolites (from 1H MRSI) were analyzed within a linear model, accounting for the effects of diagnosis and WMH, which was expressed as an index for each subject, defined as the volumes of all regions classified as WMH on MRI divided by the total volume of normal appearing WM and WMH. Age, sex, WMH index, and voxel tissue composition of MRSI were added into the model as covariates. F tests were used to determine if factors added explanatory power and were therefore appropriate for inclusion in the model. Three tested models were fitted by maximum likelihood: the first (base) model included all covariates plus the WMH index and the subsequent models added a diagnosis effect and then a further diagnosis by WMH interaction, respectively. The resulting fits were compared sequentially via F tests to determine whether diagnosis, WMH, or a diagnosis by WMH interaction added significant (P<0.05) explanatory power to the base model. No corrections for multiple comparisons were applied for the tests involving frontal and parietal WM, as the primary hypothesis of a diagnosis effect related to both regions. Regional variations were tested using repeated measures analysis of variance and post hoc Scheffe tests. An α of 0.05 was used as level of significance for all tests.
RESULTS
Demographic, clinical, and WM lesions data are displayed in Table 1. AD patients and control subjects were of similar age (P>0.05) whereas MCI patients were on average slightly older than the control subjects (P=0.05) but of similar age than AD patients (P>0.05). MMSE scores differed between each group (P<0.05), as expected. WM lesion index also differed between the groups. On average, both AD (P=0.05) and MCI (P=0.03) patients exhibited more WMH than control subjects (Table 1). The difference in WM lesion index between AD and MCI patients was not significant. Results of regional mIn differences between the groups are summarized in Table 2 and regional NAA differences in Table 3.
TABLE 1.
Demographics, Clinical Data, and White Matter Lesions
| Parameters | MCI | AD | Control |
|---|---|---|---|
| No. subjects | 14 | 17 | 16 |
| Women/men | 6/8 | 7/10 | 11/5 |
| Age, y±SD | 77.1±6.3 | 74.5±7.7 | 72.5±5.2 |
| MMSE±SD | 27.6±1.5 | 21.4±5.4 | 29.5±0.9 |
| WML index | 0.05±0.02 | 0.06±0.04 | 0.04±0.01 |
AD indicates Alzheimer disease; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination; SD, standard deviation, WML index, white matter lesions volume/(white matter volume+white matter lesions volume).
TABLE 2.
Myo-inositol Concentrations* in Frontal and Parietal Lobes in Control, Mild Cognitive Impairment, and Alzheimer Disease Patients
| Group | Region | Gray Matter | White Matter |
|---|---|---|---|
| Control | Right frontal lobe | 14.34 (3.58) | 15.84 (2.58) |
| MCI | Right frontal lobe | 13.87 (3.18) | 16.91(2.18)# |
| AD | Right frontal lobe | 15.56 (3.16)** | 18.59 (2.6)‡‡ |
| Control | Left frontal lobe | 14.91 (3.96) | 16.97 (2.65) |
| MCI | Left frontal lobe | 15.19 (3.01) | 18.43 (2.66)# |
| AD | Left frontal lobe | 16.70 (3.03)*** | 20.88 (3.99) ‡‡ |
| Control | Right parietal lobe | 14.87 (3.99) | 16.59 (2.15) |
| MCI | Right parietal lobe | 14.97 (2.05) | 18.70 (2.59)†† |
| AD | Right parietal lobe | 15.08 (2.83) | 19.80 (2.95) ‡‡ |
| Control | Left parietal lobe | 14.55 (3.67) | 14.52 (2.00) |
| MCI | Left parietal lobe | 15.28 (2.07) | 15.57 (1.85) † |
| AD | Left parietal lobe | 15.19 (2.54) | 16.17 (3.16)‡‡ |
Concentrations in arbitrary units, listed are mean and standard deviation in parenthesis.
P=0.058 MCI versus control.
P<0.04 MCI versus AD.
P<0.05 AD versus control.
P<0.05 MCI versus control.
P<0.05 AD versus control.
P=0.09 AD versus control.
AD indicates Alzheimer disease; MCI, mild cognitive impairment.
TABLE 3.
N-acetylaspartate Concentrations* in Frontal and Parietal Lobes in Control, Mild Cognitive Impairment, and Alzheimer Disease
| Group | Region | Gray Matter | White Matter |
|---|---|---|---|
| Control | Right frontal lobe | 24.68 (4.13) | 29.36 (4.55) |
| MCI | Right frontal lobe | 22.05 (4.89) | 29.02 (4.14) |
| AD | Right frontal lobe | 22.48 (4.39) | 29.53 (4.17) |
| Control | Left frontal lobe | 26.59 (4.81) | 30.31 (4.98) |
| MCI | Left frontal lobe | 25.35 (5.41) | 29.74 (5.50) |
| AD | Left frontal lobe | 26.31 (4.19) | 30.31 (6.03) |
| Control | Right parietal lobe | 27.85 (5.17) | 29.47 (3.51) |
| MCI | Right parietal lobe | 25.81 (5.06)# | 29.33 (4.95) |
| AD | Right parietal lobe | 22.11 (4.48) ‡‡ | 28.94 (5.21) |
| Control | Left parietal lobe | 27.97 (4.83) | 26.11 (3.25) |
| MCI | Left parietal lobe | 25.35 (4.93) | 25.50 (4.87) |
| AD | Left parietal lobe | 23.12 (4.91) | 24.66 (3.74) |
Concentration in arbitrary units, listed are mean and standard deviation in parenthesis.
P=0.06 AD versus control.
P=0.09 AD versus MCI.
AD indicates Alzheimer disease; MCI, mild cognitive impairment.
After accounting for age, sex, and WMH index, MCI patients showed in comparison to controls higher mIn concentrations (F3,26=4.5, P<0.05) in the right parietal WM and a trend in left parietal WM (F3,26=3.9, P=0.058), whereas differences in the frontal WM and in GM were not significant (all P>0.2). In addition, higher mIn concentration of right parietal WM in MCI was markedly different from that in frontal WM (F5,50=5.8, P<0.002), whereas left and right mIn differences in parietal WM were not significant. Surprisingly, the mIn levels of parietal WM in MCI were not significantly different from those in AD (P>0.13). However, mIn levels of frontal WM in MCI were markedly lower than those in AD (right: F3,27=4.8 P<0.04; left: F3,27=4.8 P<0.04). AD patients had in comparison to controls substantially higher mIn concentrations in both frontal and parietal WM (right frontal: F3,29=9.9, P<0.004; left frontal: F3,29=13.6, P<0.001; right parietal: F3,29=12.6, P<0.002; left parietal: F3,29=7.0, P<0.02). AD patients had also higher mIn concentrations in frontal GM compared with controls, primarily on the right side (F3,29=4.4, P<0.05) and a trend on the left side (F3,29= 3.3, P=0.09), but no significant elevations of mIn in parietal GM. Within the AD group, mIn concentration was higher in frontal WM than in parietal WM (F5,62=9.5, P<0.001), whereas regional variations of mIn in GM were not significant (P>0.3) in AD.
In contrast to mIn, NAA concentrations in WM were not significantly associated with diagnosis. Specifically, MCI subjects and AD patients had similar NAA concentrations in WM. In contrast, NAA concentration in right parietal GM showed the trend to be lower in AD than in controls (F3,29=3.7, P=0.06) and MCI (F3,29=2.9, P=0.09) (Table 3). Differences in NAA concentrations in GM between MCI and controls were also not significant. Finally, mIn and NAA variations were not significantly associated with WMH in any of the groups (P>0.5 for all tests).
DISCUSSION
The major finding of our study is increased mIn of WM in MCI and AD whereas NAA differences in WM were not significant. Furthermore, the mIn increase in MCI primarily affected parietal lobe WM, as predicted, whereas the mIn increase in AD involved both frontal and parietal lobe WM.
Vascular pathology, as assessed by WMH, did not explain increased mIn in MCI and AD.
Taken together, these results suggest that increased mIn may be an even more robust and sensitive indication for AD pathology than NAA and furthermore could have value as marker of early WM alterations in MCI.
The finding of increased mIn concentration in parietal WM in patients with MCI is consistent with the histopathologic findings of demyelination and increased gliosis in this group.28 MRS studies reported increased mIn concentrations in acquired immunodeficiency syndrome29 and in subacute sclerosis penecephalilties,30 which has been associated with inflammation. Another MRS study involving histopathology reported increased mIn concentration in MS, which furthermore correlated with the histopathologic findings of inflammation and demyelination.8 Therefore, increased mIn in MCI may indicate pathologic processes that cause disintegration of WM fibers and/or activation of glial cells. On the basis of the results from biochemical studies of inflammation, we speculate that elevation of mIn in MCI may be a biochemical manifestation of WM inflammation and degradation. In the MCI stage, however, the most affected region seems to be the parietal lobe, while frontal WM is spared. The transition from MCI to AD may therefore be accompanied by a spread of inflammation and WM damage from parietal to frontal lobe regions, though the mechanism for a regional dispersion remains unclear. Moreover, we showed that regional mIn abnormalities exist in both MCI and AD, irrespective of accompanying vascular pathologies, expressed as WMH. Although not all MCI subjects in our study may develop AD, it is well documented that MCI subjects have a higher prevalence of AD than cognitive normal subjects at the same age.10 We, therefore cautiously interpret the mIn findings in MCI as marker of early AD. Support for this view comes also from the observations of higher mIn concentration in both parietal and frontal WM in AD than in MCI.
Several theories exist concerning WM damage in AD. First, WM injury could result from anterograde Wallerian degeneration. Second, WM damage might also be induced by interruption of axonal transport processes due to the presence of neurofibrillary tangle or direct deposition of amyloid in WM. Finally, degeneration of WM could be a reaction to dysregulations of proinflammatory and anti-inflammatory activity in brain cytoskeleton.7,31,32 Differences in inflammation could help explain variable conversion to AD. It has been shown that atypical activated microglial expression and an anti-inflammatory profile can be turned into aggressive proinflammatory cells as a consequence of systemic infection.33 One might speculate that inflammatory factors are greater in those MCI patients who convert to AD.
Although increased mIn in MCI and AD has previously been reported,16,34–36 we could demonstrate more rigorously using MRSI that the abnormal elevation of mIn concentrations exhibits a systematic regional pattern. In MCI, increased mIn occurred predominantly in parietal WM while sparing frontal WM. In AD, increased mIn was found throughout WM, consistent with another MRSI study from this laboratory.18 Note, that increased mIn concentration can be interpreted as disproportionate demyelinization and/or gliosis of brain tissue in contrast to proportionate changes that should cause no change in mIn concentration. Hence, the finding of greater mIn alterations in WM than GM in MCI and AD may reflect a differential effect of demyelination/gliosis on WM. To account for potential contributions of vascular pathology to the metabolites changes, we included volume of WM lesions as confound in the analysis, but this did not alter the results implying that vascular disease cannot completely explain mIn variations.
In agreement with the paper by Kantarci et al,37 which reported increased mIn/Cr of the posterior cingulate in both AD and MI, we also found increased mIn in the parietal cortex of AD, which included the cingulate. However, we did not find increased mIn in MCI. Technical differences between our MRS method and that used by Kantarci et al may explain the discrepancy. Whereas our approach allowed estimating the concentration of mIn in ‘pure’ GM, Kantarci et al, used mIn/Cr ratios, which are intrinsically less conclusive, as mIn and Cr may both induce changes. Furthermore, Kantarci et al did not account for partial GM and WM volumes, which can mimic metabolite changes, whereas our method accounted for GM/WM variations. In summary, our results of increased mIn of parietal GM in AD agrees with the findings by Kantarci et al whereas the discrepancy for MCI may be related to technical differences between the studies that may become more relevant when mIn changes are subtle.
An interesting observation is that we did not find significant reductions of NAA in WM in both MCI and AD. Although previous MRSI studies, including from this laboratory18 reported significant NAA reductions in AD, these differences were usually relatively small in the order of 10%. Furthermore, these effects were region-specific, which may potentially vary dependent on the heterogeneity of the patient population. These factors in combination with the notorious technical difficulties of short TE MRSI may explain the discrepant findings. However, other MRS studies have also reported lack of significant NAA differences between MCI and AD in WM.38 Furthermore, another MRS study found no evidence for decreased NAA in WM AD compared with normal aging.39 Additionally, the lack of NAA WM changes in the presence of substantial mIn changes in MCI and AD patients is surprising as it suggests that NAA and mIn alterations in MCI and AD can be dissociated from each other. A similar dissociation between mIn and AD was also recently reported for AD patients.18 One explanation for the dissociation between mIn and NAA in WM is that glial changes may precede neuronal damage and therefore could be independent of neuronal damage in AD pathology. Moreover, electron microscopy study have shown an activation of glial cells in response to myelin damage but in the absence of the axonal damage.40 Decrease of NAA in parietal GM in AD patients, though small, is similar to previous finding18,41 and is consistent with pathologic and positron emission tomography studies that parietal lobe is an important site of AD pathology1,42 We also found slightly reduction of NAA in parietal GM in MCI patients, which may represent an early stage of AD pathology. Reduction of NAA concentration in GM while NAA levels in WM are normal may reflex very selective damage of neurons in AD pathology. Decrease of NAA in GM may be attributed to an impairment of metabolism or dysfunction selectively of cells body while the structure and function of axons are preserved. This observation supports the theory of GM-WM disconnection in AD pathology.
Although our results are presented in terms of brain lobes and not more regionally specific, the advantage of our approach is that partial volume effects of GM and WM on metabolite concentrations can be eliminated by evaluating metabolite variations as a function of tissue type, as described in more details,20 in contrast to the analysis of small regions which do not provide sufficient variations to eliminate partial volume effects.
This study has several limitations: AD was based on clinical diagnosis and not confirmed by autopsy. Thus, other age-related neuropathologies, especially microinfarcts that are difficult to detect on MRI may have contributed to metabolite changes in AD. Similarly, MCI was diagnosed based on clinical criteria and subjects were not followed longitudinally to determine if MRSI findings in MCI were related to conversion to AD. Another major limitation of this study is that the groups were not completely matched with respect to age and WML. Although we attempted to statistically control for a potential confounding effect of age and WML, we cannot fully exclude the possibility that age and WML influenced metabolite variations between groups.
In conclusion, this study suggests vulnerability of WM in the pathology of AD and MCI. Furthermore, increased mIn may be an even more robust and sensitive marker for MCI and AD than decreased NAA. Moreover, the dissociation between mIn and NAA alterations could provide important information regarding the role of glial and neuronal damage in MCI and AD.
Acknowledgments
Małgorzata Siger was supported by a grant from Polish Foundation of Science.
The authors thank Diana Sacrey and Shannon Buckley for MRI and MRSI scanning and Dr David Norman for evaluation of MRI scans.
References
- 1.Bartzokis G, Sultzer D, Lu PH, et al. Heterogenous age-related breakdown of white matter structural integrity: implication for cortical ‘disconnection’ in aging and Alzheimer’s disease. Neurobiol Aging. 2004;25:843–851. doi: 10.1016/j.neurobiolaging.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Braak E, Griffing K, Arai K, et al. Neuropathology of Alzheimer’s disease: what is new since A Alzheimer? Eur Arch Psychiatry Clin Neurosci. 1999;249:14–22. doi: 10.1007/pl00014168. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, Schu N, Jahng GH, et al. Diffusion tensor imaging of cingulum fibres in mild cognitive impairment and Alzheimer disease. Neurology. 2007;68:13–19. doi: 10.1212/01.wnl.0000250326.77323.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stahl R, Dietrich O, Teipel SJ, et al. White matter damage in Alzheimer disease and mild cognitive impairment: assessment with diffusion-tensor MR imaging and parallel imaging techniques. Radiology. 2007;243:483–492. doi: 10.1148/radiol.2432051714. [DOI] [PubMed] [Google Scholar]
- 5.Schu N, Amend D, Ezekiel F. Changes of hippocampal N-acetyl aspartate and volume in Alzheimer’s disease as assessed by multislice proton magnetic resonance spectroscopic imaging. Neurology. 1997;49:1513–1521. doi: 10.1212/wnl.49.6.1513. [DOI] [PubMed] [Google Scholar]
- 6.Ross BD, Bluml S, Cowan R, et al. In vivo MR spectroscopy of human dementia. Neuroimaging Clin N Am. 1998;8:809–822. [PubMed] [Google Scholar]
- 7.Minghetti L. Role of inflammation in neurodegenerative diseases. Curr Opin Neurol. 2005;18:315–321. doi: 10.1097/01.wco.0000169752.54191.97. [DOI] [PubMed] [Google Scholar]
- 8.Bitch A, Bruhn H, Vougioukas V, et al. Inflammatory CNS demyelination: histopathology correlation with in vivo quantitative proton MR spectroscopy. Am J Neuroradiol. 1999;20:1619–1627. [PMC free article] [PubMed] [Google Scholar]
- 9.Sämann PG, Schlegel J, Müller G, et al. Serial proton MR spectroscopy and diffusion imaging findings in HIV-related herpes simplex encephalitis. AJNR Am J Neuroradiol. 2003;24:2015–2019. [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen RC, Smith GE, Waring SC, et al. Mild cognitive impairment. Clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 11.Morris JC, Storandt M, Miller JP, et al. Mild cognitive impairment represent early stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- 12.Whitwell JL, Petersen RC, Negash S, et al. Patterns of atrophy differ among specific subtypes of mild cognitive impairment. Arch Neurol. 2007;64:1130–1138. doi: 10.1001/archneur.64.8.1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Es AC, van der Flier WM, Admiraal-Behloul F, et al. Magnetization transfer imaging of gray and white matter in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2006;27:1757–1762. doi: 10.1016/j.neurobiolaging.2005.09.042. [DOI] [PubMed] [Google Scholar]
- 14.Kontarci K, Weigand SD, Petersen RC, et al. Longitudinal (1)H MRS changes in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging. 2007;28:1330–1339. doi: 10.1016/j.neurobiolaging.2006.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medina D, de Toledo-Morrell L, Urresta F, et al. White matter changes in mild cognitive impairment and AD: a diffusion tensor imaging study. Neurobiol Aging. 2006;27:663–672. doi: 10.1016/j.neurobiolaging.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 16.Catani M, Cherubini A, Howard R, et al. H-MR spectroscopy differentiate mild cognitive impairment from normal brain aging. Neuroreport. 2001;12:2315–2317. doi: 10.1097/00001756-200108080-00007. [DOI] [PubMed] [Google Scholar]
- 17.Metastasio A, Rinaldi P, Tarducci R, et al. Conversion of MCI to dementia: role of proton magnetic resonance spectroscopy. Neurobiol Aging. 2006;27:926–932. doi: 10.1016/j.neurobiolaging.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Zhu X, Schu N, Kornak J, et al. Effects of Alzheimer disease on fronto-parietal brain N-acetyl aspartate and myo-inositol using magnetic resonance spectroscopic imaging. Alzheimer Dis Assoc Disord. 2006;20:77–85. doi: 10.1097/01.wad.0000213809.12553.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spielman DM, Adalsteinsson E, Lim KO. Quantitative assessment of improved homogeneity using higher-order shims for spectroscopic imaging of the brain. Magn Reson Med. 1998;40:376–382. doi: 10.1002/mrm.1910400307. [DOI] [PubMed] [Google Scholar]
- 20.Schu N, Ezekiel F, Gamst AC, et al. Region and tissue differences of metabolites in normally aged brain using multislice 1H magnetic resonance spectroscopic imaging. Magn Reson Med. 2001;45:899–907. doi: 10.1002/mrm.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiedermann D, Schu N, Matson GB, et al. Short echo time multislice proton magnetic resonance spectroscopic imaging in human brain: metabolite distribution and reliability. Magn Reson Imaging. 2001;19:1073–1080. doi: 10.1016/s0730-725x(01)00441-6. [DOI] [PubMed] [Google Scholar]
- 22.Zhu XP, Young K, Soher JB, et al. New spectral analysis of short echo time multislice 1H MRSI in human brain using Eigen spectra, baseline correction and frequency alignment. Proceedings of the 13th Annual Meeting of ISMRM; Miami Beach, USA. 2005. p. 55. [Google Scholar]
- 23.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 24.Folstein MF, Folstein SE, McHugh PR. ‘Mini mental state’ A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 25.Haupt CI, Schu N, Weiner MW, et al. Removal of lipid artifacts in 1H spectroscopic imaging by data extrapolation. Magn Reson Med. 1996;35:678–687. doi: 10.1002/mrm.1910350509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maudsley AA, Lin E, Weiner MW. Spectroscopic imaging display and analysis. Magn Reson Imaging. 1992;10:471–485. doi: 10.1016/0730-725x(92)90520-a. [DOI] [PubMed] [Google Scholar]
- 27.Soher BJ, Young K, Gavindaraju V, et al. Automated spectra analysis III: application to in vivo proton MR spectroscopy and spectroscopic imaging. Magn Reson Med. 1998;40:822–831. doi: 10.1002/mrm.1910400607. [DOI] [PubMed] [Google Scholar]
- 28.Petersen RC, Parisi JE, Dickson DW, et al. Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol. 2006;63:665–672. doi: 10.1001/archneur.63.5.665. [DOI] [PubMed] [Google Scholar]
- 29.Menon DK, Baudouin CJ, Tomlinson D, et al. Proton MR spectroscopy and imaging of the brain in AIDS: evidence of neuronal loss in regions that appear normal with imaging. J Comput Assist Tomogr. 1990;14:882–885. doi: 10.1097/00004728-199011000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Aydin K, Tatli B, Ozkan M, et al. Quantification of neurometabolites in subacute sclerosing panencephalitis by 1H-MRS. Neurology. 2006;67:911–913. doi: 10.1212/01.wnl.0000233836.16266.f5. [DOI] [PubMed] [Google Scholar]
- 31.Englund E. Neuropathology of white matter changes in Alzheimer’s disease and vascular dementia. Dement Geriatr Cogn Disord. 1998;9:6–12. doi: 10.1159/000051183. [DOI] [PubMed] [Google Scholar]
- 32.Frisoni G, Filippi M. Multiple sclerosis and Alzheimer disease through the looking glass of MRI imaging. Am J Neuroradiol. 2005;26:2488–2249. [PMC free article] [PubMed] [Google Scholar]
- 33.Combrinck M, Perry VH, Cunningham C. Peripheral infection evokes exaggerated sickness behaviour in pre-clinical murine prion disease. Neuroscience. 2002;112:7–11. doi: 10.1016/s0306-4522(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 34.Moats RA, Ernst T, Shonk TK, et al. Abnormal cerebral metabolite concentration in patients with probable Alzheimer disease. Magn Reson Med. 1994;32:110–115. doi: 10.1002/mrm.1910320115. [DOI] [PubMed] [Google Scholar]
- 35.Parnetti L, Tarducci R, Presciutti O, et al. Proton magnetic resonance spectroscopy can differentiate Alzheimer’s disease from normal aging. Mechan Ageing Dev. 1997;97:9–14. doi: 10.1016/s0047-6374(97)01877-0. [DOI] [PubMed] [Google Scholar]
- 36.Salvan AM, Ceccaldi M, Confort-Gouny S, et al. Correlation between cognitive status and cerebral inositol in Alzheimer-type dementia. J Neurol. 1998;245:686–688. doi: 10.1007/s004150050268. [DOI] [PubMed] [Google Scholar]
- 37.Kantarci K, Jack CR, Xu YC, et al. Regional metabolic patterns in mild cognitive impairment and Alzheimer’s disease. A 1H MRS study. Neurology. 2000;55:210–217. doi: 10.1212/wnl.55.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Falini A, Bozzali M, Magnani G, et al. A whole brain MR spectroscopy study from patients with Alzheimer’s disease and mild cognitive impairment. Neuroimage. 2005;26:1159–1163. doi: 10.1016/j.neuroimage.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Stoppe G, Bruhn H, Pouwels PJ, et al. Alzheimer disease: absolute quantification of cerebral metabolites in vivo using localized proton magnetic resonance spectroscopy. Alzheimer Dis Assoc Disord. 2000;14:112–119. doi: 10.1097/00002093-200004000-00009. [DOI] [PubMed] [Google Scholar]
- 40.Delacourte A, Davis JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurology. 1999;52:1158–1165. doi: 10.1212/wnl.52.6.1158. [DOI] [PubMed] [Google Scholar]
- 41.Schu N, Capizzano AA, Du AT, et al. Selective reduction of N-acetyl aspartate in medial temporal and parietal lobes in AD. Neurology. 2002;58:928–935. doi: 10.1212/wnl.58.6.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoffman JM, Welsh-Bohmer Ka, Hanson M, et al. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med. 2000;41:1920–1928. [PubMed] [Google Scholar]