

SUMMARY
Although deficient CD8 T cell responses have long been associated with chronic viral infections, the underlying mechanisms are still unclear. Here we report that enhanced and sustained TGF-β/Smad signaling is a distinctive feature of virus-specific CD8 T cells during chronic versus acute viral infections in vivo. The result is TGF-β-dependent up-regulation of the pro-apoptotic protein Bim that relates to cell-intrinsic apoptosis and significantly reduced numbers of virus-specific CD8 T cells. Moreover, selective attenuation of TGF-β signaling on T cells increases the function of CD8 T cells in an indirect fashion, rapidly eradicates the persistence-prone virus and enables the generation of an effective memory response. Our findings reveal persisting TGF-β/Smad signaling as a hallmark and key regulator of virus-specific CD8 T cell responses during chronic viral infections in vivo.
Keywords: virus, chronic infection, LCMV, TGF-β, T cell –immune regulation
INTRODUCTION
Microorganisms have evolved sophisticated mechanisms to interfere with the ensuing immune response, leading to a race between microbe (virulence) and immune system (resistance) that dictates whether the pathogen establishes chronically or is eliminated. Dramatic examples of human viruses wining this race are human immunodeficiency virus (HIV), hepatitis B virus (HBV) and hepatitis C virus (HCV), which currently infect more than 500 million people worldwide (Letvin and Walker, 2003; Rehermann and Nascimbeni, 2005). During several viral infections, progression into the chronic phase has been associated with a varying degree of T cell dysfunction or “exhaustion”, ranging from diminished cytotoxicity to decreased production of anti-viral and growth cytokines and T cell deletion (Gruener et al., 2001; Klenerman and Hill, 2005; Kostense et al., 2002; Shankar et al., 2000; Wherry et al., 2003; Zajac et al., 1998). Although these parameters of exhaustion have long been noted, the causative factors and the relative contributions of T-cell-intrinsic and T-cell-extrinsic inhibitory signals in determining viral persistence are not completely understood. Complete dissection of these pathways represents a major biomedical challenge and has the potential to unveil novel therapeutic strategies to treat persistently infected individuals. Recent studies using lymphocytic choriomeningitis virus (LCMV) infection in mice identified programmed cell death-1 (PD-1) and interleukin-10 (IL-10) as key inhibitory factors responsible for T cell suppression and viral establishment during persistent infection (Barber et al., 2006; Brooks et al., 2006; Ejrnaes et al., 2006). Although it is still unclear whether these inhibitory signals act intrinsically on virus-specific CD8 T cells or indirectly through other cell types, these reports indicate that viruses can exploit immune-regulatory pathways to counteract the ensuing immune response. Importantly, these findings were rapidly extended to or independently reported in humans (Boni et al., 2007; Clerici et al., 1994; Day et al., 2006; Landay et al., 1996; Petrovas et al., 2006; Radziewicz et al., 2007; Rigopoulou et al., 2005; Trautmann et al., 2006; Urbani et al., 2006; Zhang et al., 2007), suggesting that multiple conserved T cell inhibitory pathways are shared between distinct chronic viral infections in different hosts.
TGF-β is an ancient cytokine that regulates cell proliferation, differentiation, survival and adhesion in multiple cell types (Li et al., 2006b; Massague, 2008). The critical function of TGF-β in suppressing autoreactive T cells is evident in TGF-β1 deficient mice and mice with complete ablation of TGF-β signaling on T cells, which die from a multi-organ autoimmune disease at 3-4 weeks of age (Kulkarni et al., 1993; Li et al., 2006a; Marie et al., 2006; Shull et al., 1992). A milder phenotype was observed in mice expressing a dominant negative form of TGF-β receptor II from a modified CD4 promoter (hereafter referred as dnTGFBRII) that results in attenuation of TGF-β signaling in CD4 and CD8 T cells (Gorelik and Flavell, 2000). The dnTGFBRII mice live without any detectable problems until 3-4 months of age when they began showing signs of sickness, wasting and diarrhea. This was associated with multifocal inflammatory infiltrates and autoantibody secretion (Gorelik and Flavell, 2000). As expected for such a pleiotropic factor, the activities of TGF-β are subject to multiple levels of regulation. This growth factor is normally secreted in a latent configuration and is subsequently activated in the vicinity of target cells to exert its function in a paracrine/autocrine manner (Annes et al., 2003; Jenkins, 2008; Wipff and Hinz, 2008). TGF-β binding to its receptor induces phosphorylation of Smad proteins, which itself is tightly regulated by a delicate balance between kinases, phosphatases and ubiquitin ligases (Rubtsov and Rudensky, 2007; Shi and Massague, 2003). As with many highly tuned biological processes, alteration of TGF-β/Smad signaling has been associated with tumorigenesis (Massague, 2008), atherosclerosis (Topper, 2000) and autoimmunity (Li et al., 2006b; Rubtsov and Rudensky, 2007) but its role in regulating T cells responses and viral persistence during in vivo chronic infection remained unknown. Here, we report that virus-specific CD8 T cells exhibit increased Smad-2 phosphorylation and TGF-β expression during chronic compared to acute viral infection in vivo. Selective attenuation of TGF-β pathway on T cells decreases the expression of the pro-apoptotic protein Bim and increases survival and numbers of virus-specific CD8 T cells. Under these conditions, T cells exhibit enhanced cytotoxicity, increased production of anti-viral cytokines, and down-regulation of the inhibitory molecules, PD1 and IL-10. Notably, while TGF-β effect on CD8 T cell numbers and survival is cell-intrinsic, its impact on CD8 T cell function and PD-1 expression is indirect or cell-extrinsic. The end result is rapid virus eradication and generation of an effective memory T cell response that protects the host upon subsequent challenge.
RESULTS
Enhanced Smad-2 phosphorylation and TGF-β expression in virus-specific CD8 T cells during chronic LCMV infection
To investigate the role of TGF-β in T cell suppression and viral persistence, we infected mice with two genetically related LCMV isolates that induce either acute or chronic viral infections, but share identical T cell epitopes. The parental virus strain, Armstrong 53b (ARM), induces a strong CD8 T cell response responsible for acute viral clearance after 8-10 days post-infection (pi) (Ahmed et al., 1984). In contrast, LCMV clone 13 (Cl 13) initiates an abortive CD8 T cell program that results in a persistent infection for 60 to 90 days in blood and most tissues (Ahmed et al., 1988; Moskophidis et al., 1993; Wherry et al., 2003; Zajac et al., 1998). In particular, Cl 13 infection induces CD8 T cell deletion and exhaustion characterized by compromised effector functions (Moskophidis et al., 1993; Ou et al., 2001; Wherry et al., 2003; Zajac et al., 1998) and data not shown). To examine downstream TGF-β signaling on virus-specific CD8 T cells during acute versus chronic LCMV infection, the levels of phospho(p)-Smad-2/3 were analyzed on LCMV GP33-41 tetramer+ CD8+ T cells by FACS. A significant increase in p-Smad-2/3 was consistently observed in virus-specific CD8 T cells from Cl 13-infected compared to ARM-infected mice (Figure 1A). To further evaluate Smad signaling on virus-specific CD8 T cells, we adoptively transferred naïve CD45.2+ P14 TCR transgenic CD8+ T cells that are specific for the GP33-41 LCMV epitope into CD45.1+ WT mice one day before infection with LCMV ARM or Cl 13 (Figure 1B). Consistent with the FACS data, immunoblot analysis of P14 cells isolated at day 8-10 pi showed enhanced Smad-2 phosphorylation during chronic compared to acute LCMV infection (Figure 1C). Moreover, a slight increase in Smad-2 phosphorylation was observed by day 5 after Cl 13 infection. Minimal or no difference was observed in levels of total Smad-2 after ARM versus Cl 13 infection. Further studies addressing the source of this enhanced Smad-2 signaling during chronic infection revealed an elevated expression of TGF-β1 protein in virus-specific CD8 T cells from Cl 13- compared to ARM-infected mice (Figure 1D). In contrast, no differences were observed in the expression of TGF-β receptor during either infection (Supplementary Figure 1). Together, these results indicate that TGF-β/Smad signaling is turned on in antiviral CD8 T cells during both acute and chronic LCMV infection but becomes enhanced and sustained only during persistent infection.
Figure 1. TGF-β/Smad signaling and accumulation of virus-specific CD8 T cells during LCMV infection.
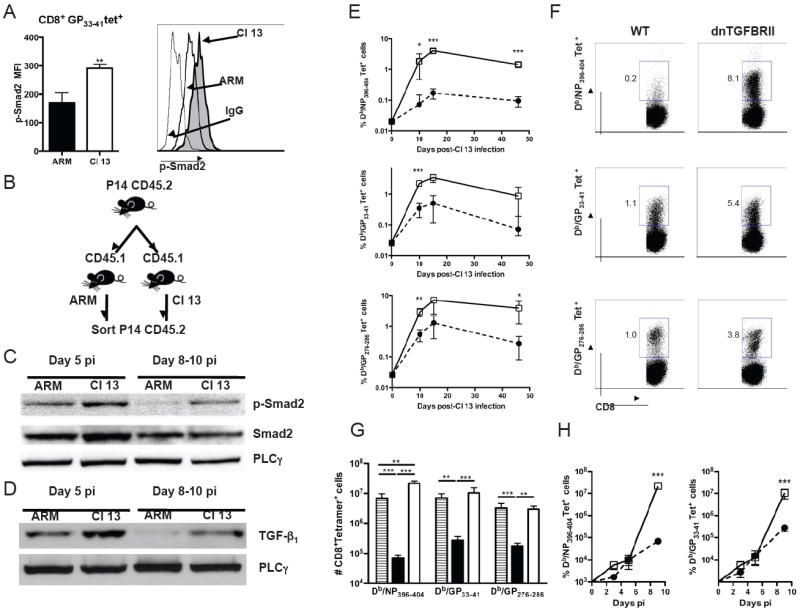
A) WT mice were infected with LCMV ARM or Cl 13, splenocytes were isolated at day 10 pi and stained with anti-CD8, Db/GP33-41 tetramers and anti-p-Smad-2/3 mAb. p-Smad-2/3 levels in Db/GP33-41 tetramer+CD8+ T cells were determined by FACS. Bar graphs indicate the average mean fluorescence intensity (MFI) ± standard deviation (sd) of 4 mice per group. Histograms depict one representative mouse per group. Thin line, Isotype control. B-D) Donor CD45.2+ P14 TCR transgenic CD8+ T cells were transferred into WT CD45.1+ recipients, hosts were infected with LCMV ARM or Cl 13, and pooled P14 cells FACS-purified at day 5 or 8-10 pi and processed for immunoblot blot. B) Sketch of experimental design. c) Levels of p-Smad-2, total Smad-2 in P14 cells. D) Levels of TGF-β1 in P14 cells. Phospholipase-γ (PLC-γ) was used as loading control. E-H) WT (black circles or bars) or dnTGFBRII (white squares or bars) mice were infected with LCMV Cl 13. WT-ARM infected mice were processed as controls (striped bars). Virus-specific CD8+ T cells from blood (E) or spleen (F-H) were stained with H2Db/NP396-404, Db/GP33-41 and/or Db/GP276-286 tetramers. E) Log-scaled plots showing average frequencies ± sd of tetramer+CD8+ blood cells at the indicated times pi. F) Dot plots displaying tetramer staining profiles in splenocytes from a representative mouse per group at day 9 pi. Numbers indicate % tetramer+CD8+ cells in the respective gate. G-H) Total numbers of tetramer+CD8+ cells per spleen at day 9 (G) or days 1 through 9 (H) pi. Note that numbers of tetramer+ T cells were similar to background levels at day 1 and 3 pi. All results are representative of two or three independent experiments with three to four mice per group each. (LCMV ARM vs Cl 13 or WT vs dnTGFBRII as indicated, *p<0.05, **p<0.005 and ***p<0.0005)
Selective attenuation of TGF-β signaling in T cells enables accumulation of virus-specific CD8 T cells during chronic Cl 13 infection
To investigate the biological significance of TGF-β signaling in T cells during chronic viral infection, we infected dnTGFBRII transgenic, which display selective attenuation of TGF-β signaling in T cells (Gorelik and Flavell, 2000), with LCMV Cl 13. Levels of peripheral blood CD8 T cells specific for three different MHC class I restricted LCMV epitopes were monitored by tetramer staining throughout the course of infection (Figure 1E). Cl 13-infected dnTGFBRII mice showed significantly higher frequencies of GP33-41 and GP276-286 specific T cells than WT controls. More impressively, the proportion of NP396-404 specific CD8 T cells, which are largely deleted during Cl 13 infection in WT mice (Ou et al., 2001) was also enhanced in dnTGFBRII mice. The increased proportions of peripheral blood tetramer-positive CD8 T cells persisted for at least 45 days pi. Analysis of splenic T cells at day 9 pi also revealed elevated frequencies and numbers of NP396-404, GP33-41 and GP276-286-specific CD8+ T cells in dnTGFBRII mice to similar extend as WT-ARM infected mice and up to 25 times higher than WT-Cl 13 infected mice (Figure 1F and 1G). Early kinetics indicate that the initial expansion of CD8 T cells is similar in WT and dnTGFBRII Cl 13 infected mice (Figure 1H), suggesting that the negative impact of TGF-β on the numbers of virus-specific CD8 T cells becomes effective at later time points after infection. Thus, reduced TGF-β receptor signaling in the dnTGFBRII T cells increases the accumulation of virus-specific CD8 T cells during chronic Cl 13 infection.
TGF-β signaling induces apoptosis of virus-specific CD8 T cells and enhances Bim expression
Heightened accumulation of virus-specific CD8 T cells in the Cl 13-infected dnTGFBRII mice could result from increased proliferation and/or a reduction in cell death. To discriminate between these possibilities, we first monitored proliferation in vivo by analyzing BrdU incorporation on uninfected and Cl 13 infected mice at days 7 and 9 pi. While increased proliferation of total CD8 T cells was observed before infection (Supplementary Figure 2A), no enhancement in the proliferation of GP33-41 and GP276-286 specific T cells from dnTGFBRII versus WT mice was detected after infection (Figure 2A and Supplementary Figure 3A, respectively). We then monitored apoptosis by Annexin V staining at the same time points. Notably, in contrast to the higher percentages of apoptotic CD8 T cells detected in dnTGFBRII compared to WT mice before infection (Supplementary Figure 2B), a lower frequency of Annexin V+ cells was determined within GP33-41 or GP276-286 specific CD8 T cells after Cl 13 infection (Figure 2B and Supplementary Figure 3B, respectively). Thus, dnTGFBRII mice show increase CD8 T cell-proliferation and death before infection but similar proliferation and enhanced survival of virus-specific CD8 T cells during chronic LCMV infection.
Figure 2. dnTGFBRII mice show increased survival and reduced levels of Bim in virus-specific CD8 T cells.
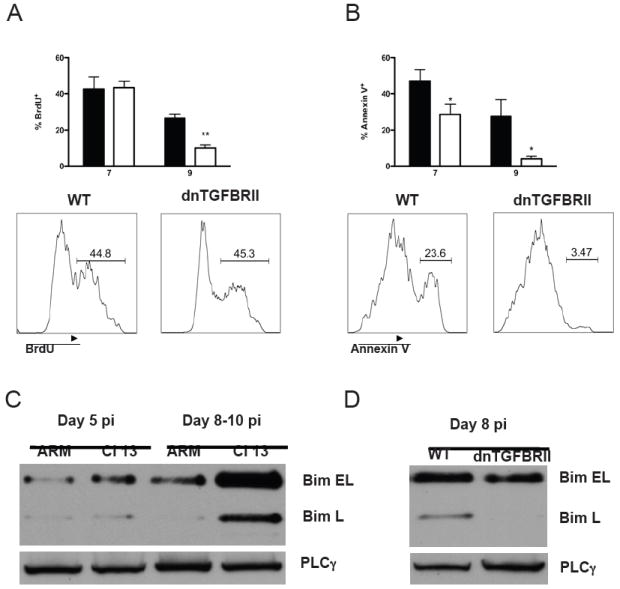
A-B) WT (black bars) or dnTGFBRII (white bars) mice were infected with LCMV Cl 13. Spleen cells were isolated at day 7 and 9 pi and BrdU incorporation (A) and Annexin V staining (B) of H2Db GP33-41 tetramer+CD8+ T cells were determined. Histograms display a representative mouse per group and numbers indicate the frequency of cells within regions. C-D) Virus-specific CD8 T cells were FACS-isolated from WT mice infected with LCMV ARM or Cl 13 (C) or dnTGFBRII mice infected with Cl 13 (D) at days 8-10 pi. Cells were processed by immunoblot and the levels of Bim EL and Bim L isoforms are shown. Phospholipase-γ (PLC-γ) was used as loading control. Results are representative of 2-3 independent experiments with 3-5 mice per group (WT vs dnTGFBRII, *p<0.05 and **p<0.005)
The BH3-only protein Bim has been implicated in the killing of virus-specific CD8 T cells during chronic infection with LCMV Cl 13 and murine γ-herpesvirus (Grayson et al., 2006; Hughes et al., 2008). Accordingly, we observed that the three isoforms of Bim (i.e., BIM-EL, L and S) were markedly up-regulated after Cl 13 versus ARM LCMV infection (Figure 2C and data not shown). Notably, up-regulation of Bim coincided with enhanced Smad-2 phosphorylation in virus-specific CD8 T cells (Figure 1C), suggesting that TGF-β signaling could induce Bim up-regulation. To test this possibility, we analyzed Bim expression in LCMV-specific CD8 T cells isolated from WT or dnTGFBRII Cl 13-infected mice. We found that the expression of both Bim-EL and Bim-L isoforms were dramatically decreased in dnTGFBRII virus-specific CD8 T cells compared to those from WT controls (Figure 2D). Altogether, these data indicate that TGF-β signaling promotes apoptosis of virus-specific CD8 T cells and is a pre-requisite for Bim up-regulation during chronic viral infection in vivo.
CD8 T cell dysfunction is dependent on TGF-β signaling in T cells during Cl 13 infection
In the next series of experiments, we investigated whether diminished TGF-β receptor signaling in T cells enabled the acquisition of effector CD8 T cell functions. Production of IFN-γ, TNF-α and IL-2 was determined in CD8 T cells from uninfected and day 9-Cl 13-infected WT or dnTGFBRII mice by ex-vivo stimulation. We observed that uninfected dnTGFBRII mice exhibited about 2 fold increase in the percentage of IFN-γ+ cells but no increased frequencies of IL-2 and TNF-α producing cells after PMA-ionomicyn stimulation (Supplementary Figure 2, C and D). As previously reported during Cl 13 infection (Wherry et al., 2003; Zajac et al., 1998), a large proportion of virus-specific-CD8 T cells from WT mice were unable to produce anti-viral cytokines (Figure 3A). In stark contrast, CD8 T cells from dnTGFBRII mice display functional effector activity to similar extend as WT-ARM infected mice, as indicated by the dramatically elevated frequencies of epitope-specific CD8 T cells that produce IFN-γ, TNF-α and/or IL-2 upon re-stimulation with NP396-404, GP33-41 or GP276-286 LCMV peptides (Figure 3A and Supplementary Figure 4). Moreover, substantial numbers of multi-producer CD8 T cells, which simultaneously secrete two or three of the cytokines studied, were detected in dnTGFBRII mice (Supplementary Figure 5). Cytotoxic activity towards target cells loaded with either NP396-404 or GP33-41 peptides was markedly enhanced in CD8 T cells from Cl 13-infected dnTGFBRII versus WT mice at day 9 pi (Figure 3B). Consistently, a larger number of virus-specific CD8 T cells from dnTGFBRII mice exhibited surface expression of the lysosomal markers CD107a/b upon GP33-41 peptide re-stimulation, indicating their increased cell degranulation compared to WT controls (Figure 3C). Finally, we observed that the previously described inhibitory molecules PD-1 in virus-specific CD8 T cells (Barber et al., 2006; Boni et al., 2007; Day et al., 2006; Petrovas et al., 2006; Radziewicz et al., 2007; Trautmann et al., 2006; Urbani et al., 2006; Zhang et al., 2007) and IL-10 in splenocytes (Brooks et al., 2006; Ejrnaes et al., 2006), were reduced in dnTGFBRII mice compared to WT controls at day 9 and later after Cl 13 infection (Figure 3D, 3E and Supplementary Figure 6), but were similar at day 5 pi or slightly increased in uninfected dnTGFBRII mice (Figure 3D, 3E and Supplementary Figure 2E).
Figure 3. Functional virus-specific CD8 T cells in LCMV Cl 13 infected dnTGFBRII mice.
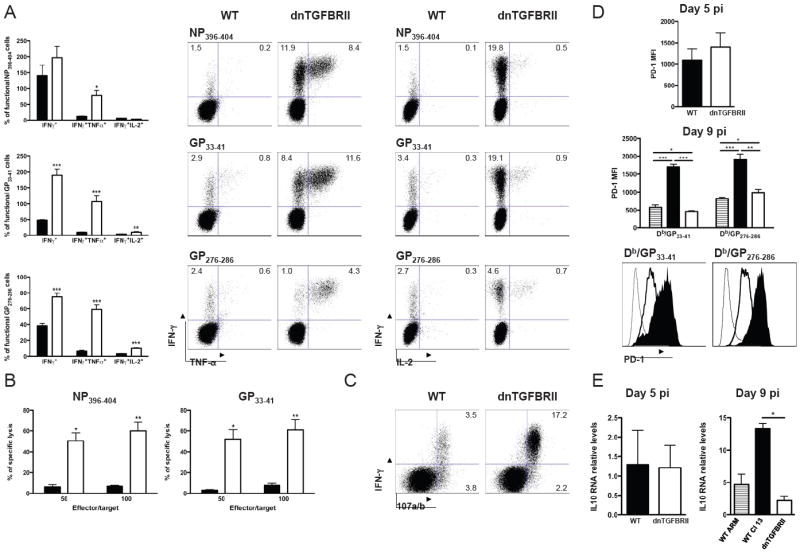
WT (black bars or histograms) or dnTGFBRII (white bars or histograms) mice were infected with LCMV Cl 13 and splenocytes obtained at day 9 pi unless otherwise stated. A) Cells were stimulated with NP396-404, GP33-41 and GP276-286 LCMV peptides and production of IFN-γ, TNF-α and IL-2 by CD8 T cells was analyzed. Bar graphs depict the average % ± sd of cytokine-producing CD8+ T cells normalized to the number of tetramer+ cells in the same spleen. Dot plots display a representative mouse for each peptide. Numbers indicate the % cells within the indicted gates. B) The cytotoxic capacity of virus-specific CD8 T cells was quantified by 51Cr release assay against targets cells loaded with LCMV peptides at the indicated ratios. The graphs show average % specific lysis ± sd. C) Cells were stimulated with GP33-41 peptide in the presence of FITC-labeled-anti-CD107a/b mAb. Dot plots represent one representative mouse per group D) PD-1 expression was quantified in Db/GP33-41+CD8+ and Db/GP276-286+CD8+ splenocytes. Bar graphs indicate the average PD-1 MFI ± sd. The histograms show representative data from one mouse per group. Shown are PD-1 expression on total CD8+ cells from uninfected controls (thin line) or on the indicated tetramer-positive dnTGFbRII (thick line) or WT (filled histogram) CD8 T cells from Cl 13 infected mice. E) IL-10 mRNA levels were quantified by real time RT-PCR and are shown as average ± sd normalized to GAPDH mRNA levels. WT-ARM infected mice at day 9 pi were processed as controls (striped bars). Results are representative of two or three independent experiments with three to four mice each. (WT vs dnTGFBRII, *p<0.05 and **p<0.005)
These results indicate that attenuation of TGF-β signaling in T cells results in a functional virus-specific CD8 T cell response and prevents the sustained up-regulation of PD-1 and IL-10. Notably, the similar levels of PD-1 and IL-10 early after infection of dnTGFBRII mice suggest that their latter reduction is due to indirect mechanisms rather than direct TGF-β modulation.
Reduced TGF-β signaling in T cells accelerates Cl 13 clearance
We next assessed whether the enhanced CD8 T cell responses observed in dnTGFBRII mice during Cl 13 infection were sufficient to eradicate the virus. For this purpose, we determined virus titers in the blood of WT or dnTGFBRII mice at days 8, 15, 45 and 49 after Cl 13 infection. In contrast to the sustained high titers in WT controls, dnTGFBRII mice had less LCMV in their blood at day 8 pi, and their viremia cleared by day 15 pi without recurrence within the 50-day-period investigated (Figure 4A). At day 15 pi, liver and brain tissues from WT mice exhibited substantial virus titers of ~107 PFU/g. In stark contrast, dnTGFBRII mice had completely purged Cl 13 from these tissues and had mostly cleared the virus from their lungs (Figure 4B). Importantly, kinetics of early infection in spleen indicate that viral growth at days 1, 3 and 5 pi was indistinguishable in WT versus dnTGFBRII mice and only became reduced or undetectable by days 9 and 15 pi, respectively (Figure 4C and 4D). Interestingly, the LCMV titers in dnTGFBRII mice at day 8 pi showed a strong negative correlation with the frequency of CD8 T cells producing IFN-γ and TNF-α upon ex-vivo GP33-41 stimulation (Supplementary Figure 7). Moreover, depletion of CD8 T cells in dnTGFBRII mice prevented virus containment (Figure 4E), providing a causal link between improved CD8 T cell responses and Cl 13 eradication in dnTGFBRII mice.
Figure 4. dnTGFBRII mice exhibit accelerated clearance of persistent LCMV.
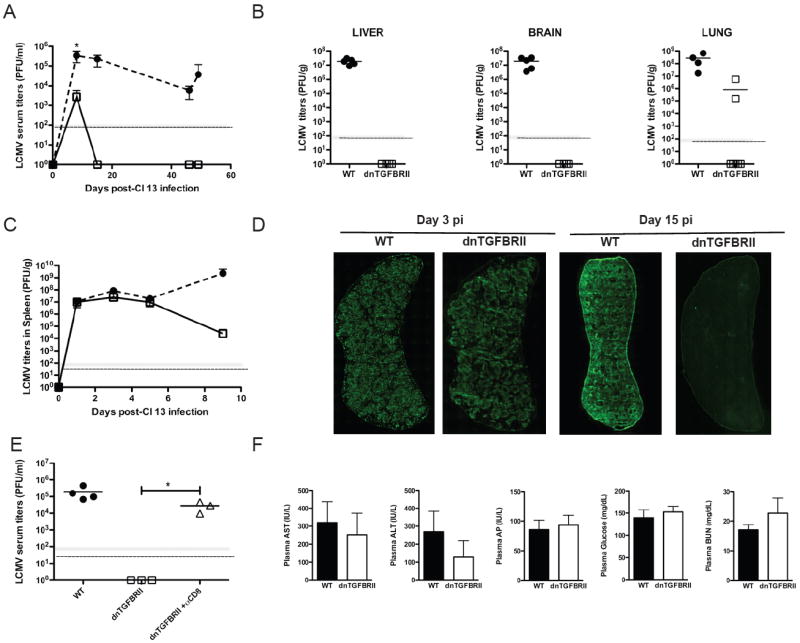
WT (black circles or bars) or dnTGFBRII (white squares or bars) mice were infected with LCMV Cl 13. A-C) Virus titers were determined by plaque assay in blood (A) and spleen (C) at the indicated time points and average LCMV titers ± sd are shown. B) The average viral titers in liver, brain and lung at day 15 pi are indicated by the lines and individual mice represented by each symbol. D) Spleen sections from WT or dnTGFBRII at day 3 or 15 pi were stained with anti-LCMV Ab. Panoramic spleen images are 10x magnification. E) dnTGFBRII mice were injected with depleting anti-CD8 mAb (white triangles) and virus titers in blood determined at day 12 pi. F) Plasma levels of the indicated molecules were determined at day 10 pi. Bar graphs show mean values ± sd. Results are representative of two or three independent experiments with three or five mice per group each. (WT vs dnTGFBRII or as indicated, *p<0.05)
Given that dnTGFBRII mice exhibit signs of auto-immunity (Gorelik and Flavell, 2000), we evaluate whether TGF-β attenuation of T cells could increase or accelerate tissue damage during viral infection. To this end, we first compared histological sections of liver, lung and stomach from ~2 month-old WT and dnTGFBRII mice at day 10 pi (Supplementary Figure 8). Cl 13 infection increased mononuclear cell infiltration in all these organs of WT as well as dnTGFBRII mice compared to uninfected controls. Consistent with their more potent T cell response, the cell infiltrate was larger in dnTGFBRII than in WT infected mice. However, no enhancement in tissue damage was observed in liver, lung or stomach of infected dnTGFBRII mice compared to WT controls. In fact, fewer vacuolated hepatocytes were observed in Cl 13-infected dnTGFBRII versus WT mice, likely due to the reduced viral burden described above. Moreover, in agreement with the 100% survival of Cl 13-infected dnTGFBRII mice until at least 66 days pi (data not shown), plasma levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (AP), glucose and blood urea nitrogen (BUN) were comparable in dnTGFBRII and WT mice at day 10 pi (Figure 4F). Creatinine levels were undetectable in all mice tested (data not shown).
These findings demonstrate that attenuation of TGF-β signaling in T cells allows for a strong CD8 T cell response in Cl 13-infected mice that prevents persistent infection without apparent enhancement of immunopathology.
Effective memory CD8 T cell response in Cl 13-infected dnTGFBRII mice
The fact that LCMV-specific CD8 T cells were detected in dnTGFBRII mice long after viral elimination (Figure 1E, day 45 pi) suggests that early control of Cl 13 infection enables the successful generation of T cell memory under conditions of constrained TGF-β signaling. To further explore this possibility, we evaluated the functional properties of these long-lasting virus-specific CD8 T cells by re-stimulation with NP396-404, GP33-41 or GP276-286 LCMV peptides at day 49 after Cl 13 infection (Figure 5A). Frequencies of IFNγ- and/or TNFα-producing clonotypic CD8 T cells were significantly elevated in dnTGFBRII compared to WT mice. Moreover, at 2 months pi, GP33-41 and GP276-286 specific T cells from dnTGFBRII Cl 13-infected mice exhibited a more differentiated memory phenotype, comparable to the one observed in WT-ARM infected mice, as indicated by up-regulation of CD127, Ly6C and CD122 (Figure 5B and Supplementary Figure 9). Finally, in contrast to primary infection, Cl 13 re-challenge in dnTGFBRII mice resulted in complete viral clearance from blood and dramatic virus reduction in the liver already by day 5 pi (Figure 6C), similar to the phenotype observed upon Cl 13 re-challenge of ARM-immune mice (data not shown). Thus, attenuated TGF-β receptor signaling in T cells allows generation of a functional and effective memory anti-viral CD8 T cell response.
Figure 5. Memory CD8 T cell response in Cl 13 infected dnTGFBRII mice.
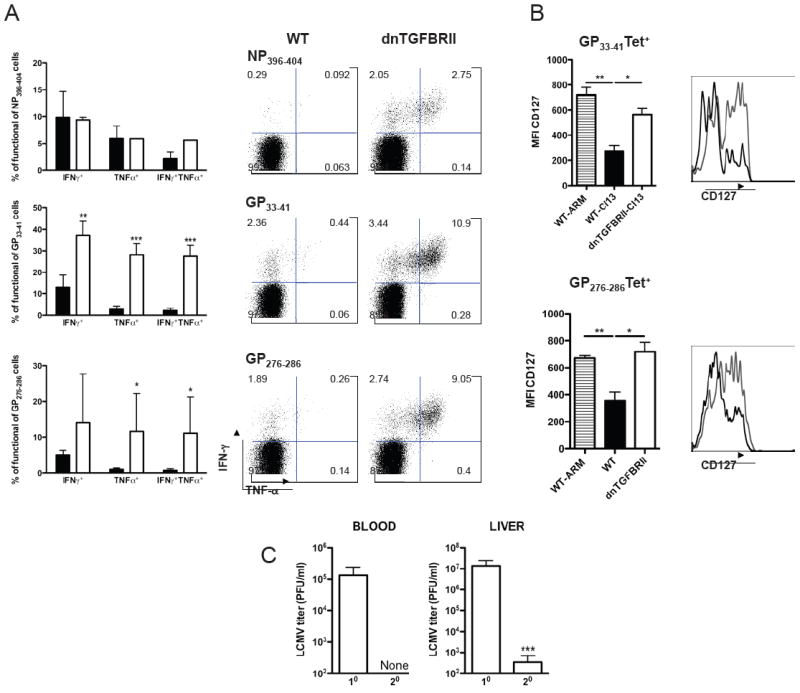
WT (black bars or histograms) or dnTGFBRII (white bars or grey histograms) mice were infected with LCMV Cl 13. A) Splenocytes were obtained at day 49 pi, stimulated with NP396-404, GP33-41 and/or GP276-286 LCMV peptides and production of IFN-γ and TNF-α by CD8 T cells was analyzed. Bar graphs depict average % ± sd of cytokine-producing CD8+ T cells normalized to the number of corresponding tetramer+ cells in the same spleen. Dot plots display one representative mouse per group; Numbers indicate the % of cells within the respective gate. B) CD127 expression was quantified in Db/GP33-41+CD8+ and Db/GP276-286+CD8+ blood cells after 2 months pi. Bar graphs indicate the average CD127 MFI ± sd. ARM infected mice were processed as controls (striped bars). Histograms depict one representative mouse per group. C) dnTGFBRII mice were re-challenged with LCMV Cl 13 (secondary, 20) and processed in parallel to primary-infected dnTGFBRII mice (10). Average viral titers in blood and liver at day 5 pi ± sd are depicted. Results are representative of two experiments with three to five mice per group. (WT vs dnTGFBRII or 10 vs 20, *p<0.05, **p<0.005 and ***p<0.0005)
Figure 6. Direct and indirect TGF-β effects on virus-specific CD8 T cells.
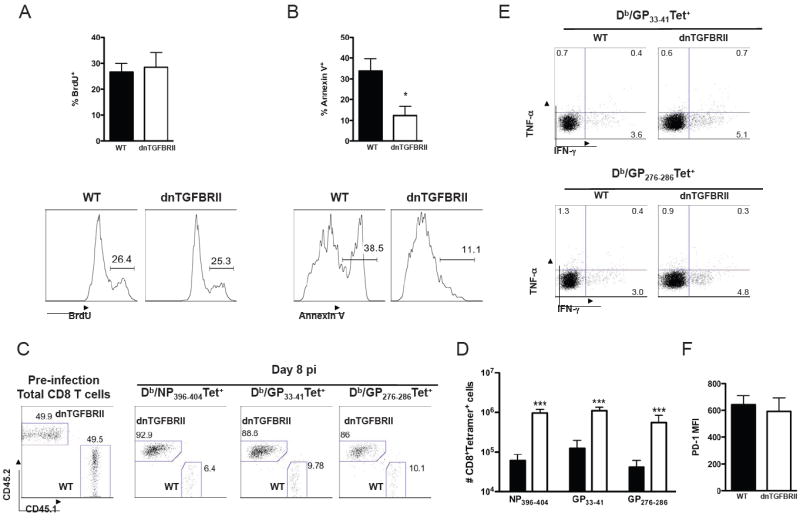
A-B) P14-WT (black bars) and P14-dnTGFBRII (white bars) CD8+ T cells were co-transfered into WT mice 1 day before LCMV Cl 13 infection. BrdU incorporation (A) and Annexin V staining (B) of P14 cells were determined at day 8 pi. Bar graphs depict the average frequency of positive cells ± sd. Histograms display a representative mouse and numbers indicate the frequency of cells within regions. C-F) WT:dnTGFBRII mixed BM chimeras were processed to analyze CD45.1+ WT and CD45.2+ dnTGFBRII CD8 T cells before (pre-infection) and at day 8 after Cl 13 infection. C) Total CD8 T cells in blood before infection and within spleen Db/NP 396-404 and Db/GP33-41 tetramer+ cells at day 8 pi. D) Total numbers of tetramer+CD8+ cells per spleen at day 8 pi. E) Production of IFN-γ and TNF-α after GP33-41 and GP276-286 LCMV peptide-stimulation at day 8 pi. F) PD-1 expression within Db/GP33-41 tetramer+ cells at day 8 pi. Dot plots display a representative mouse and numbers indicate the frequency of cells within regions. Results are representative of two independent experiments with 4-8 mice per group.
Intrinsic and extrinsic effects of TGF-β on survival and function during Cl 13 infection
The results described above indicate that CD8 T cell deletion and dysfunction during chronic infection are dependent on (and not necessary a direct consequence of) TGF-β signaling in T cells. To discriminate between direct (cell-intrinsic) versus indirect (cell-extrinsic) TGF-β effects on virus-specific CD8 T cells, we first co-injected WT (CD45.1+CD45.2+) or dnTGFBRII (CD45.2+) P14-TCR tg CD8 T cells into CD45.1+ WT recipients, infected these mice with Cl 13 one day after cell transfer and evaluated BrdU incorporation and Annexin V staining (Figure 6A and 6B, respectively). Again, we detected similar extents of proliferation, but lower Annexin V binding in P14-dnTGFBRII versus P14-WT CD8 T cells. These data were validated in WT/dnTGFBRII bone marrow (BM) mixed chimeras showing similar BrdU incorporation and reduced Annexin V staining in dnTGFBRII compared to WT DbGP33-41 and DbGP276-286-specific CD8 T cells (data not shown). Moreover, the ratio of dnTGFBRII/WT CD8 T cells in the BM chimeras was ≤1 before infection and dramatically increased up to 16 times within DbNP396-404, DbGP33-41 or DbGP276-286, -specific CD8 T cell populations at day 8 after Cl 13 infection (Figure 5C). Importantly, although dnTGFBRII mice and most of the BM chimeras showed enhanced expression of some activation markers in dnTGFBRII CD8 T cells compared to WT counterparts before infection (Supplementary Fig 2F and data not shown), similar expression of activation markers were detected in few chimeric mice before infection (Supplementary Figure 10A). Even in the later-mentioned scenario, we observed a significant increase in dnTGFBRII:WT ratio in virus-specific CD8 T cells at day 8 pi compared to uninfected total CD8 T cell (Supplementary Figure 10B and 10C). These data clearly dissociate the disparate activation of WT and dnTGFBRII total CD8 T cells before infection and the increased numbers of virus-specific CD8 T cells in dnTGFBRII-Cl13 infected mice at day 8 pi. Notably, the total numbers of virus-specific CD8 T cells recovered from BM mixed chimeras were ~10 times lower than the ones recovered in non-irradiated dnTGFBRII mice after Cl 13 infection (Figure 5D vs. Figure 1G) and apparently insufficient to control viremia by day 9 pi (7.2 × 105 ± 2.0 × 105 PFU/ml; n=8 mice), which was as high as in WT mice (Figure 4C). Similarly, ~10 times lower numbers of splenocytes and delayed viral-clearance were observed in previous experiments using WT:WT BM chimeras compared to non-irradiated WT mice during Cl 13 infection (unpublished results), suggesting an immunocompromised response of BM chimeras during Cl 13 infection. Interestingly, under the aforementioned conditions, WT and dnTGFBRII virus-specific CD8 T cells from BM mixed chimeras showed comparable functional exhaustion as judged by their ability to produce IFN-γ and TNF-α upon GP33-41 or GP276-286 peptide stimulation (Figure 5E). In addition, PD-1 expression was also comparable in WT and dnTGFBRII DbGP33-41-specific CD8 T from the mixed chimeras (Figure 5F).
Together these data demonstrate that TGF-β acts directly on virus-specific CD8 T cell to induce apoptosis and decrease cell-numbers but its effect on CD8 T cell function and PD-1 expression is indirect during Cl 13 infection.
Functional CD4 T cell response in dnTGFBRII mice infected with Cl 13
In the following series of experiments, we investigated the CD4 T cell response in dnTGFBRII and WT mice during Cl 13 infection. Assessing the numbers and frequencies of CD4 T cells specific for the IAb/GP66-77 LCMV epitope at day 9 pi revealed a 2.5-fold elevation in the numbers and a modest increase in the frequency of clonotypic LCMV-specific-CD4 T cells in dnTGFBRII compared to WT Cl 13-infected mice (Figure 7A and 7B, respectively). Furthermore, a greater number and fraction of virus-specific CD4 T cells from dnTGFBRII mice produced IFN-γ, TNF-α and IL-2 when re-stimulated with the LCMV GP66-77 peptide compared to WT controls (Figure 7A, 7C and 7D). Determination of viremia in CD4-depleted dnTGFBRII mice showed partial or complete viral containment (Figure 7E), indicating that the persistent-prone virus was still repressed in the absence of CD4 T cells.
Figure 7. dnTGFBRII mice mount a potent anti-viral CD4 T cell response.
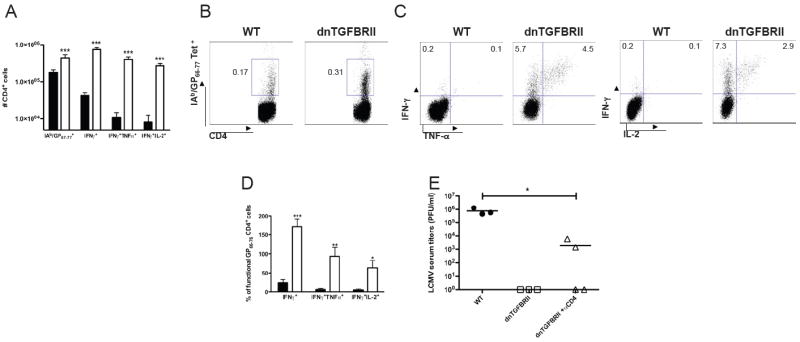
WT (black bars or black circles) or dnTGFBRII (white bars or white squares) mice were infected with LCMV Cl 13 and splenocytes obtained at day 9 pi. (A-D) Virus-specific CD4+ T cells were stained with IAb/GP66-77 tetramers or stimulated with GP66-77 LCMV peptide to analyze the production of IFN-γ TNF-α and IL-2. (A) Average number ± sd of total IAb/GP66-77+ or cytokine producing CD4+ cells per spleen. (B, C) Dot plots represent one representative mouse per group; Numbers denote the % cells within each gate. (D) Mean percentages ± sd of cytokine-producing CD4+ T cells normalized to the number of IAb/GP66-77 tetramer+ cells in the same spleen. E) dnTGFBRII mice were injected with depleting anti-CD4 mAb (white triangles) and virus titers in blood determined at day 12 pi. Data are representative of two or three independent experiments with three to four mice each. (WT vs dnTGFBRII, or dnTGFBRII-anti-CD4, *p<0.05, **p<0.005 and ***p<0.0005)
These data indicate that diminished TGF-β signaling in T cells during LCMV Cl 13 infection promotes a strong virus-specific CD4 T cell response that contributes to but is not essential for viral control.
DISCUSSION
Viral persistence in humans and mice has been previously associated with insufficient numbers and/or function of CD8 T cells (Gruener et al., 2001; Klenerman and Hill, 2005; Kostense et al., 2002; Moskophidis et al., 1993; Shankar et al., 2000; Shin and Wherry, 2007; Zajac et al., 1998). Our study reported here demonstrates for the first time that enhanced and sustained TGF-β/Smad signaling is a key molecular feature that directly compromises the magnitude of CD8 T cell responses during chronic viral infection in vivo. Remarkably, by itself, the attenuation of TGF-β signaling on T cells prevents CD8 T cell dysfunction in a cell extrinsic fashion and dramatically switches the outcome of the infection enabling rapid viral eradication instead of persistence. Considering that numerous other features of chronic infections are shared between mice and humans (Klenerman and Hill, 2005; Shi and Massague, 2003), it is likely that the effect of TGF-β on T cells represents a common pathway leading to CD8 T cell deletion, exhaustion and viral persistence in different hosts. Indeed, previous reports indicate that TGFβ blockade in vitro restores the response of T cells from HCV- or HIV-infected humans, or from simian immunodeficiency virus (SIV)-infected macaques (Alatrakchi et al., 2007; Cumont et al., 2007; Garba et al., 2002). Although these cell-culture-based studies cannot assess the impact of TGF-β on viral persistence or T cell responses within the immunosuppressive environment in vivo, they suggest that inhibition of anti-viral T cells by TGF-β is a generalized event during several chronic infections.
Consistently with previous studies demonstrating TGF-β production after acute LCMV infection (Su et al., 1993; Su et al., 1991), we found that TGF-β/Smad signaling is initiated early in virus-specific CD8 T cells after both acute and chronic viral infections. However, while Smad-2 phosphorylation decreases in functional CD8 T cells during ARM infection it is conversely enhanced and sustained in exhausted CD8 T cells during chronic Cl 13 infection. The levels of active Smad-2 correlate with enhanced TGF-β1 protein expression in virus-specific CD8 T cells, suggesting an autocrine feedback loop that perpetuates TGF-β/Smad signaling during chronic infection. Similarly, autocrine/paracrine sources of TGF-β are critical for silencing autoreactive T cells in uninfected mice (Longenecker et al., 2002; McGeachy and Cua, 2007). On the other hand, TGF-β can be produced by a variety of cell types (Rubtsov and Rudensky, 2007) and therefore other cell sources could contribute to the enhanced and persisting Smad signaling during chronic infection. A previous study indicates that TGF-β RNA and biological activity are induced upon T cell activation (Kehrl et al., 1986). Consistently, TGF-β promoter contains binding sites for NFAT and AP1 (Nakano et al., 2007), two transcription factors that become activated after TCR stimulation. Thus, it is likely that TGF-β expression is induced by TCR stimulation of virus-specific CD8 T cells during infection. In this line, the enhanced and persisting TGF-β expression during Cl13 infection could result from high and continuous antigenic stimulation of virus-specific CD8 T cells.
A recent in vivo study documented that a low number of CD8 effector versus target cells after SIV or LCMV infection correlates with partial or poor viral control and persistence (Li et al., 2009), highlighting the need to indentify the molecular factors that limit the magnitude of the ensuing immune response. We demonstrate here that enhanced TGF-β/Smad signaling induces apoptosis in a cell-intrinsic fashion and reduces the number of effector CD8 T cells available to fight the infection. Moreover, we found that attenuation of TGF-β signaling on T cells reduces the expression of the pro-apoptotic protein Bim in virus-specific CD8 T cell during chronic infection. Previous studies have demonstrated that Bim is involved in limiting anti-viral CD8 T cell responses, particularly the profound deletion of NP396-404-specific CD8 T cells, during Cl 13 infection (Grayson et al., 2006), indicating that up-regulation of Bim is at least in part responsible for TGF-β induced deletion of virus-specific CD8 T cells. However, some LCMV specific CD8 T cells are more effectively rescued in dnTGFBRII mice than in Bim-deficient mice during Cl 13 infection (Figure 1E-G and (Grayson et al., 2006)). Therefore, other TGF-β-induced apoptotic mechanisms may be in place or become active in the absence of Bim. Interestingly, the proliferation of virus-specific CD8 T cells was not enhanced in dnTGFBRII-Cl 13-infected mice. This finding contrasts with the inhibitory effect of TGF-β on the proliferation of (presumably auto-reactive) CD8 T cells under steady-state conditions ((Li et al., 2006b) and Supplementary Figure 2A). This discrepancy indicates a differential outcome of TGF-β signaling before and after infection and could reflect differential cell cycle stages at the time when TGF-β signaling becomes effective as has been described in epithelial cells (Guasch et al., 2007; Massague, 2008; Nguyen and Pollard, 2000).
Besides noting a reduction in the extent of cell death, we found that the attenuation of TGF-β signaling globally improves the functions of virus-specific T cells during chronic LCMV infection. In particular, the generation of CD8 T cells that simultaneously secrete IFN-γ, TNF-α and IL-2 has important implications, since multi-cytokine-producing T cells have been associated with better control of several human infections (Betts et al., 2006; Kannanganat et al., 2007; Seder et al., 2008). Importantly, the data obtained from WT:dnTGFBRII mixed chimeras indicate that TGF-β suppression of CD8 T cell function is mediated through indirect or cell extrinsic mechanisms during Cl 13 infection. Two recent reports demonstrate that high and persisting antigen levels cause CD8 T cell exhaustion (Bucks et al., 2009; Mueller and Ahmed, 2009). Therefore, it is likely that the increased function of CD8 T cells in dnTGFBRII-infected mice mostly results from the reduced viremia attained by the sufficiently elevated numbers of virus-specific CD8 T cell. In addition, TGF-β dependent CD8 T cell-extrinsic regulatory elements, which are absent in dnTGFBRII mice but compensated by WT cells in mixed chimeras, may also contribute to the enhanced CD8 T cell function in dnTGFBRII mice. In this regard, TGF-β exerts immunosuppressive activity on autoreactive T cells through regulatory T cell (Treg)-dependent and independent mechanisms (Li et al., 2006a; Marie et al., 2006). Given that interference with TGF-β signaling results in decreased numbers of Foxp3+CD4+ Treg cells (data not shown and (Li et al., 2006a; Marie et al., 2006)), the improved anti-viral T cell responses in dnTGFBRII-mice could result partly from reduced Treg numbers or function.
Suppression of CD8 T cells seems to result from multiple non-overlapping regulatory circuits during chronic viral infection (Barber et al., 2006; Blackburn et al., 2009; Brooks et al., 2008; Brooks et al., 2006; Ejrnaes et al., 2006; Wherry et al., 2007). It is intriguing that the functional differentiation of virus-specific T cells in dnTGFBRII mice coincides with down-regulation of previously characterized inhibitory molecules such as PD-1 and IL-10 at day 9 pi (Barber et al., 2006; Brooks et al., 2006; Ejrnaes et al., 2006). However, this is likely secondary to the decreased viral titers observed in dnTGFBRII mice since under conditions of similar viral load (day 5 pi), attenuation of TGF-β signaling in T cells does not affect the expression of IL-10 or PD-1. Consistently, WT and dnTGFBRII virus-specific CD8 T cells exposed to the same infectious environment in mixed chimeras showed comparable PD-1 expression.
The transient but detectable levels of TGF-β and p-Smad-2, as well as other inhibitory molecules (Barber et al., 2006; Blackburn et al., 2009; Brooks et al., 2006; Wherry et al., 2007), early during ARM and Cl 13 infection suggest that regulatory signals are a default mechanism that co-exist with positive T cell signals during both acute and chronic viral infections. It is interesting that, even when TGF-β/Smad signaling is clearly detected at day 5 after Cl 13 infection, numbers of virus-specific CD8 T cells are similar in WT and dnTGFBRII mice. This observation suggests that there is an initial window of time when TGF-β signaling is ongoing but its negative effect on the magnitude of CD8 T cell response is overcome or modulated by the contextual signaling. It is tempted to hypothesize that if the virus (e.g. ARM) is at least partially controlled by the ensuing immune response during such initial window of time, CD8 T cells would benefit from less antigenic stimulation and limited TGF-β signaling during the later phase of the response, when the negative effect of TGF-β is no longer restrained. On the other hand, during infection with a persistence-prone virus, such as Cl 13, which infects a large number of antigen presenting and fribroreticular cells (Li et al., 2009; Matloubian et al., 1993; Mueller et al., 2007; Sevilla et al., 2003), the virus cannot be rapidly repressed providing continuous antigenic stimulation and TGF-β induction in the later phase of the infection, when the negative role of TGF-β is fully effective. Under the later conditions, the brake imposed by TGF-β on T cells represents a determinant factor for viral persistence. Interestingly, we recently reported that inhibition of type I IFN produced by plasmacytoid dendritic cells does not correlate with the virus’ ability to replicate in such cells, suggesting that regulatory pathways may impose a brake on the innate immune system as well (Zuniga et al., 2008). It is important to note, that immunoregulatory signals may have evolved to high-tune the magnitude of the ensuing anti-viral response to prevent excessive tissue damage. However, the fact that attenuation of certain regulatory circuits during chronic infection could enhance CD8 T cells responses without drastic immunopathology (Barber et al., 2006; Blackburn et al., 2009; Brooks et al., 2008; Brooks et al., 2006; Ejrnaes et al., 2006; Velu et al., 2009), implies that there may be a window of therapeutic opportunity to boost antiviral responses during chronic infections. The potent T cell responses and rapid clearance of a persistence-prone virus that we described upon genetic attenuation of TGF-β signaling in T cells not only brings to light a novel molecular mechanism by which persistent viruses curtail immune responses but also offers promising novel opportunities for the much-needed improvement of treatment for chronic viral diseases.
EXPERIMENTAL PROCEDURES
Mice and viruses
dnTGFBRII mice (Gorelik and Flavell, 2000) were purchased at The Jackson laboratory or generously provided by Dr. Richard Flavell (School of Medicine, Yale University). C57BL/6 mice or control littermates were used as WT controls. C57BL/6 CD45.1+ and DbGP33-41 TCR-tg (P14) mice were a generous gift from Dr. Stephen Hedrick (University of California San Diego-UCSD). Mice were bred and maintained in a closed breeding facility and mouse handling conformed to the requirements of the National Institutes of Health and the Institutional Animal Care and Use Guidelines of UCSD. Unless otherwise stated, mice (6-8 weeks) were infected intravenously (iv) with 2 × 106 PFU of LCMV ARM or Cl 13. Re-challenge with LCMV Cl 13 was performed with the same dose and route of infection as primary infections. Viruses were grown, identified and quantified as described (Ahmed et al., 1984; Borrow et al., 1995).
Adoptive transfer and CD4 depletion
Where indicated CD8+ T cells were purified from the spleens of naive P14xWT (CD45.1+CD45.2+) and/or P14xdnTGFBRII (CD45.2+) mice by negative selection (StemCell Technologies), and equal number (1-10 × 103) Vβ8+ cells from each population were adoptively transferred iv into C57BL/6 CD45.1+ recipient mice 1-2 days before LCMV Cl 13 infection. Where indicated P14 cells were purified after the infection by FACs sort as described before (Zuniga et al., 2004). For CD4 T cell depletion, dnTGFBRII mice were intraperitoneally injected with control rat IgG or anti-CD4 mAb (clone GK1.5; 300 μg/mouse) at day 0, 2.5 and 9 pi. This treatment resulted in >95% reduction in the numbers of CD4 T cells at day 5 and 10 pi (data not shown). For CD8 depletion mice were injected with anti-CD8 mAb (clone 53-6.72; 200 μg/mouse) at day -2, -1, 0, and 5 after infection.
Generation of mixed BM chimeras
To obtain mixed BM chimeras, WT CD45.1+ C57BL/6 recipient mice were lethally irradiated with 1000 rads and reconstituted one day after with a mixture of BM cells from CD45.1+ WT mice and CD45.2+ dnTGFBRII mice. Bone-marrow cells were isolated from femurs and tibia of donor mice and 10 million total cells were i.v. transferred into the irradiated recipient mice. Recipient mice were treated with antibiotics (Trimethoprim 8 mg/ml and Sulfamethoxazole 40 mg/ml supplied in the drinking water) for three weeks to prevent infection and allow immune reconstitution. Reconstitution was analyzed 6-8 weeks after bone-marrow transfer and the ratio of WT:dnTGFBRII cells was determined to be ≤1 for CD8 positive cells. At this point mice were infected with LCMV Cl 13 as indicated above.
Flow cytometry
The following antibodies purchased from E-bioscience or BD-bioscience were used to stain blood or spleen cells: anti-IFN-γ--APC, anti-TNF-α-FITC, anti-IL-2-PE, anti-PD-1-PE, anti-Annexin-V-PE, anti CD45.1-PE-Cy7, anti-CD45.2 APC-Alexa-750, anti-CD44-PECy7, anti-CD62L-FITC, anti-CD4-Alexa-700, anti-CD127-PE, Anti-CD122-FITC, anti-TGFBR-PE, anti-Ly6C-biotin, Streptavidin-PercP-Cy5.5 and isotype-control-IgG-PE. Anti CD8-pacific blue was purchased from Caltag. Anti p-Smad-2/3 (Ser 423/425) was purchased from Santa Cruz Biotechnology and its control goat IgG from Jackson Immunoresearch. For staining with IAb/GP66-77, Db/NP396–404, Db/GP276–286 (provided by NIH Tetramer Core Facility; Atlanta, GA) or Db/GP33–41 (Beckman Coulter; Fullerton, CA) tetramers, cells were incubated for 1 h and 15 min at room temperature. To quantify incorporation of BrdU by tetramer+ CD8 T cells, mice were injected with 1 mg of BrdU (Sigma-Aldrich) 16 h before analysis and splenocytes stained with BrdU Flow kit (BD Biosciences) following the manufacturer instructions. Cells were acquired using the Digital LSR II flow cytometer (Becton Dickinson, San Jose, CA). Flow cytometric data were analyzed with FlowJo software.
Ex vivo-T cell stimulation
Splenocytes were stimulated with 2 μg/ml of the MHC class-I-restricted LCMV NP396–404, GP33–41 or GP276–286 peptides (all >99% pure; Synpep) in the presence of 50 U/ml recombinant murine IL-2 (R&D Systems) or PMA (10 ng/ml) and ionomicyn (0.5 μg/ml). Cells were cultured for 5 h in the presence of brefeldin A (1 μg/ml; Sigma) and stained for surface expression of CD8, fixed, permeabilized and stained with Abs to IFN-γ, TNF-α and IL-2. To evaluate cell degranulation splenocytes were incubated in the presence of anti-CD107a-FITC and anti-CD107b-FITC (BD-Biosciences). All cultures without peptide performed in parallel show no production of cytokines or degranulation.
51Cr release assays
LCMV-specific CTL activity was evaluated in splenocytes isolated at day 9 pi by standard 51Cr release assay. MHC-matched MC57 (H-2b) cells either unloaded or loaded with 1 μg/ml of LCMV NP396–404 or GP33–41 were used as targets. Cells were mixed at a 50:1 and 100:1 effector/target ratio. Samples were performed in triplicate and 51Cr release was measured in the supernatant after 5 h as previously described (Borrow et al., 1995).
Immunofluorescence Microscopy
Spleens were removed, frozen in OCT, and cut in 6-μm sections with a Leica CM3050 S Cryostat. Slides were fixed with 4% PFA for 4 min, washed in PBS, and then incubated for 1 h in 5% Normal Donkey Serum flowed by 1h incubation at RT with a guinea pig anti-LCMV Ab (1:1,500). Tissues were washed and incubated for 1 h at RT with a FITC labeled anti-guinea pig Ab (Jackson Immuno; 1:200). Fluorescence was captured at 10x using an Olympus DSU Disk Scanning Confocal Microscope, and a montage was created using SlideBook software.
SDS-PAGE and Western blotting
Cells were lysed in a Ripa buffer (Thermo Scientific) containing protease and phosphatase inhibitors (Calbiochem). Protein homogenates were run on 4-12% SDS-PAGE gels (Invitrogen) and transferred to a polyvinylidene difluoride membrane (Millipore) using a semidry transfer cell (Bio-Rad). Blots were blocked in blocking buffer (Phosphate buffer saline (PBS) containing 0.1 % tween-20 and 5% non-fat milk) and incubated with primary anti-pSmad-2, anti-Smad-2/3, anti-TGF-β1, anti-Bim or anti-phospholipase C γ (PLC- γ) mAbs (All from Cell Signaling; 1/1000 in blocking buffer) at 4°C overnight, or at room temperature for 2 h. HRP-conjugated anti-rabbit IgG mAb (Cell Signaling; 1/5000 in blocking buffer) was then added for 45 min at room temperature and ECL (GE Healthcare) was used to visualize the proteins.
Histology and biochemical blood tests
Liver, lung and stomach were obtained and fixed in 10% formalin, stained with hematoxylin-eosin and processed for histopathological analysis at the Histology and Immunohistochemistry Shared Resource (UCSD). For evaluation of AST, ALT, AP, Glucose, BUN and creatinine, plasma samples were processed at the UCSD Chemical and Coagulation Core Laboratory using a Beckman CX-7 analyzer.
Real-time RT-PCR
Total RNA was extracted from splenocytes using RNeasy kits (Qiagen), digested with DNase I (RNase-free DNase set; Qiagen) and reverse transcribed into cDNA. cDNA quantification was performed using SYBR Green PCR kits (Applied Byosistems) and a Real-Time PCR Detection System (ABI). The RNA levels of the IL-10 gene (Forward: GGT TGC CAA GCC TTA TCG GA; Reverse: ACC TGC TCC ACT GCC TTG CT) were normalized to cellular glyceraldehyde 3-phosphate dehydrogenase (GAPDH) RNA levels.
Statistical analysis
Unpaired student’s t-tests or ANOVA tests were performed using the InStat 3.0 software (GraphPad, CA.).
Supplementary Material
Acknowledgments
We thank Dr. S Hedrick and Dr. A Goldrath for critically reading the manuscript. We acknowledge Dr. M Oldstone for his generous support. We express gratitude to M. Poling, E. Lee and L. Mack for technical help. This work was supported by grants from the University of California San Diego Srce06/07 (EZ), Hellman Faculty Award (EZ) and the National Institutes of Health (AI072752 and AI081923 to EZ; AI09484 and AI45927 to MO; AI070845 to KS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmed R, Butler LD, Bhatti L. T4+ T helper cell function in vivo: differential requirement for induction of antiviral cytotoxic T-cell and antibody responses. J Virol. 1988;62:2102–2106. doi: 10.1128/jvi.62.6.2102-2106.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160:521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alatrakchi N, Graham CS, van der Vliet HJ, Sherman KE, Exley MA, Koziel MJ. Hepatitis C virus (HCV)-specific CD8+ cells produce transforming growth factor beta that can suppress HCV-specific T-cell responses. J Virol. 2007;81:5882–5892. doi: 10.1128/JVI.02202-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni C, Fisicaro P, Valdatta C, Amadei B, Di Vincenzo P, Giuberti T, Laccabue D, Zerbini A, Cavalli A, Missale G, et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J Virol. 2007;81:4215–4225. doi: 10.1128/JVI.02844-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Evans CF, Oldstone MB. Virus-induced immunosuppression: immune system-mediated destruction of virus-infected dendritic cells results in generalized immune suppression. J Virol. 1995;69:1059–1070. doi: 10.1128/jvi.69.2.1059-1070.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Ha SJ, Elsaesser H, Sharpe AH, Freeman GJ, Oldstone MB. IL-10 and PD-L1 operate through distinct pathways to suppress T-cell activity during persistent viral infection. Proc Natl Acad Sci U S A. 2008;105:20428–20433. doi: 10.1073/pnas.0811139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med. 2006;12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucks CM, Norton JA, Boesteanu AC, Mueller YM, Katsikis PD. Chronic antigen stimulation alone is sufficient to drive CD8+ T cell exhaustion. J Immunol. 2009;182:6697–6708. doi: 10.4049/jimmunol.0800997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Wynn TA, Berzofsky JA, Blatt SP, Hendrix CW, Sher A, Coffman RL, Shearer GM. Role of interleukin-10 in T helper cell dysfunction in asymptomatic individuals infected with the human immunodeficiency virus. J Clin Invest. 1994;93:768–775. doi: 10.1172/JCI117031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumont MC, Monceaux V, Viollet L, Lay S, Parker R, Hurtrel B, Estaquier J. TGF-beta in intestinal lymphoid organs contributes to the death of armed effector CD8 T cells and is associated with the absence of virus containment in rhesus macaques infected with the simian immunodeficiency virus. Cell Death Differ. 2007 doi: 10.1038/sj.cdd.4402192. [DOI] [PubMed] [Google Scholar]
- Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- Ejrnaes M, Filippi CM, Martinic MM, Ling EM, Togher LM, Crotty S, von Herrath MG. Resolution of a chronic viral infection after interleukin-10 receptor blockade. J Exp Med. 2006;203:2461–2472. doi: 10.1084/jem.20061462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garba ML, Pilcher CD, Bingham AL, Eron J, Frelinger JA. HIV antigens can induce TGF-beta(1)-producing immunoregulatory CD8+ T cells. J Immunol. 2002;168:2247–2254. doi: 10.4049/jimmunol.168.5.2247. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity. 2000;12:171–181. doi: 10.1016/s1074-7613(00)80170-3. [DOI] [PubMed] [Google Scholar]
- Grayson JM, Weant AE, Holbrook BC, Hildeman D. Role of Bim in regulating CD8+ T-cell responses during chronic viral infection. J Virol. 2006;80:8627–8638. doi: 10.1128/JVI.00855-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruener NH, Lechner F, Jung MC, Diepolder H, Gerlach T, Lauer G, Walker B, Sullivan J, Phillips R, Pape GR, Klenerman P. Sustained dysfunction of antiviral CD8+ T lymphocytes after infection with hepatitis C virus. J Virol. 2001;75:5550–5558. doi: 10.1128/JVI.75.12.5550-5558.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E. Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell. 2007;12:313–327. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PD, Belz GT, Fortner KA, Budd RC, Strasser A, Bouillet P. Apoptosis regulators Fas and Bim cooperate in shutdown of chronic immune responses and prevention of autoimmunity. Immunity. 2008;28:197–205. doi: 10.1016/j.immuni.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins G. The role of proteases in transforming growth factor-beta activation. Int J Biochem Cell Biol. 2008;40:1068–1078. doi: 10.1016/j.biocel.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Kannanganat S, Ibegbu C, Chennareddi L, Robinson HL, Amara RR. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J Virol. 2007;81:8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrl JH, Wakefield LM, Roberts AB, Jakowlew S, Alvarez-Mon M, Derynck R, Sporn MB, Fauci AS. Production of transforming growth factor beta by human T lymphocytes and its potential role in the regulation of T cell growth. J Exp Med. 1986;163:1037–1050. doi: 10.1084/jem.163.5.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenerman P, Hill A. T cells and viral persistence: lessons from diverse infections. Nat Immunol. 2005;6:873–879. doi: 10.1038/ni1241. [DOI] [PubMed] [Google Scholar]
- Kostense S, Vandenberghe K, Joling J, Van Baarle D, Nanlohy N, Manting E, Miedema F. Persistent numbers of tetramer+ CD8(+) T cells, but loss of interferon-gamma+ HIV-specific T cells during progression to AIDS. Blood. 2002;99:2505–2511. doi: 10.1182/blood.v99.7.2505. [DOI] [PubMed] [Google Scholar]
- Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landay AL, Clerici M, Hashemi F, Kessler H, Berzofsky JA, Shearer GM. In vitro restoration of T cell immune function in human immunodeficiency virus-positive persons: effects of interleukin (IL)-12 and anti-IL-10. J Infect Dis. 1996;173:1085–1091. doi: 10.1093/infdis/173.5.1085. [DOI] [PubMed] [Google Scholar]
- Letvin NL, Walker BD. Immunopathogenesis and immunotherapy in AIDS virus infections. Nat Med. 2003;9:861–866. doi: 10.1038/nm0703-861. [DOI] [PubMed] [Google Scholar]
- Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006a;25:455–471. doi: 10.1016/j.immuni.2006.07.011. [DOI] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006b;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- Li Q, Skinner PJ, Ha SJ, Duan L, Mattila TL, Hage A, White C, Barber DL, O’Mara L, Southern PJ, et al. Visualizing antigen-specific and infected cells in situ predicts outcomes in early viral infection. Science. 2009;323:1726–1729. doi: 10.1126/science.1168676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker G, Thyagarajan T, Nagineni CN, Flanders KC, Factor V, Miller G, Ward JM, Nalca A, Rangnekar VM, Thorgeirsson S, Kulkarni AB. Endocrine expression of the active form of TGF-beta1 in the TGF-beta1 null mice fails to ameliorate lethal phenotype. Cytokine. 2002;18:43–50. doi: 10.1006/cyto.2002.1025. [DOI] [PubMed] [Google Scholar]
- Marie JC, Liggitt D, Rudensky AY. Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity. 2006;25:441–454. doi: 10.1016/j.immuni.2006.07.012. [DOI] [PubMed] [Google Scholar]
- Massague J. TGFbeta in Cancer. Cell. 2008;134:215–230. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Kolhekar SR, Somasundaram T, Ahmed R. Molecular determinants of macrophage tropism and viral persistence: importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J Virol. 1993;67:7340–7349. doi: 10.1128/jvi.67.12.7340-7349.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Cua DJ. T cells doing it for themselves: TGF-beta regulation of Th1 and Th17 cells. Immunity. 2007;26:547–549. doi: 10.1016/j.immuni.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. 1993;362:758–761. doi: 10.1038/362758a0. [DOI] [PubMed] [Google Scholar]
- Mueller SN, Ahmed R. High antigen levels are the cause of T cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2009;106:8623–8628. doi: 10.1073/pnas.0809818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SN, Matloubian M, Clemens DM, Sharpe AH, Freeman GJ, Gangappa S, Larsen CP, Ahmed R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc Natl Acad Sci U S A. 2007;104:15430–15435. doi: 10.1073/pnas.0702579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano N, Hosokawa H, Kohyama M, Hozumi N. NF-AT-mediated expression of TGF-beta1 in tolerant T cells. J Immunol. 2007;178:3067–3075. doi: 10.4049/jimmunol.178.5.3067. [DOI] [PubMed] [Google Scholar]
- Nguyen AV, Pollard JW. Transforming growth factor beta3 induces cell death during the first stage of mammary gland involution. Development. 2000;127:3107–3118. doi: 10.1242/dev.127.14.3107. [DOI] [PubMed] [Google Scholar]
- Ou R, Zhou S, Huang L, Moskophidis D. Critical role for alpha/beta and gamma interferons in persistence of lymphocytic choriomeningitis virus by clonal exhaustion of cytotoxic T cells. J Virol. 2001;75:8407–8423. doi: 10.1128/JVI.75.18.8407-8423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, Schacker T, Roederer M, Douek DC, Koup RA. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B, Nascimbeni M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat Rev Immunol. 2005;5:215–229. doi: 10.1038/nri1573. [DOI] [PubMed] [Google Scholar]
- Rigopoulou EI, Abbott WG, Haigh P, Naoumov NV. Blocking of interleukin-10 receptor--a novel approach to stimulate T-helper cell type 1 responses to hepatitis C virus. Clin Immunol. 2005;117:57–64. doi: 10.1016/j.clim.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–258. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- Sevilla N, Kunz S, McGavern D, Oldstone MB. Infection of dendritic cells by lymphocytic choriomeningitis virus. Curr Top Microbiol Immunol. 2003;276:125–144. doi: 10.1007/978-3-662-06508-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar P, Russo M, Harnisch B, Patterson M, Skolnik P, Lieberman J. Impaired function of circulating HIV-specific CD8(+) T cells in chronic human immunodeficiency virus infection. Blood. 2000;96:3094–3101. [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Shin H, Wherry EJ. CD8 T cell dysfunction during chronic viral infection. Curr Opin Immunol. 2007 doi: 10.1016/j.coi.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su HC, Ishikawa R, Biron CA. Transforming growth factor-beta expression and natural killer cell responses during virus infection of normal, nude, and SCID mice. J Immunol. 1993;151:4874–4890. [PubMed] [Google Scholar]
- Su HC, Leite-Morris KA, Braun L, Biron CA. A role for transforming growth factor-beta 1 in regulating natural killer cell and T lymphocyte proliferative responses during acute infection with lymphocytic choriomeningitis virus. J Immunol. 1991;147:2717–2727. [PubMed] [Google Scholar]
- Topper JN. TGF-beta in the cardiovascular system: molecular mechanisms of a context-specific growth factor. Trends Cardiovasc Med. 2000;10:132–137. doi: 10.1016/s1050-1738(00)00061-x. [DOI] [PubMed] [Google Scholar]
- Trautmann L, Janbazian L, Chomont N, Said EA, Gimmig S, Bessette B, Boulassel MR, Delwart E, Sepulveda H, Balderas RS, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. J Virol. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - An intimate relationship. Eur J Cell Biol. 2008 doi: 10.1016/j.ejcb.2008.01.012. [DOI] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JY, Zhang Z, Wang X, Fu JL, Yao J, Jiao Y, Chen L, Zhang H, Wei J, Jin L, et al. PD-1 up-regulation is correlated with HIV-specific memory CD8+ T-cell exhaustion in typical progressors but not in long-term nonprogressors. Blood. 2007;109:4671–4678. doi: 10.1182/blood-2006-09-044826. [DOI] [PubMed] [Google Scholar]
- Zuniga EI, Liou LY, Mack L, Mendoza M, Oldstone MB. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe. 2008;4:374–386. doi: 10.1016/j.chom.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga EI, McGavern DB, Pruneda-Paz JL, Teng C, Oldstone MB. Bone marrow plasmacytoid dendritic cells can differentiate into myeloid dendritic cells upon virus infection. Nat Immunol. 2004;5:1227–1234. doi: 10.1038/ni1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.