

Abstract
T cell-mediated adaptive immune response is controlled by both positive costimulation and negative coinhibition, generated mainly by the interaction between the B7 family and their receptor CD28 family. Coinhibition is exploited by prostate cancer as an immune evasion pathway. Overexpression of coinhibitory B7x and B7-H3 in prostate cancer correlates with poor disease outcome, whereas tumor-infiltrating immune cells have enhanced expression of PD-L1 and its receptor PD-1. New insights into the complex mechanisms governing B7 expression in the tumor microenvironment have been reported and therapies aimed at overcoming T cell coinhibition with antagonistic monoclonal antibodies are emerging as effective tumor immunotherapies. Therapies that block B7x and B7-H3, either as monotherapies or in synergism with traditional therapies, should be pursued.
Keywords: T cell coinhibition, prostate cancer, immune evasion, B7x, B7-H3, PD-L1, anti-CTLA-4 therapy, anti-PD-1 therapy, signaling pathway, microRNA
T cell-based immunotherapy in prostate cancer
Prostate cancer is a serious public health problem. It is the most common invasive cancer among men and the second and third leading cause of cancer-related death in males in the U.S. and Europe [1, 2], respectively. Standard treatment for prostate cancer includes surgery, radiation, hormonal therapy and chemotherapy in various combinations. Although treatment usually eliminates the primary tumor, it has been largely unsuccessful in controlling metastatic disease and is often accompanied by negative side effects [3]. New therapeutic strategies are therefore needed. A promising new approach in metastatic prostate cancer treatment is T cell-based immunotherapy, a varied group of treatments attempting to combat cancer by initiating or augmenting T cell responses against tumor cells.
The majority of T cell immunotherapies are vaccines derived from tumor associated antigens (TAAs) and have been designed to prime T cells to respond to TAA-expressing tumor cells. Several TAAs have been identified in prostate cancer, making prostate cancer a prime candidate for immunotherapy. Among these are prostate specific antigen (PSA) [4],prostate-specific membrane antigen (PSMA) [5], prostatic acid phosphatase (PAP) [6] and prostate stem cell antigen (PSCA) [7]. Early prostate cancer vaccines consisted of purified TAA peptides and adjuvant [8, 9], but in an effort to increase immunogenicity, most current strategies deliver these peptides in viral vectors or plasmids, presented on dendritic cells (DCs) or as genetically modified whole tumor cells [10].
Some prostate cancer vaccines have been in development for over a decade and are just now maturing for use in the clinic. The first and currently only FDA-approved vaccine is a DC-based vaccine known as sipuleucel-T (Provenge; Dendreon Seattle, WA, US) [11]. As DCs are professional antigen presenting cells (APCs) capable of cross-presenting TAAs to cognate T cells, several vaccines have been developed using DCs pulsed in vitro with TAA-derived peptides. In sipuleucel-T, monocytes from a patient's blood are differentiated into mature DCs, cultured with a fusion protein containing PAP and the proinflammatory cytokine granulocyte-macrophage colony-stimulating factor (GM-CSF) and then returned to the patient as PAP-presenting DCs [12]. This personalized vaccine modestly increases survival in patients with metastatic prostate cancer [11].
Other vaccines use viral vectors encoding TAAs. The most advanced development in virus-based vaccines is PROSTVAC-VF. It consists of a vaccinia vector encoding both PSA and a triad of costimulatory molecules as well as a fowlpox vector as a booster encoding the same molecules [13]. Phase II trials have been completed for PROSTVAC-VF [14, 15] and a phase III trial is being planned [10]. In addition to viral vectors, plasmids encoding PSMA [16], PSA [17] and most recently PAP [18] have completed phase I/II trials, demonstrating safety but modest efficacy. Another approach uses whole tumor cells to supply tumor antigens. GM-CSF-gene transduced allogenic prostate cancer immunotherapy (GVAX), a vaccine composed of prostate cancer cells engineered to express GM-CSF, showed promise in phase II trials [19] but did not successfully complete phase III trials [10].
Although advances in tumor vaccines are encouraging, progress has been slow and clinical benefit in terms of survival and tumor regression has been limited. Despite its promise, immunotherapy shares the same challenges that prevent the immune system from eliminating cancer on its own, namely, cancer immune evasion. By the time a tumor is established, the cells have usually evolved multiple immune evasion mechanisms that allow them to escape the immune responses generated by the body as well as those employed in immunotherapy. Although many cancer vaccines are designed to bolster tumor antigen presentation, most do not address the immune evasion pathways that suppress T cell function.
T cell coinhibition as immune evasion mechanisms in prostate cancer
Prostate cancer can escape immune responses via a variety of mechanisms [20]. A prime example is defective antigen presentation. Although tumor cells can theoretically present TAAs with major histocompatibility complex (MHC) class I molecules, the expression of these MHC molecules and antigen processing machinery are often downregulated, allowing tumors to hide their malignant identity from immune cells. This loss of expression has been noted in some human prostate cancer cell lines and in primary tumor tissues [21, 22]. The immunosuppressive environment surrounding prostate tumors, marked by increased levels of nitric oxide synthase and arginase [23, 24], the anti-inflammatory cytokines interleukin-10 (IL-10) [25] and transforming growth factor β (TGF-β) [26] as well as the infiltration of suppressor cell populations such as FoxP3+ regulatory T cells (Tregs) [27], also contributes to immune escape.
Another immunosuppressive process targets T cell activation and function directly. A prerequisite for an effective T cell response, T cell activation requires two signals: binding of the T cell receptor (TCR) by a cognate peptide presented on the MHC of an APC and a costimulatory signal, mainly generated between members of the B7 ligand family on the APC and the CD28 receptor family on the T cell. By contrast, coinhibitory signaling between these two families acts to downgrade T cell activation, resulting in T cell exhaustion, deletion, or anergy/tolerance (Box 1). In the tumor environment, the balance in T cell activation and function is often skewed towards coinhibition [28]. Coinhibitory ligands, such as PD-L1/B7-H1(programmed death-ligand 1 or B7 homolog 1), B7-H3 and B7x (B7-H4 or B7S1) are frequently upregulated within the tumor microenvironment [28]. Costimulatory signaling might also be decreased owing to loss of costimulatory molecules or increased competition from coinhibitory signaling [28]. These immune evasion mechanisms appear to play a prominent role in prostate cancer immunity.
The PD-L1/PD-1 pathway in prostate cancer
PD-L1/PD-1-mediated T cell coinhibition is involved in immune evasion in prostate cancer. PD-1 (programmed death 1) has two ligands, PD-L1 [30, 31] and PD-L2 (B7-DC) [32, 33] and functions to maintain peripheral tolerance; PD-1 knockout mice suffer from spontaneous autoimmune diseases [34, 35]. This coinhibitory pathway also appears to play a role in prostate cancer. PD-1 is upregulated on immune cell clusters surrounding prostate cancer lesions but not in healthy prostate or benign hyperplastic prostate [36]. These immune cells include both CD4 and CD8 T cells which lack perforin and interferon-γ (IFN-γ) expression [37], and thus appear quiescent. A high percentage of prostate tumor-infiltrating CD8 T cells, close to 90% in some patients, express PD-1 [38]. Interestingly, these CD8 T cells exhibit restricted TCR Vß gene usage [38], indicating only a limited number of T cell clones infiltrate and/or expand in prostate cancer, a phenomenon that might be linked to PD-1 expression.
The involvement of the PD-L1/PD-1 pathway in prostate cancer immunity is further supported by reports of PD-L1 expression within prostate tumors. In healthy tissue, PD-L1 protein is limited to monocytes in peripheral blood and macrophages in the lungs, liver and tonsils [39]. By contrast, PD-L1 protein is highly expressed in some cancers and might correlate with negative clinical outcome [28]. Although PD-L1 has been reported in fewer than 1% of prostate tumor samples [40], one study found that although prostate cancer cells were PD-L1-negative, they were surrounded by PD-1-positive and PD-L1-positive immune cells [36]. A separate study reported PD-L1 in some prostate cancer cell lines and a limited number of prostate cancer samples [41]. Expression correlated with activated phosphoinositide 3-kinase (PI3K) [41], suggesting a relationship between PD-L1 expression in prostate tumors and the PI3K signaling pathway.
PD-L1/PD-1 signaling is now well established as a T cell coinhibitory pathway and both PD-1 and PD-L1 have been found in the prostate cancer microenvironment; it is therefore likely that these molecules play a role in downgrading antitumor immunity. Prostate cancer-infiltrating immune cells, more so than the cancer cells, express high levels of PD-L1 and PD-1, suggesting that T cell suppression might occur mainly in the context of signaling between APCs cross-presenting tumor antigens rather than through direct signaling by tumor cells. Prostate cancer-infiltrating CD8 T cells have restricted TCR Vß gene usage, indicating that only a few dominant tumor antigens are able to induce specific CD8 T cell clonal expansion, perhaps as a consequence of immune evasion, but even these cells might be incapable of mounting an effective antitumor immune response owing to their high levels of PD-1 and PD-L1. Although prostate tumors are surrounded by Tregs [36] and PD-L1 signaling can aid the development and function of induced Tregs [42], it is currently unknown if PD-L1/PD-1 signaling is linked to Treg induction or expansion in prostate cancer. Further research will help to reveal clinical associations between PD-L1/PD-1 signaling and prostate cancer progression and how this pathway controls dynamic interactions between tumors and the immune system.
B7-H3 and B7x in prostate cancer
B7-H3 and B7x, the most recently discovered members of the B7 family, are also key players in prostate cancer immunity. B7-H3 has low expression in many normal lymphoid and peripheral tissues but is elevated in many cancers [28, 43]. B7-H3 binds activated T cells, leading to costimulation in some cases and to coinhibition in others [43, 44] (Box 2). The function of B7-H3 in cancer is controversial as well, but the majority of clinical data reveal a positive correlation with tumor progression, suggesting that coinhibitory signaling might be dominant in this context [45-49] (Box 2). Also highly expressed in cancer is B7x. Although B7x mRNA is present in a variety of lymphoid and nonlymphoid tissues [29, 50], protein expression is scarce in healthy human tissue but abundant in malignancy, including cancer of the breast [51, 52], ovary [45, 52], lung [53], kidney [54], brain [55] and pancreas [56]. An unambiguously coinhibitory ligand, binding of B7x to its putative receptor on activated T cells suppresses T cell proliferation and cytokine production [29, 50, 57], and is likely exploited by cancer cells to evade T cell immune responses.
The presence and coinhibitory effects of B7-H3 and B7x in prostate cancer were highlighted by a large study of 823 prostate cancer patients treated with radical retropubic prostatectomy (RRP) [63]. Although B7-H3 is weakly expressed in normal prostate epithelium [40], intensified B7-H3 as well as widespread B7x expression were found in prostate cancer with 93% and 99% of tumors expressing B7-H3 and B7x, respectively [63]. Strong B7-H3 and B7x staining was found in 26% and 15% of patients (Figure 1), respectively, and seven years of patient follow-up data revealed that patients with high expression of either ligand were significantly more likely to have disease spread at the time of surgery, cancer recurrence and death from cancer [63]. In a separate study of 338 men treated with RRP, B7-H3 was found in 100% of prostate adenocarcinoma samples and the level of expression significantly correlated with pathological features [40]. 19% of samples tested had high intensity B7-H3 staining, conferring a greater than four-fold risk of cancer progression after surgery [40]. The similarity in the findings of these two independent studies reinforces the significance of B7-H3 in prostate cancer. As for B7x, clinical observations in prostate cancer are limited to the 823 patient study, but the pathological associations of B7x in this study are consistent with clinical data from kidney cancer [54]. In both prostate and kidney cancer, B7x is abundant on tumor cells, as well as tumor vasculature in the case of kidney cancer, but is not detectable on tumor-infiltrating immune cells [54, 63].
Figure 1. T cell coinhibition is exploited by prostate cancer as immune evasion pathways.
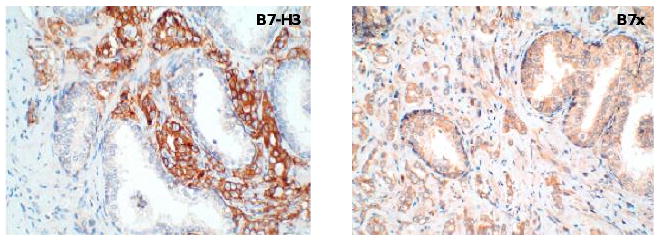
T cell coinhibitory B7 family members B7x and B7-H3 are overexpressed by prostate cancer cells as revealed by immunohistochemical staining. Tissue microarrays of human prostate cancer were stained with antibodies against B7-H3 (left) or B7x (right). B7-H3 and B7x bind unidentified receptors on activated T cells to downregulate TCR-mediated signaling, which might contribute to poor clinical outcome. In addition, many prostate cancer patients have soluble B7 molecules in the blood, but the mechanism of production and the function of soluble B7 are currently unknown
Key among the pathological features associated with both B7-H3 and B7x in prostate cancer is metastasis. Intense staining of either ligand in primary tumor correlates with extracapsular extension, seminal vesicle invasion, and nonorgan-confined disease [40]; primary tumor B7x intensity correlates with positive lymph nodes [63], and bone metastases express B7-H3 [49], suggesting B7-H3 and B7x could be biomarkers of metastasis. B7-H3 might be a valuable prognostic marker for additional reasons. B7-H3 levels within the primary tumor and in bone metastases are largely unaffected by neoadjuvant hormonal therapy (NHT), suggesting that B7-H3 can persist even after NHT [49]. Furthermore, B7-H3 level in the primary tumor can predict the efficacy of salvage radiation therapy (SRT) after prostatectomy, as higher B7-H3 intensity correlates with biochemical recurrence (i.e. serum PSA incline) after SRT [67]. A link between B7-H3 and radiation was also noted in the prostate cancer cell line 22Rv1, which secreted B7-H3-laden exosomes following irradiation-induced senescence [68].
Taken together, these studies reveal a crucial role for B7-H3 and B7x in prostate cancer immunity with potential clinical applications. Patients with strong B7-H3 or B7x expression have a higher risk for metastasis and recurrence and consequently, are more likely to die from prostate cancer. Therefore, B7-H3 and B7x represent prognostic markers and therapeutic targets in primary and metastatic prostate cancer. B7-H3 and B7x most likely function through T cell coinhibition, and the identification of their receptors will provide important new insights into how these pathways work.
Regulation of the B7 family
As B7 family expression is altered with clinical impact in many cancers, the question of how the tumor microenvironment induces these changes is a subject of much interest. There is no consensus regarding B7 regulation in prostate cancer; however, recent studies in a variety of cell lines have revealed a number of regulatory pathways that might be involved (Figure 2).
Figure 2. Regulatory mechanisms of PD-L1 and B7-H3 expression.

The PD-L1 promoter has binding sites for transcription factors IRF-1 and NF-κB, so IFN-γ-mediated signaling through the JAK/STAT-1/IRF-1 pathway and TLR ligand-mediated signaling through the MEK/ERK/NF-κB pathway can upregulate PD-L1 gene transcription. Signaling through the PI3K/Akt/mTOR pathway, usually inhibited by PTEN, can activate S6K1, which regulates the 5′ UTR of PD-L1. This results in recruitment of PD-L1 transcripts to polysomes and associated PD-L1 translation. By contrast, miR-513, which can be inhibited by IFN-γ signaling, prevents PD-L1 translation by binding to the PD-L1 3′ UTR. B7-H3 expression can also be enhanced by IFN-γ signaling, perhaps through blocking miR-29, which otherwise represses B7-H3 translation. It seems that different types of cells use overlapping yet divergent pathways to regulate the expression of PD-L1 and B7-H3.
PD-L1 regulation
There is a close relationship between proinflammatory cytokine signaling and the expression of B7 family members. IFN-γ can induce the expression of PD-L1 mRNA [30, 69] and protein on a variety of cell types including tumor cells [39, 70-72]. As IFN-γ production by T cells is inhibited by PD-L1 signaling [30], PD-L1 and IFN-γ are connected by a regulatory feedback loop, highlighting the role of PD-L1 in dampening the immune response. This is especially relevant in cancer, where PD-L1 overexpression on tumor cells could be a mechanism of immune evasion from cytoxic T lymphocytes (CTLs) secreting IFN-γ.
IFN-γ can regulate PD-L1 at the level of transcription by initiating the synthesis of interferon regulatory factor-1 (IRF-1), a transcription factor that has two binding sites on the PD-L1 promoter [72], via the JAK (Janus kinase)/STAT (signal transducers and activators or transcription) pathway. The MyD88/MEK/ERK (MyD88/mitogen-activated protein kinase kinase/extracellular signal-regulated kinase) pathway also can result in downstream PD-L1 transcription and can be initiated both by IFN-γ and Toll-like receptor (TLR) ligands [73-75]. The PD-L1 promoter also contains a binding site for nuclear factor (NF)-κB, a transcription factor involved in PD-L1 transcription in at least one cell type [70].
PI3K is another important pathway in PD-L1 regulation, particularly in prostate cancer [41, 73, 76, 77]. PD-L1 expression on human prostate cancer cells depends on PI3K activation, either owing to loss of tumor suppressor phosphatase and tensin homolog (PTEN) or other mutations [41]. Similarly, PD-L1 expression in glioma cells is linked to PTEN loss and active Akt, a kinase activated by PI3K [77]. In glioma as well as in trophoblasts, PI3K activation correlates with recruitment of PD-L1 transcripts to polysomes, leading to increased PD-L1 translation [76, 77]. S6 kinase 1 (S6K1), a translational regulator downstream of the PI3K/Akt/mTOR (mammalian target of rapamycin) pathway, is likely involved [77].
Another post-transcriptional mechanism regulating PD-L1 expression involves microRNAs (miRs). miRs regulate gene expression by binding to the 3′-untranslated region (UTR) of target mRNAs and generally decrease translation [78, 79]. Aberrant expression of miRs is common in cancer and is associated with altered gene expression, leading to tumor initiation, growth and resistance to treatment [80]. The influence of miRs on PD-L1 expression was demonstrated in human cholangiocytes, where stimulation with IFN-γ both increased PD-L1 and changed the expression of miRs, one of which, miR-513, is complementary to the PD-L1 3′-UTR. miR-513 repressed the translation of PD-L1, whereas IFN-γ treatment decreased miR-513 and induced PD-L1 translation [70]. Thus, regulation of PD-L1 appears to result from complex interactions between environmental stimuli, intracellular signaling pathways, and both transcriptional and translational control mechanisms.
B7-H3 regulation
Proinflammatory cytokines also regulate B7-H3 in a cell type-specific manner. Treatment of colon cancer cell lines with tumor necrosis factor-α (TNF-α) did not increase the expression of membrane B7-H3 but increased B7-H3 transcription as well as the expression of soluble B7-H3 [46]. Although IFN-γ induced expression of B7-H3 in DCs [81], it did not increase B7-H3 transcription, cell surface expression or secretion in colon cancer cells [46] and decreased B7-H3 expression on bronchial and alveolar epithelial cells [71].
Similar to PD-L1, B7-H3 can also be regulated by miRs. Normal human tissues express high levels of miR-29, but miR-29 is downregulated in human tumor cell lines and biopsies of solid tumors, including sarcomas and brain tumors [82]. In all of the tumor cell lines and tissues tested, expression of B7-H3 protein was inversely correlated with miR-29 levels, and direct targeting of miR-29 to the 3′-UTR of B7-H3 and control of B7-H3 protein expression has been confirmed [82].
Additional B7 regulation
The regulation of B7 family members in general and in prostate cancer specifically is only beginning to be investigated. Most studies have focused on PD-L1 and are limited to specific cell types in an in vitro setting. It is therefore unclear how broadly the pathways identified apply and to what extent they are distinct or overlap, particularly with regard to transcriptional versus post-transcriptional control. B7-H3 studies are even more limited and regulatory mechanisms of B7x have yet to be reported. B7x has been observed within the cytoplasm and on the cell membrane in cancers including those of the prostate and the ovary [45, 63]. Because only cell-surface proteins can interact with surrounding immune cells, B7x trafficking constitutes an additional level of regulation. As PD-L1, B7-H3 and B7x might play important roles in limiting the immune responses to prostate cancer, the regulatory pathways leading to their expression might be attractive targets for therapeutic intervention. More research in this area will provide new insights into the possibility of such an approach.
Blockade of T cell coinhibition for prostate cancer therapy
Costimulation and coinhibition are critical for initiating and regulating T cell activation and function. They are altered in prostate cancer corresponding to clinical outcome [49, 63, 67], highlighting these pathways as potential targets for therapeutic design, which is currently underway. In one approach, costimulatory signaling to T cells is boosted by incorporating B7-1 into the vaccine. The addition of B7-1-expressing vaccinia to a TAA-expressing vaccinia vaccine resulted in enhanced T cell proliferation and antitumor protection compared to the TAA-expressing vaccine alone [83]. Vaccinia vaccines have further been developed to include three costimulatory molecules, B7-1, LFA-3 (lymphocyte function-associated antigen-3) and ICAM-1 (intercellular adhesion molecule-1) (TRICOM), the combination of which improves prostate cancer specific responses in clinical trials [14]. A second, complementary approach to enhance T cell-mediated antitumor immunity, blocking coinhibitory signaling pathways, has received much more attention (Figure 3). Blockade of T cell coinhibition for prostate cancer therapy is currently in clinical trials.
Figure 3. Blockade of T cell coinhibition as an emerging therapeutic approach for prostate cancer.
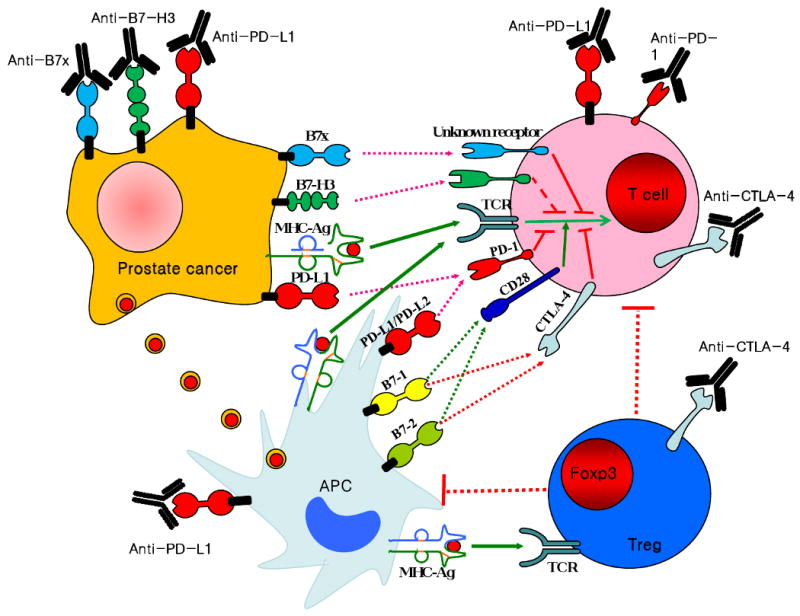
APCs (light blue) take up antigens (red circles) released from tumor cells and present them to T cells (pink) in the context of B7-1 and B7-2 costimulation. Tumor cells (orange) can also present tumor antigens to T cells in the absence of costimulation. Upon T cell activation, CTLA-4 and PD-1 are expressed and inhibit immune responses. Therefore, specific blockade of CTLA-4, leaving TCR and CD28 signaling intact, enhances antitumor immunity. Blockade of CTLA-4 on Tregs (blue) might also reduce Treg-mediated immunosuppression. Immune cells infiltrating prostate cancer have enhanced expression of PD-L1 and PD-1. Therefore immunotherapies blocking the PD-1/PD-L1 pathway are being tested. Finally, both B7x and B7-H3 inhibit T cell functions and are overexpressed by many human cancers including prostate cancer. Consequently, blockade of tumor associated B7-H3 and B7x could be another attractive approach in tumor immunotherapy.
Blockade of CTLA-4
CTLA-4(cytoxic T lymphocyte antigen-4), the coinhibitory homolog of CD28, is upregulated on T cells in lymphoid organs following initial T cell activation and can bind B7-1 and B7-2 on APCs with a higher affinity than CD28, resulting in T cell suppression. Inhibition of CTLA-4 was therefore investigated as a way of boosting antitumor immune responses to cancer. CTLA-4 blockade in prostate cancer was first explored with a murine model of prostate cancer [84]; treatment with anti-CTLA-4 antibody had profound effects both on the primary tumor and metastasis. Complete tumor regression was observed in 42% of the mice and most of the remaining mice displayed significantly delayed tumor growth [84]. A second study showed that CTLA-4 blockade reduced metastatic outgrowths by 50% following primary tumor resection [85]. A fully humanized monoclonal anti-CTLA-4 antibody, ipilimumab, was developed (Bristol-Myers Squibb and Medarex) and is currently undergoing clinical trials for treatment of advanced prostate cancer (www.clinicaltrials.gov). The antibody has shown some antitumor effects. In one early trial including 12 patients with various advanced malignancies, no objective responses were seen in the four prostate cancer patients enrolled [86]. In another trial, however, two of 14 patients with metastatic hormone-refractory prostate cancer experienced a ≥50% decline in serum PSA, a decline large enough to suggest antitumor activity [87].
Combination of anti-CTLA-4 with other therapies increases clinical responses. A phase I trial in metastatic castration-resistant prostate cancer (mCRPC) patients combined ipilimumab with GM-CSF. Three of six patients receiving the largest dose of ipilimumab together with GM-CSF experienced >50% declines in PSA, one showed a partial response in visceral metastases and most patients had an expansion of activated circulating CD8 T cells [88]. An even higher response rate was seen in a phase I trial combining ipilimumab with GVAX. Among six patients receiving the highest dose of ipilimumab, five had PSA declines of >50%, including four who maintained declines for over a year and three who experienced tumor regression at multiple sites, including bone and abdominal lymph nodes, and pain reduction [89]. The improved antitumor responsiveness in these combination regimens highlights the importance of a synergetic approach in immunotherapy. Several trials for anti-CTLA-4 in combination with other treatments are currently underway (www.clinicaltrials.gov), including an ongoing phase III trial of 800 mCRPC patients receiving either radiation therapy alone or in combination with ipilimumab (clinical trial identifier NCT00861614) with the primary endpoint being overall survival.
The results thus far in anti-CTLA-4 therapy are encouraging; however, the testing of anti-CTLA-4 therapies is still in early stages. The more developed vaccines PROSTVAC-VF and sipuleucel-T have benefited from extended trials, which ultimately revealed significant survival advantages in patients receiving the vaccines, even though this was not initially apparent [12, 14, 15]. For anti-CTLA-4 approaches, additional trials and time will allow more complete evaluation of the effects of this therapy, including survival, spectrum of toxicity and the profiles of patients most likely to respond. The use of anti-CTLA-4 approaches in combination therapy is also an important area of investigation. At this time, combining anti-CTLA-4 with GM-CSF or GVAX appears to be beneficial. It will be interesting to see if anti-CTLA-4 will produce synergistic effects in combination with vaccines such as PROSTVAC-VF (currently in trials [90]) or sipuleucel-T.
Although CTLA-4 blockade holds much promise for cancer therapy, the precise mechanism of action for this therapy has not been completely delineated. Blockade of CTLA-4 was developed in the hope of enhancing effector T cell responses by preventing B7-1/B7-2/CTLA-4 inhibitory signaling to T cells and eliminating competition for the costimulatory CD28 receptor [28]. Alternatively, CTLA-4 blockade might indirectly augment antitumor T cell responses by reducing Treg-mediated immunosuppression, as CTLA-4 is constitutively expressed on Tregs [88, 91]. Tregs are abundant in patients with prostate cancer [36], within the tumor microenvironment as well as in their blood [92]. Treg numbers were therefore monitored during several CTLA-4 blockade studies, which reported a range of results, including (i) a brief Treg decline following treatment [88]; (ii) an increase in both Tregs and activated effector CD4 T cells in a dose-dependent manner [93]; and (iii) induced colitis despite stable intramucosal Treg level [94]. Collectively, these studies suggest that anti-CTLA-4 does not function primarily through Treg reduction, however, this does not exclude the possibility that CTLA-4 blockade interferes with Treg function. Recent studies suggest that Tregs, through the action of CTLA-4, can inhibit T cell responses by inducing the downregulation of B7-1 and B7-2 on DCs [95]. Further research is necessary to fully elucidate the mechanisms by which CTLA-4 blockade enhances antitumor T cell immunity.
Blockade of PD-1/PD-L1
A second potential target for coinhibitory blockade in prostate cancer is the PD-1/PD-L1 pathway. Although PD-1/PD-L1 blockade has not yet been studied in a prostate cancer model, blockade studies in other cancers have reported increased survival, reduced tumor growth or metastasis, increased effector T cells, and/or decreased Tregs [96-98]. As PD-1 and PD-L1 are present in the prostate cancer microenvironment, prostate cancer could be a candidate for PD-1/PD-L1 pathway blockade.
Research into the effects of PD-1 and PD-L1 blockade is only just beginning. Humanized monoclonal anti-PD-1 antibodies have recently entered clinical trials, two of which have been published. One studied the safety and efficacy of CT-011 in the treatment of 17 patients with hematologic malignancies. The treatment was safe, relatively tolerated and resulted in a complete response in one non-Hodgkin's lymphoma patient [99]. The other study evaluated MDX-1106 in 39 patients with treatment-refractory solid tumors and showed similar results, including safety, tolerability, one complete response and two partial responses [100]. Included in this study were several patients with mCRPC; however, no objective response was seen in these patients [100]. PD-L1 blockade testing is also underway, including an ongoing phase I trial for monoclonal anti-PD-L1 antibody MDX-1105 in patients with advanced or recurrent solid tumors (clinical trial identifier NCT00729664). It is too early to know whether PD-1/PD-L1 blockade can be effective in treatment of prostate and other cancers. However, given its safety and limited but promising antitumor effects, PD-1/PD-L1 blockade continues to be developed and evaluated.
Blockade of B7x and B7-H3
Blockade of the CTLA-4 and PD-1 coinhibitory pathways for the treatment of prostate cancer are currently underway; however, the real jackpot in overcoming immune evasion in prostate cancer might be yet to come. B7x and B7-H3 are the most highly expressed B7 family members in prostate cancer and the most strongly correlated with clinical outcome. Blocking B7-H3 or B7x might have the advantage of lower immune-related adverse events than anti-CTLA-4 and anti-PD-1 treatments, because the loss of B7-H3 and B7x in knockout mice does not result in spontaneous autoimmunity and these mice appear to be healthy. Development of neutralizing antibodies against B7-H3 and B7x is therefore warranted.
Concluding remarks
Prostate cancer is amenable to immunological intervention, which offers hope for an effective treatment, especially in mCRPC where there are currently no curative options. Although many clinical approaches are being explored, understanding of the immunology of prostate cancer lags behind other cancers such as melanoma, producing a slow and arduous road for the development of effective immunotherapy. B7 family-mediated T cell coinhibition is an emerging immune evasion pathway in prostate cancer that likely limits the effectiveness of immunotherapies. Blocking these pathways could be an important step towards providing new therapeutic options and improving current ones. Combination therapy appears to be key. Effective combinations might include blocking multiple immune evasion pathways, adding coinhibitory blockade to cancer vaccines or combining traditional approaches such as chemotherapy or radiation with immunotherapy. Further studies are needed to fully understand the mechanisms of T cell costimulation, coinhibition and tumor immune evasion, so rational design and implementation of successful immunotherapy against prostate cancer will be possible.
Box 1. The B7 and CD28 families.
T cell activation, proliferation, differentiation to effector function and memory generation depend on two signals; one signal is provided via the TCR through interactions with specific peptide/MHC complexes, whereas signal two is antigen-independent costimulation or coinhibition. The B7 family on APCs or other cells and their receptor CD28 family on T cells are largely responsible for generating signal two. We have divided the members of the B7 and CD28 families into three groups by phylogenetic analysis [28, 29]: group I includes the pathway of B7-1/B7-2/CD28/CTLA-4, and the pathway of B7h (ICOS-L)/ICOS (inducible costimulator); group II consists of the pathway of PD-L1/PD-L2/PD-1; and group III contains B7-H3 and B7x, whose receptors are currently unknown. The B7-1/B7-2/CD28 and B7h/ICOS pathways are costimulatory, whereas B7-1/B7-2/CTLA-4, PD-L1/PD-L2/PD-1 and B7x are coinhibitory. B7-H3 has a contrasting role as both costimulator and coinhibitor.
Box 2. B7-H3 as costimulator or coinhibitor.
B7-H3 binds activated T cells [43], but the physiological role of this signaling is unclear, as both costimulatory and coinhibitory effects have been observed [44].
Several studies in murine models of cancer suggest a costimulatory function for B7-H3. In a mastocytoma model, B7-H3-transfected P815 cells led to rapid expansion of tumor-specific CTLs, tumor regression in almost half of the mice injected, and extended survival and slower tumor growth compared to mice receiving control P815 cells [58]. Similarly, mice implanted with B7-H3-transfected colon cancer cells survived significantly longer than mice implanted with wild-type tumor cells [59]. Furthermore, intratumoral administration of B7-H3 plasmid and arsenic trioxide, but neither agent alone, achieved complete eradication of established hepatocellular tumors [60].
In contrast to mouse tumor studies, clinical association of B7-H3 expression with human cancers vary, with the majority of studies suggesting a coinhibitory role. Although B7-H3 expression was associated with increased survival in gastric carcinoma patients [61] and one study of pancreatic cancer [62], B7-H3 correlated with advanced pathological state and lymph node metastasis in another study of pancreatic cancer [47], as well as reduced survival, increased cancer recurrence, adverse clinical features, and/or decreased tumor infiltrating lymphocytes in lung cancer [53], prostate cancer [40, 63], neuroblastoma [64], renal cell carcinoma [48], colorectal carcinoma [46], and ovarian cancer [45]. In addition to controversy over B7-H3 function, there is no consensus regarding the B7-H3 receptor. TREM-like transcript 2 has been reported to be a receptor for B7-H3 [65], but another report did not detect interaction between these two molecules [66].
Acknowledgments
Y.S.B is supported by a National Institutes of Health (NIH) training grant T32GM007491. X.Z. is supported in part by NIH Type 1 Diabetes Pathfinder Award DP2DK083076 and Department of Defense Prostate Cancer Research Program New Investigator Award PC094137.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production processrrors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, et al. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, et al. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer. 2010;46:765–781. doi: 10.1016/j.ejca.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 3.Antonarakis ES, et al. Novel targeted therapeutics for metastatic castration-resistant prostate cancer. Cancer Lett. 2010;291:1–13. doi: 10.1016/j.canlet.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riegman PH, et al. Characterization of the prostate-specific antigen gene: a novel human kallikrein-like gene. Biochem Biophys Res Commun. 1989;159:95–102. doi: 10.1016/0006-291x(89)92409-1. [DOI] [PubMed] [Google Scholar]
- 5.Kawakami M, Nakayama J. Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res. 1997;57:2321–2324. [PubMed] [Google Scholar]
- 6.Solin T, et al. Gene expression and prostate specificity of human prostatic acid phosphatase (PAP): evaluation by RNA blot analyses. Biochim Biophys Acta. 1990;1048:72–77. doi: 10.1016/0167-4781(90)90024-v. [DOI] [PubMed] [Google Scholar]
- 7.Kiessling A, et al. Prostate stem cell antigen: Identification of immunogenic peptides and assessment of reactive CD8+ T cells in prostate cancer patients. Int J Cancer. 2002;102:390–397. doi: 10.1002/ijc.10713. [DOI] [PubMed] [Google Scholar]
- 8.Noguchi M, et al. Induction of cellular and humoral immune responses to tumor cells and peptides in HLA-A24 positive hormone-refractory prostate cancer patients by peptide vaccination. Prostate. 2003;57:80–92. doi: 10.1002/pros.10276. [DOI] [PubMed] [Google Scholar]
- 9.Meidenbauer N, et al. Generation of PSA-reactive effector cells after vaccination with a PSA-based vaccine in patients with prostate cancer. Prostate. 2000;43:88–100. doi: 10.1002/(sici)1097-0045(20000501)43:2<88::aid-pros3>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 10.Drake CG, Antonarakis ES. Update: immunological strategies for prostate cancer. Curr Urol Rep. 2010;11:202–207. doi: 10.1007/s11934-010-0106-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 12.Small EJ, et al. Placebo-controlled phase III trial of immunologic therapy with sipuleucel-T (APC8015) in patients with metastatic, asymptomatic hormone refractory prostate cancer. J Clin Oncol. 2006;24:3089–3094. doi: 10.1200/JCO.2005.04.5252. [DOI] [PubMed] [Google Scholar]
- 13.DiPaola RS, et al. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J Transl Med. 2006;4:1. doi: 10.1186/1479-5876-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kantoff PW, et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:1099–1105. doi: 10.1200/JCO.2009.25.0597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gulley JL, et al. Immunologic and prognostic factors associated with overall survival employing a poxviral-based PSA vaccine in metastatic castrate-resistant prostate cancer. Cancer Immunol Immunother. 2010;59:663–674. doi: 10.1007/s00262-009-0782-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mincheff M, et al. Naked DNA and adenoviral immunizations for immunotherapy of prostate cancer: a phase I/II clinical trial. Eur Urol. 2000;38:208–217. doi: 10.1159/000020281. [DOI] [PubMed] [Google Scholar]
- 17.Pavlenko M, et al. A phase I trial of DNA vaccination with a plasmid expressing prostate-specific antigen in patients with hormone-refractory prostate cancer. Br J Cancer. 2004;91:688–694. doi: 10.1038/sj.bjc.6602019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Becker JT, et al. DNA vaccine encoding prostatic acid phosphatase (PAP) elicits long-term T-cell responses in patients with recurrent prostate cancer. J Immunother. 2010;33:639–647. doi: 10.1097/CJI.0b013e3181dda23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higano CS, et al. Phase 1/2 dose-escalation study of a GM-CSF-secreting, allogeneic, cellular immunotherapy for metastatic hormone-refractory prostate cancer. Cancer. 2008;113:975–984. doi: 10.1002/cncr.23669. [DOI] [PubMed] [Google Scholar]
- 20.Miller AM, Pisa P. Tumor escape mechanisms in prostate cancer. Cancer Immunol Immunother. 2007;56:81–87. doi: 10.1007/s00262-005-0110-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bander NH, et al. MHC class I and II expression in prostate carcinoma and modulation by interferon-α and -γ. Prostate. 1997;33:233–239. doi: 10.1002/(sici)1097-0045(19971201)33:4<233::aid-pros2>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 22.Sanda MG, et al. Molecular characterization of defective antigen processing in human prostate cancer. J Natl Cancer Inst. 1995;87:280–285. doi: 10.1093/jnci/87.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bronte V, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. 2005;201:1257–1268. doi: 10.1084/jem.20042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, et al. Expression of inducible nitric oxide synthase in paired neoplastic and non-neoplastic primary prostate cell cultures and prostatectomy specimen. Urol Oncol. 2003;21:117–122. doi: 10.1016/s1078-1439(02)00208-9. [DOI] [PubMed] [Google Scholar]
- 25.Elsasser-Beile U, et al. Different basal expression of type T1 and T2 cytokines in peripheral lymphocytes of patients with adenocarcinomas and benign hyperplasia of the prostate. Anticancer Res. 2003;23:4027–4031. [PubMed] [Google Scholar]
- 26.Cardillo MR, et al. Transforming growth factor-β expression in prostate neoplasia. Anal Quant Cytol Histol. 2000;22:1–10. [PubMed] [Google Scholar]
- 27.Yokokawa J, et al. Enhanced functionality of CD4+CD25(high)FoxP3+ regulatory T cells in the peripheral blood of patients with prostate cancer. Clin Cancer Res. 2008;14:1032–1040. doi: 10.1158/1078-0432.CCR-07-2056. [DOI] [PubMed] [Google Scholar]
- 28.Zang X, Allison JP. The B7 family and cancer therapy: costimulation and coinhibition. Clin Cancer Res. 2007;13:5271–5279. doi: 10.1158/1078-0432.CCR-07-1030. [DOI] [PubMed] [Google Scholar]
- 29.Zang X, et al. B7x: a widely expressed B7 family member that inhibits T cell activation. Proc Natl Acad Sci U S A. 2003;100:10388–10392. doi: 10.1073/pnas.1434299100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong H, et al. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 32.Tseng SY, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med. 2001;193:839–846. doi: 10.1084/jem.193.7.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura H, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 35.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 36.Ebelt K, et al. Prostate cancer lesions are surrounded by FOXP3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur J Cancer. 2009;45:1664–1672. doi: 10.1016/j.ejca.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 37.Ebelt K, et al. Dominance of CD4+ lymphocytic infiltrates with disturbed effector cell characteristics in the tumor microenvironment of prostate carcinoma. Prostate. 2008;68:1–10. doi: 10.1002/pros.20661. [DOI] [PubMed] [Google Scholar]
- 38.Sfanos KS, et al. Human prostate-infiltrating CD8+ T lymphocytes are oligoclonal and PD-1+ Prostate. 2009;69:1694–1703. doi: 10.1002/pros.21020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 40.Roth TJ, et al. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res. 2007;67:7893–7900. doi: 10.1158/0008-5472.CAN-07-1068. [DOI] [PubMed] [Google Scholar]
- 41.Crane CA, et al. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene. 2009;28:306–312. doi: 10.1038/onc.2008.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–3029. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chapoval AI, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-γ production. Nat Immunol. 2001;2:269–274. doi: 10.1038/85339. [DOI] [PubMed] [Google Scholar]
- 44.Hofmeyer KA, et al. The contrasting role of B7-H3. Proc Natl Acad Sci U S A. 2008;105:10277–10278. doi: 10.1073/pnas.0805458105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zang X, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol. 2010;23:1104–1112. doi: 10.1038/modpathol.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun J, et al. Clinical significance and regulation of the costimulatory molecule B7-H3 in human colorectal carcinoma. Cancer Immunol Immunother. 2010;59:1163–1171. doi: 10.1007/s00262-010-0841-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamato I, et al. Clinical importance of B7-H3 expression in human pancreatic cancer. Br J Cancer. 2009;101:1709–1716. doi: 10.1038/sj.bjc.6605375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crispen PL, et al. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res. 2008;14:5150–5157. doi: 10.1158/1078-0432.CCR-08-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chavin G, et al. Expression of immunosuppresive B7-H3 ligand by hormone-treated prostate cancer tumors and metastases. Clin Cancer Res. 2009;15:2174–2180. doi: 10.1158/1078-0432.CCR-08-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sica GL, et al. B7-H4, a molecule of the B7 family, negatively regulates T cell immunity. Immunity. 2003;18:849–861. doi: 10.1016/s1074-7613(03)00152-3. [DOI] [PubMed] [Google Scholar]
- 51.Tringler B, et al. B7-h4 is highly expressed in ductal and lobular breast cancer. Clin Cancer Res. 2005;11:1842–1848. doi: 10.1158/1078-0432.CCR-04-1658. [DOI] [PubMed] [Google Scholar]
- 52.Salceda S, et al. The immunomodulatory protein B7-H4 is overexpressed in breast and ovarian cancers and promotes epithelial cell transformation. Exp Cell Res. 2005;306:128–141. doi: 10.1016/j.yexcr.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 53.Sun Y, et al. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer. 2006;53:143–151. doi: 10.1016/j.lungcan.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 54.Krambeck AE, et al. B7-H4 expression in renal cell carcinoma and tumor vasculature: associations with cancer progression and survival. Proc Natl Acad Sci U S A. 2006;103:10391–10396. doi: 10.1073/pnas.0600937103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yao Y, et al. B7-H4 is preferentially expressed in non-dividing brain tumor cells and in a subset of brain tumor stem-like cells. J Neurooncol. 2008;89:121–129. doi: 10.1007/s11060-008-9601-x. [DOI] [PubMed] [Google Scholar]
- 56.Awadallah NS, et al. Detection of B7-H4 and p53 in pancreatic cancer: potential role as a cytological diagnostic adjunct. Pancreas. 2008;36:200–206. doi: 10.1097/MPA.0b013e318150e4e0. [DOI] [PubMed] [Google Scholar]
- 57.Prasad DV, et al. B7S1, a novel B7 family member that negatively regulates T cell activation. Immunity. 2003;18:863–873. doi: 10.1016/s1074-7613(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 58.Luo L, et al. B7-H3 enhances tumor immunity in vivo by costimulating rapid clonal expansion of antigen-specific CD8+ cytolytic T cells. J Immunol. 2004;173:5445–5450. doi: 10.4049/jimmunol.173.9.5445. [DOI] [PubMed] [Google Scholar]
- 59.Lupu CM, et al. An orthotopic colon cancer model for studying the B7-H3 antitumor effect in vivo. J Gastrointest Surg. 2006;10:635–645. doi: 10.1007/BF03239969. [DOI] [PubMed] [Google Scholar]
- 60.Luo L, et al. Arsenic trioxide synergizes with B7H3-mediated immunotherapy to eradicate hepatocellular carcinomas. Int J Cancer. 2006;118:1823–1830. doi: 10.1002/ijc.21557. [DOI] [PubMed] [Google Scholar]
- 61.Wu CP, et al. Relationship between co-stimulatory molecule B7-H3 expression and gastric carcinoma histology and prognosis. World J Gastroenterol. 2006;12:457–459. doi: 10.3748/wjg.v12.i3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loos M, et al. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer. 2009;9:463. doi: 10.1186/1471-2407-9-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zang X, et al. B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc Natl Acad Sci U S A. 2007;104:19458–19463. doi: 10.1073/pnas.0709802104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gregorio A, et al. Small round blue cell tumours: diagnostic and prognostic usefulness of the expression of B7-H3 surface molecule. Histopathology. 2008;53:73–80. doi: 10.1111/j.1365-2559.2008.03070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashiguchi M, et al. Triggering receptor expressed on myeloid cell-like transcript 2 (TLT-2) is a counter-receptor for B7-H3 and enhances T cell responses. Proc Natl Acad Sci U S A. 2008;105:10495–10500. doi: 10.1073/pnas.0802423105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Leitner J, et al. B7-H3 is a potent inhibitor of human T-cell activation: No evidence for B7-H3 and TREML2 interaction. Eur J Immunol. 2009;39:1754–1764. doi: 10.1002/eji.200839028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parker AS, et al. Evaluation of B7-H3 expression as a biomarker of biochemical recurrence after salvage radiation therapy for recurrent prostate cancer. Int J Radiat Oncol Biol Phys. 2010;1:1–7. doi: 10.1016/j.ijrobp.2010.01.061. [DOI] [PubMed] [Google Scholar]
- 68.Lehmann BD, et al. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008;68:7864–7871. doi: 10.1158/0008-5472.CAN-07-6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee SK, et al. IFN-γ regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005;40:95–103. doi: 10.1016/j.jdermsci.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 70.Gong AY, et al. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-γ-induced B7-H1 expression in cholangiocytes. J Immunol. 2009;182:1325–1333. doi: 10.4049/jimmunol.182.3.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stanciu LA, et al. Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. J Infect Dis. 2006;193:404–412. doi: 10.1086/499275. [DOI] [PubMed] [Google Scholar]
- 72.Lee SJ, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-γ-induced upregulation of B7-H1 (CD274) FEBS Lett. 2006;580:755–762. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- 73.Karakhanova S, et al. ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur J Immunol. 2010;40:254–266. doi: 10.1002/eji.200939289. [DOI] [PubMed] [Google Scholar]
- 74.Liu J, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-γ and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- 75.Qian Y, et al. TLR4 signaling induces B7-H1 expression through MAPK pathways in bladder cancer cells. Cancer Invest. 2008;26:816–821. doi: 10.1080/07357900801941852. [DOI] [PubMed] [Google Scholar]
- 76.Holets LM, et al. Differentiation-induced post-transcriptional control of B7-H1 in human trophoblast cells. Placenta. 2009;30:48–55. doi: 10.1016/j.placenta.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13:84–88. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 78.Filipowicz W, et al. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 79.Lai EC. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet. 2002;30:363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 80.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 81.Suh WK, et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol. 2003;4:899–906. doi: 10.1038/ni967. [DOI] [PubMed] [Google Scholar]
- 82.Xu H, et al. MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: potential implications for immune based therapy of human solid tumors. Cancer Res. 2009;69:6275–6281. doi: 10.1158/0008-5472.CAN-08-4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hodge JW, et al. Admixture of a recombinant vaccinia virus containing the gene for the costimulatory molecule B7 and a recombinant vaccinia virus containing a tumor-associated antigen gene results in enhanced specific T-cell responses and antitumor immunity. Cancer Res. 1995;55:3598–3603. [PubMed] [Google Scholar]
- 84.Kwon ED, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94:8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kwon ED, et al. Elimination of residual metastatic prostate cancer after surgery and adjunctive cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) blockade immunotherapy. Proc Natl Acad Sci U S A. 1999;96:15074–15079. doi: 10.1073/pnas.96.26.15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.O'Mahony D, et al. A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin Cancer Res. 2007;13:958–964. doi: 10.1158/1078-0432.CCR-06-1974. [DOI] [PubMed] [Google Scholar]
- 87.Small EJ, et al. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2007;13:1810–1815. doi: 10.1158/1078-0432.CCR-06-2318. [DOI] [PubMed] [Google Scholar]
- 88.Fong L, et al. Potentiating endogenous antitumor immunity to prostate cancer through combination immunotherapy with CTLA4 blockade and GM-CSF. Cancer Res. 2009;69:609–615. doi: 10.1158/0008-5472.CAN-08-3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gerritsen W, et al. A dose-escalation trial of GM-CSF-gene transduced allogeneic prostate cancer cellular immunotherapy in combination with a fully human anti-CTLA antibody (MDX-010, ipilimumab) in patients with metastatic hormone-refractory prostate cancer In. J Clin Oncol ASCO Annual Meeting Proceedings Part I. 2006;24(No 18S) [Google Scholar]
- 90.Theoret MR, et al. Phase I trial of an enhanced prostate-specific antigen-based vaccine and anti-CTLA-4 antibody in patients with metastatic androgen-independent prostate cancer. Clin Genitourin Cancer. 2007;5:347–350. doi: 10.3816/CGC.2007.n.017. [DOI] [PubMed] [Google Scholar]
- 91.Peggs KS, et al. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206:1717–1725. doi: 10.1084/jem.20082492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miller AM, et al. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J Immunol. 2006;177:7398–7405. doi: 10.4049/jimmunol.177.10.7398. [DOI] [PubMed] [Google Scholar]
- 93.Kavanagh B, et al. CTLA4 blockade expands FoxP3+ regulatory and activated effector CD4+ T cells in a dose-dependent fashion. Blood. 2008;112:1175–1183. doi: 10.1182/blood-2007-11-125435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lord JD, et al. Refractory colitis following anti-CTLA4 antibody therapy: analysis of mucosal FOXP3+ T cells. Dig Dis Sci. 2010;55:1396–1405. doi: 10.1007/s10620-009-0839-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 96.Curran MA, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hirano F, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- 98.Iwai Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Berger R, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic malignancies. Clin Cancer Res. 2008;14:3044–3051. doi: 10.1158/1078-0432.CCR-07-4079. [DOI] [PubMed] [Google Scholar]
- 100.Brahmer JR, et al. Phase I Study of Single-Agent Anti-Programmed Death-1 (MDX-1106) in Refractory Solid Tumors: Safety, Clinical Activity, Pharmacodynamics, and Immunologic Correlates. J Clin Oncol. 2010;28:3167–3175. doi: 10.1200/JCO.2009.26.7609. [DOI] [PMC free article] [PubMed] [Google Scholar]