

Abstract
To investigate the role of Arginine 276 in the conserved leucine-loop of human uracil-DNA glycosylase (UNG), the effects of six R276 amino acid substitutions (C, E, H, L, W, and Y) on nucleotide flipping and enzyme conformational change were determined using transient and steady state, fluorescence-based, kinetic analysis. Relative to UNG, the mutant proteins exhibited a 2.6- to 7.7-fold reduction in affinity for a doubled-stranded oligonucleotide containing a pseudouracil residue opposite 2-aminopurine, as judged by steady-state DNA binding-base flipping assays. An anisotropy binding assay was utilized to determine the Kd of UNG and the R276 mutants for carboxyfluorescein-labeled uracil-containing single- and double-stranded oligonucleotides; the binding affinities varied 11-fold for single-stranded uracil-DNA, and 43-fold for double-stranded uracil-DNA. Productive uracil-DNA binding was monitored by rapid quenching of UNG intrinsic protein fluorescence. Relative to UNG, the rate of intrinsic fluorescence quenching of five mutant proteins for binding double-stranded uracil-DNA was reduced approximately 50%; the R276E mutant exhibited 1% of the rate of fluorescence quenching of UNG. When reacted with single-stranded uracil-DNA, the rate of UNG fluorescence quenching increased. Moreover, the rate of fluorescence quenching for all the mutant proteins, except R276E, was slightly faster than UNG. The kcat of the R276 mutants was comparable to UNG on single-stranded DNA and differentially affected by NaCl; however, kcat on double-stranded DNA substrate was reduced 4–12-fold and decreased sharply at NaCl concentrations as low as 20 mM. Taken together, these results indicate that the effects of mutations at Arg276 were largely limited to enzyme interactions with double-stranded uracil-containing DNA, and suggested that mutations at Arg276 effectively transformed UNG into a single-stranded DNA-specific uracil-DNA glycosylase.
Keywords: Arginine 276, Single-stranded DNA, Uracil-DNA glycosylase
1. Introduction
Uracil residues are introduced into DNA by misincorporation of dUMP in place of dTMP by DNA polymerase during the DNA replication, and by deamination of existing cytosine residues [1]. If left unrepaired, uracil-DNA may disturb protein–DNA interactions (U·A base pairs) or result in G·C to A·T transition mutations (U·G base pairs) [2,3]. Repair of uracil residues in DNA is initiated by uracil-DNA glycosylase, which catalyzes the hydrolysis of the N-glycosylic bond joining the uracil base to the deoxyribose phosphate backbone of DNA to produce a free uracil base and an apyrimidinic/apurinic (AP) site in DNA [4]. Removal of the uracil base constitutes the first step in the multi-step DNA repair pathway referred to as uracil-initiated base excision repair (BER) [1].
The nuclear (UNG2) and mitochondrial (UNG1) forms of human uracil-DNA glycosylase are generated by alternative splicing and transcription from different positions in the first exon of UNG gene. Thus, the nuclear and mitochondrial enzymes have different N-termini but share an identical core catalytic domain (UNG) [5]. The X-ray crystal structure of UNG has been solved and the enzyme’s properties characterized [6–11]. Based on the structural studies of UNG in complex with DNA structures (see Fig. 1A), it was proposed that UNG excises uracil residues from DNA via a “pinch–push–pull” catalytic mechanism [9,10]. The proposed mechanism consisted of three steps: (1) UNG compresses the duplex DNA phosphate backbone via the action of two conservative motifs, 4-Pro-Ser loop and the Gly-Ser loop, while scanning the DNA minor groove for uracil residues using the minor groove reading head, which consists of Tyr275 and Arg276 residues; (2) upon locating a uracil residue, the side-chain of conserved leucine residue of the leucine-loop (268-HPSPLSVYR-276 of UNG) inserts into the DNA minor groove, pushes the uracil nucleotide out of the DNA helix, and occupies its helical space; (3) the extrahelical uracil nucleotide is then pulled into the conserved uracil specificity pocket, where the uracil base is subjected to conformational strain which is relieved by glycosylic bond cleavage. X-ray crystallographic studies of free UNG and UNG in complex with DNA revealed that the enzyme underwent a global conformational change upon binding uracil-DNA [10]. Concerted loop movements clamp the enzyme around the flipped-out uracil nucleotide, driving the transition from an open unbound state to a closed bound state. This discrete uracil-DNA-induced conformation change was accompanied by the insertion of the Leu272 side chain into the DNA minor groove [11].
Fig. 1.

Tertiary structure of human uracil-DNA glycosylase in complex with DNA. (A) Co-crystal structure of UNG in complex with DNA containing the non-hydrolyzable uracil analog 2′-deoxypseudouridine (dψU) based on the coordinates (PDB 1EMH) submitted by Parikh et al. [24] The ball-and-stick DNA structure is located in the upper portion of panel A. The uracil-containing DNA strand and its complement are colored yellow and silver, respectively. The view is looking into the DNA minor groove. UNG, which contains four central β-sheets (grey strands) surrounded by α-helixes (green cylinders), is located in the lower portion of panel A. Several of the conserved motifs of the UNG polypeptide backbone critical to the proposed “pinch–pull–push” catalytic mechanism are indicated. (1) Two motifs (orange), 4-Pro-Ser (165-PPPPS-169) and Gly-Ser (246-GS-247) loops, compress the DNA phosphate backbone flanking the uracil residue from the 5′ and 3′ directions, respectively. (2) The Leu272 (purple), located in the conserved leucine-loop (268-HPSPLSVYR-276), penetrates the DNA base stack (“push”) into the DNA minor groove and occupies the helical space of the flipped-out dψU residue (ψU in the figure). (3) The uracil-specificity pocket of UNG, which contains conserved amino acid residues Gln144, Asn204, and His268 (not shown), captures (“pull”) and stabilizes the expelled extrahelical dψU. The location of Arg276 (red) is indicated by an arrow. (B) The leucine-loop residues 271-PLSVYR-276 are shown in yellow, except for Arg276, which is orange. A portion of the oligonucleotide sequence 3′-CTAψU-5′ is shown in silver. The ηN of the Arg276 guanidinium side chain (nitrogen atoms, blue balls) is shown to interact (black rippled lines) with the 3′ phosphate of the thymine residue (oxygen atom, red ball). The εN participates in water-bridged (water, black ball) hydrogen bonds (black rippled lines) with the N3 of adenine (blue ball) and the carbonyl group (red ball) of Leu272. Structures were drawn with the Cn3D 4.1 software program obtained from the Molecular Modeling Database of the National Center for Biotechnology Information. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
Mutational studies have indicated that the amino acid residues His268, Ser270, and Leu272 in the leucine-loop are critical to UNG activity. His268 appears to be involved in enzyme active site chemistry, whereas Ser270 is required for DNA phosphate interactions [6]. Substitution of Leu272 with alanine altered DNA binding affinity as well as catalytic activity on both U·G- and U·A-containing DNAs [10]. Although the roles of His268, Ser270, and Leu272 in the leucine-loop in enzyme catalysis have been studied [6,10], the function of conserved Arg276, which is located at the C-terminal end of the leucine-loop, remains unknown. As illustrated in Fig. 1B, the ε and η nitrogens of the Arg276 guanidinium side chain are structurally competent to interact with the third DNA phosphate 3′ to the uracil residue, and to participate in water-bridged hydrogen bonds with the N3 of the purine adjacent to the uracil nucleotide as well as with the carbonyl group of Leu272 [9–11]. These observations suggest that Arg276 may stabilize the leucine-loop and Leu272 side chain either before or after it is inserted into the DNA minor groove. Recent mutational analysis of Arg276 demonstrated that substitutions at Arg276 resulted in reduction of steady-state uracil-DNA binding, base flipping, and enzyme catalysis [12].
To further investigate the role of Arg276 in leucine-loop stabilization, we have studied the effect of mutating R276 to C, E, H, L, W, and Y on uracil-DNA binding, uracil flipping, and enzyme conformational change, using fluorescence-based transient and steady-state kinetic analysis. In addition, the relative catalytic activity of the enzymes toward single-and double-stranded uracil-containing DNA was determined. The experiments have provided additional insights into the function of Arg276 in the UNG leucine intercalation loop, and demonstrate that mutations at Arg276 effectively transform UNG into a single-stranded DNA-specific uracil-DNA glycosylase.
2. Materials and methods
2.1. Materials
Plasmid pET-22b was purchased from Novagen, and plasmid pRP (Cmr) from Stratagene. E. coli strain BLR was purchased from Novagen. Chloramphenicol was obtained from Sigma, and ampicillin from Fisher Scientific, Inc. E. coli Endo IV was obtained from B. Demple (Harvard University).
2.2. Oligonucleotides
Commercial gel-purified synthetic deoxyribonucleotides, 2′-deoxyuridine-25-mer (U-25-mer; 5′-GGGGCTCGTAUAAGGAATTCGTACC-3′), 2′-deoxypseudouridine-25-mer (ψU-25-mer, 5′-GGGGCTCGTAψUAAGGAATTCGTACC-3′), 2′-deoxyadenine-25-mer (A-25-mer, 5′-GGTACGAA-TTCCTTATACGAGCCCC-3′), and 2-aminopurine-25-mer (2AP-25-mer, 5′-GGTACGAATTCCTT2APTACGAGCCCC-3′) were obtained from TriLink Biotechnologies, Inc. Duplex oligonucleotides, dsU·A-25-mer and ψU·2AP-25-mer were hybridized as previously described by Chen et al. [12]. Annealing was confirmed by 15% native polyacrylamide gel electrophoresis analysis and the decrease of 2-aminopurine fluorescent emission at wavelength 370 nm. Single-stranded carboxyfluorescein (FAM) 5′-end labeled oligonucleotide 5′-FAM-U-25-mer (5′-FAM-GGGGCTCGTAUAAGGAATTCGTACC-3′) was obtained from Invitrogen, Inc., and Integrated DNA Technologies. Duplex 5′-FAM-U·A-25-mer was prepared as described previously [12].
2.3. Protein purification
The core catalytic domain of human uracil-DNA glycosylase (UNG) containing six histidine residues on the N-terminus was cloned into pET22b, overproduced in BLR/pRP, and purified as described by Chen et al. [12]. Arg276 mutant proteins (R276C, R276E, R276H, R276L, R276W, and R276Y) were also overproduced and purified as described [12]. The protein concentrations were determined by the Bradford reaction [13] using Bio-Rad Protein Assay.
2.4. Steady-state fluorescence measurements
Excitation and emission spectra of the 2AP-containing oligonucleotides were measured using a SLM-Aminco model 8100 series 2 fluorescence spectrometer equipped with a quartz cell cuvette (1 cm × 1 cm). Reactions (2 ml) were performed in buffer A (30 mM Tris–HCl (pH 7.4), 1 mM EDTA, 1 mM DTT, 5% glycerol, w/v) at 25 °C. The excitation spectra of 2AP-containing oligonucleotides were recorded over the wavelength range of 250–350 nm with an excitation wavelength at 310 nm, while the emission spectra were recorded over the wavelength range of 330–500 nm with an excitation wavelength at 310 nm. The spectral band-pass slit was 8 nm for the emission spectra. For the time-based scans of 2AP-containing oligonucleotide samples, the excitation wavelength was set to 310 nm and the emission was monitored at 370 nm with sampling at 1 s intervals for 60 s. Samples were incubated 10 min at 25 °C prior to fluorescence measurements, which were taken in triplicate.
The dissociation constant (Kd) for duplex dψU·2AP-25-mer (100 nM) was determined by measuring 2AP fluorescence enhancement with varying amounts (0–2 μM) of UNG or Arg276 mutant protein. The binding data were fitted to Eq. (1) [14] after subtracting the background UNG or Arg276 mutant protein fluorescence intensity from each measurement:
| (1) |
In Eq. (1), F represents the percentage of fluorescence enhancement, Lo and Ro the total concentrations of DNA substrate and UNG or Arg276 mutant protein, respectively, Kd the overall dissociation constant for UNG or mutant protein binding to dψU·2AP-25-mer, and Z represents the ratio of UNG- or R276X-bound dψU·2AP-25-mer fluorescence to free dψU·2AP-25-mer fluorescence.
2.5. Pre-steady-state fluorescence measurements
Stopped-flow fluorescence experiments were performed using an Applied Photophysics SX18MV Stopped Flow Spectrophotometer (dead time = 1.0 ms). All experiments were performed in the buffer A at 25 °C. To measure changes in protein tryptophan fluorescence, samples were excited at 290 nm, slit width 7 nm, and the fluorescence emission was monitored using a 320-nm long pass filter (Applied Photophysics). For each preparation, 10 kinetic traces were averaged for kinetic analysis. All kinetic experiments were performed under pseudo-first-order conditions with DNA added at a 10-fold molar excess over the enzyme. Values for the various parameters were derived by non-linear curve fitting using the computer program Origin Version 7.0 (MicroCal Software Inc.). The observed rate constant of enzyme conformational change was determined using a fixed amount of UNG or Arg276 mutant protein combined with an excess amount of DNA substrate. The averaged tryptophan fluorescence traces were fit to Eq. (2):
| (2) |
In Eq. (2), Ft represents the fluorescence at time t, and ΔF1, ΔF2, and ΔF3 represent the amplitudes of the fast fluorescence decrease, slow fluorescence decrease, and recovery of fluorescence kinetic phases, respectively. The observed rate constants of these kinetic phases are given by k1, k2, and k3. C is the constant offset and t is the reaction time. Due to the multiphasic (>3) nature of the typical tryptophan fluorescence recovery phase, accurate resolution of the individual exponentials of this process was not possible, and only the fits for the fastest recovery phase (kR) are given.
2.6. Anisotropy binding assay
A steady-state fluorescence anisotropy binding assay was developed to measure the binding of UNG and the R276 mutant enzymes to single-stranded U-25-mer and double-stranded U·A-25-mer DNA. The uracil-containing strand was 5′-end labeled with 6-carboxyfluorescein (FAM). The steady-state anisotropy of FAM was measured at 25 °C with a Cary Eclipse fluorescence spectrophotomer equipped with a Peltier thermostatted single-cell holder by exciting at 495 nm and monitoring emission at 518 nm with 5 nm slit widths. Binding reactions (350 μl) were prepared in buffer B (25 mM Hepes·KOH, 0.5 mM EDTA, 1 mM DTT, 25 μg/ml acetylated bovine serum albumin) and contained 100 nM FAM-labeled DNA. For reactions containing single-stranded DNA, anisotropy was measured at R276 enzyme concentrations of 0, 100, 200, 400, 600, 900, and 1200 nM; for reactions containing double-stranded DNA, anisotropy was measured at enzyme concentrations of 0, 200, 400, 600, 800, 1200, and 1800 nM. Reaction mixtures were equilibrated at 25 °C for three minutes prior to fluorescence measurements. Anisotropy values (r) were calculated using Eq. (3), where IVV and IVH are the vertically and horizontally polarized fluorescence intensities, respectively, and g is the grating factor used to correct for wavelength-dependent polarization bias:
| (3) |
The anisotropy values obtained from Eq. (3) at each enzyme concentration were expressed as the fraction increase (Z) relative to the anisotropy value of the DNA only reaction and fitted to Eq. (1) in order to determine the dissociation constant (Kd).
2.7. Uracil-DNA glycosylase assays
The uracil-DNA glycosylase activities of UNG and the R276E mutant enzyme were compared on single-stranded 5′-FAM-U-25-mer and double-stranded 5′-FAM-U·A-25-mer DNA substrates. Reaction mixtures (20 μl) were prepared in buffer B and contained 1.6 pmol–4.4 fmol of UNG or R276X mutant protein (diluted in buffer B) and 5 pmol of either single-stranded 5′-FAM-U-25-mer or double-stranded 5′-FAM-U·A-25-mer. Following incubation at 37 °C for 15 min, enzyme reactions were stopped with the addition of Ugi (1 μg) and placed on ice. Reaction mixtures containing single-stranded 5′-FAM-U-25-mer were supplemented with 2.4 μl of buffer containing 3 M NaOH and 0.3 M EDTA, subjected to heat treatment at 90 °C for 6 min, and neutralized by the addition of 2.4 μl of 3 M HCl. Reaction mixtures containing duplex 5′-FAM-U·A-25-mer were supplemented with 2.4 μl of 0.5 M KCl to make a final [KCl] = 50 mM, and then subjected to E. coli endonuclease IV (1 unit) treatment at 37 °C for 30 min. Enzyme reactions were terminated by heating reaction mixtures at 70 °C for 5 min and then placed on ice for 3 min. Samples of the reaction mixtures were combined with an equal volume of 2× denaturing formamide dye buffer containing 95% de-ionized formamide, 1 mM EDTA, and 0.05% bromphenol blue, heated at 97 °C for 3 min, and resolved by 12% polyacrylamide, 8.3 M urea, gel electrophoresis. Gels were scanned with an FMBioII fluorescence imaging system (Hitachi Genetic Systems) and the fluorescent bands quantified by ImageQuant software (Amersham Bioscience). The percentage of product formed was calculated by dividing the amount of 5′-FAM-10-mer (product) by that of 5′-FAM-10-mer plus 5′-FAM-25-mer (substrate) and multiplying by 100.
To determine the kcat of UNG and the R276 mutant enzymes, time course reactions (70 μ1) were conducted at 37 °C in buffer B that contained 35 pmol of 5′-FAM-U-25-mer or 5′-FAM-U·A-25-mer DNA substrate, and either 0, 20, 40, 60, or 100 nM NaCl. In reactions containing 5′-FAM-U-25-mer, 1.75 fmol of wild-type or mutant enzyme was added to the reaction mixtures, and aliquots (10 μl) were removed at 0, 1, 2, 3, 4, and 5 min, stopped with Ugi (0.5 μg), and placed on ice. In reactions that contained 5′-FAM-U·A-25-mer, 14 fmol of R276 mutant enzyme was added, and aliquots (10 μl) were removed (and treated with Ugi) as follows: (1) at 0 mM NaCl, 0, 1, 2, 3, 4, and 5 min; (2) at 20 mM NaCl, 0, 2, 4, 6, 8, and 10 min; (3) at 40, 60, and 100 mM NaCl, 0, 5, 10, 15, 20, and 25 min. However, for 5′-FAM-U·A-25-mer reactions that contained UNG, 0.25 fmol of enzyme was added and aliquots were removed 0, 1, 2, 3, 4, and 5 min. All reaction time points were treated with NaOH as described above and analyzed by denaturing polyacrylamide gel electrophoresis and scanning fluorometry as described. The dependence of product formation on reaction time was determined by graphic analysis to be linear for all reactions, and kcat (min−1) was calculated by multiplying the slope (percent product formed per min) by the amount of substrate in the aliquot (5 pmol) and dividing by the amount enzyme (0.25 or 2 fmol) per aliquot.
3. Results
3.1. Effect of Arg276 mutations on UNG–DNA interactions
In order to determine the effect of mutations at Arg276 on UNG–DNA interactions, six Arg276 mutant enzymes were selected for analysis: R276C, R276L, R276H, R276E, R276W, and R276Y. These six mutant proteins were chosen based on our previous findings [12] that showed: (i) the efficiency of R276C and R276L UV-crosslinking to [32P]dψU-25-mer was comparable to that of UNG; (ii) the activity of R276H and R276E was highest and lowest, respectively, of the 18 mutant enzymes; (iii) defects in the catalytic activity of the aromatic R276W and R276Y mutant proteins were moderate, and the fluorescent properties of the R276W side chain might serve to report on changes in the Arg276 environment.
Equilibrium binding measurements were conducted using dψU·2AP-25-mer (100 nM) and increasing amounts (0–2 μM) of UNG. The average fluorescence at each UNG concentration was acquired. The net increase in 2AP fluorescence was then obtained by subtracting the fluorescence of the enzyme acquired in the absence of dψU·2AP-25-mer DNA (Fig. 2A, open triangles) from the total fluorescence intensity (Fig. 2A, open squares). As shown in Fig. 2A, 2AP fluorescence saturated at approximately 500 nM UNG. The net fluorescence enhancement (filled circles) at each UNG concentration (Fig. 2A, filled circles) was then fitted to Eq. (1), and the Kd value for UNG binding to dψU·2AP-25-mer was determined to be 185 ± 7 nM. Similarly, when dψU·2AP-25-mer (100 nM) was titrated with increasing amounts (0–2 μM) of R276C, a concentration-dependent increase of 2AP fluorescence was also observed (Fig. 2B). However, the 2AP fluorescence curve saturated at ~1500 nM R276C, and the magnitude of 2AP fluorescence enhancement at saturation was significantly reduced as compared to that of UNG (Fig. 2B). The R276C net fluorescence enhancement data were fitted to Eq. (1) and the Kd value for R276C binding to dψU·2AP-25-mer was determined to be 490 ± 49 nM. Therefore, the reduction in 2AP fluorescence intensity by R276C correlated with an increased Kd, reflecting both weaker binding and reduced forward partitioning. Identical dψU binding/2AP fluorescence enhancement experiments were conducted for the R276E, R276H, R276L, R276W, and R276Y mutant proteins, and the Kd values determined were 1428, 686, 945, 824, and 799 nM, respectively. As shown in Fig. 2C, the R276 mutant proteins displayed a 2.6–7.7-fold reduction in affinity (1/Kd) for binding-base flipping dψU·2AP-25-mer compared to UNG; R276E exhibited the largest reduction in binding affinity. Thus, mutations at Arg276 reduce UNG–DNA interactions as monitored by binding and base flipping dψU·2AP-25-mer.
Fig. 2.

DNA binding affinity of UNG and Arg276 mutants for dψU·2AP-25-mer. Changes in the 2AP fluorescence intensity of dψU·2AP-25-mer at 370 nm were determined as a function of UNG (panel A) or R276C (panel B) concentration. dψU·2AP-25-mer (100 nM) was combined with 0–2 μM UNG or R276C mutant protein in buffer A (2 ml), and the total fluorescence intensity at each addition (open squares) was monitored at λem = 370 nm (λex = 310 nm) after mixing at 25 °C for 5 min. The fluorescence intensity of each protein concentration (open triangles) without DNA substrate was also recorded under the same conditions. The net fluorescence intensity (closed circles) at each protein concentration was obtained by subtracting the protein fluorescence intensity from the total fluorescence intensity. The net fluorescence intensities were a best fit to Eq. (1), and the obtained Kd values for UNG and R276C were 185 ± 7 and 490 ± 49 nM, respectively. The Kd values for the remaining Arg276 mutant proteins were also determined. (C) The binding affinity of each Arg276 mutant protein, indicated by the corresponding single letter amino acid abbreviation, for dψU·2AP-25-mer was obtained by calculating 1/Kd for each protein and graphed relative to wild-type UNG (WT).
3.2. Effect of mutations at Arg276 on fluorescent DNA binding monitored by steady-state anisotropy
The binding affinity of the R276 mutant enzymes for single-stranded and double-stranded DNA was determined in anisotropy equilibrium binding assays that utilized 6-carboxyfluorescein (FAM) 5′-end labeled U-25-mer DNA or dψU-25-mer as described in Section 2. The anisotropy of the fluorescent DNA substrate was enhanced in the presence of increasing concentrations of R276 protein. Since the total fluorescent intensity measured at each R276 concentration did not change, the differences in anisotropy values among the R276 enzymes could be interpreted as being caused simply by differences in rotational motion, i.e., binding affinity [15]. The dependence of anisotropy enhancement as a function of R276 protein concentration was analyzed graphically, and the curves were fitted using Eq. (1) in order to determine the Kd of UNG and each R276 mutant enzyme for 5′-FAM-dψU-25-mer, 5′-FAM-U-25-mer, and 5′-FAM-U·A-25-mer (Table 1). The data presented in Table 1 show that, without exception, all R276 enzymes bound single-stranded DNA with considerably greater affinity than double-stranded DNA. The binding affinity of the R276 proteins for 5′-FAM-dψU-25-mer was generally very similar to that for 5′-FAM-U-25-mer, whereas the Kds for 5′-FAM-U·A-25-mer were higher than those determined for 5′-dψU·AP-25-mer (see above). The binding affinity of the R276L mutant for single-stranded DNA was greater (lowest Kd) than the other R276 mutants and UNG, while the R276E mutant exhibited the highest Kd (weakest binding) for both single- and double-stranded DNA substrates.
Table 1.
Determination of Kd for wild-type UNG and R276 mutants by steady-state anisotropy
| Substrate | Enzyme | Kd (nM) ± S.D.a |
|---|---|---|
| 5′-FAM-dψU-25-mer | UNG | 358 ± 80 |
| R276C | 254 ± 50 | |
| R276E | 2029 ± 594 | |
| R276H | 159 ± 13 | |
| R276L | 104 ± 13 | |
| R276W | 505 ± 114 | |
| R276Y | 242 ± 22 | |
| 5′-FAM-U-25-mer | UNG | 265 ± 36 |
| R276C | 400 ± 52 | |
| R276E | 1958 ± 514 | |
| R276H | 253 ± 31 | |
| R276L | 176 ± 18 | |
| R276W | 446 ± 55 | |
| R276Y | 430 ± 36 | |
| 5′-FAM-U·A-25-mer | UNG | 734 ± 125 |
| R276C | 4082 ± 1000 | |
| R276E | 31918 ± 18127 | |
| R276H | 1394 ± 171 | |
| R276L | 1170 ± 141 | |
| R276W | 2291 ± 664 | |
| R276Y | 1163 ± 155 |
Standard deviation.
3.3. Pre-steady-state kinetic analysis of UNG intrinsic fluorescence change
Crystal structures of free UNG and UNG in complex with uracil-DNA indicated that the transition from the open free state to the closed uracil-bound state was accompanied by insertion of the conserved leucine-loop into the DNA minor groove [9,11]. These movements effectively clamped the enzyme around the flipped-out uracil nucleotide, created the enzyme active site by bringing key residues into functional position, and inserted the leucine-loop into the DNA minor groove to hinder return of the uracil nucleotide to the base stack [11]. In pre-steady-state studies of DNA damage recognition and uracil flipping by E. coli uracil-DNA glycosylase (Ung), Stivers et al. [16] observed transient quenching of E. coli Ung intrinsic tryptophan fluorescence associated with uracil flipping. The authors proposed that the fluorescence quenching reflected an enzyme conformational change associated with the leucine-loop insertion step [16].
To determine whether a change in UNG conformation could be detected in real time, UNG was mixed with excess dsU·A-25-mer DNA and the intrinsic protein fluorescence was monitored. The kinetic trace of protein fluorescence showed a rapid fluorescence quenching in the first 15 ms followed by a slower fluorescence recovery over 2 s (Fig. 3B). This kinetic pattern was uracil-DNA-specific, since no fluorescence change was observed when UNG was reacted with dsU·A-25-mer DNA (Fig. 3A). Hence, the change in UNG intrinsic fluorescence upon uracil-DNA binding most likely reflected the structural transition of UNG from an open conformation to a closed, uracil-DNA-bound conformation. Analysis of the intrinsic fluorescence time trace revealed that rapid fluorescence quenching was a combination of two different fluorescence decreases. The faster observed rate of fluorescence quenching (kQa) was ~7-fold more rapid than the slower observed rate (kQb). Initial fluorescence recovery was monophasic, and exhibited an observed rate (kR) similar to kQb. Dependence of the observed rate constants on dsU·A-25-mer DNA concentration is shown in Fig. 3C. The results showed that the rapid rate of fluorescence quenching (kQa) (filled squares) was dependent on DNA concentration and reached the maximum rate (~150 s−1) at ≥250 nM dsU·A-25-mer. The slow rate of fluorescence quenching (kQb) (closed triangles) and fluorescence recovery (kR, open circles) appeared to be independent of DNA concentration, and remained at ~20 s−1 (Fig. 3C).
Fig. 3.

Stopped-flow time trace of UNG intrinsic protein fluorescence upon binding double-stranded DNA. The intrinsic protein fluorescence of UNG (100 nM) was monitored in a stopped-flow spectrometer after mixing with 1 μM dsT·A-25-mer (panel A) or dsU·A-25-mer (panel B). Each trace represents an average often individual acquisitions. The solid lines represent the best-fit curves to Eq. (2). Top of panel (B) residual analysis of each trace to the fit curve. (C) The intrinsic protein fluorescence change of UNG (100 nM) was monitored in a stopped-flow spectrometer after mixing with 0–2 μM dsU·A-25-mer, respectively. The observed rate constants of individual kinetic trace were obtained by fitting each trace to Eq. (2) as described in Section 2. The filled squares and filled triangles represent the rates of rapid (kQa) and slow (kQb) fluorescence quenching, respectively. The open circles represent the rate of slow fluorescence recovery (kR).
3.4. Effect of NaCl on UNG intrinsic fluorescence change
Previous reports suggested that disturbance of UNG–DNA electrostatic interactions influenced enzyme catalysis [17,18]. To examine the effect of monovalent ions (NaCl) on the change in UNG intrinsic fluorescence associated with uracil-DNA binding, UNG was reacted with dsU·A-25-mer DNA in the presence of 0–150 mM NaCl, and the change in UNG fluorescence was monitored. As shown in Fig. 4A, the magnitudes of fluorescence quenching and fluorescence recovery were gradually reduced in the presence of NaCl. The reduction in fluorescence quenching correlated with the decrease in kQa (Fig. 4B, closed squares), but both kQb and kR were insensitive to NaCl (Fig. 4B, closed triangles and open circles, respectively). The value of kQa was increased by ~34% in the presence of 25 mM of NaCl. This result was consistent with previous reports that low concentrations of NaCl stimulated uracil-DNA glycosylase activity, whereas high concentrations were inhibitory [7,19]. These results suggested that perturbation of UNG–DNA electrostatic interactions influenced both enzyme catalysis and the fast phase of enzyme conformational change in a similar manner. Thus, catalysis and conformational change were closely associated.
Fig. 4.
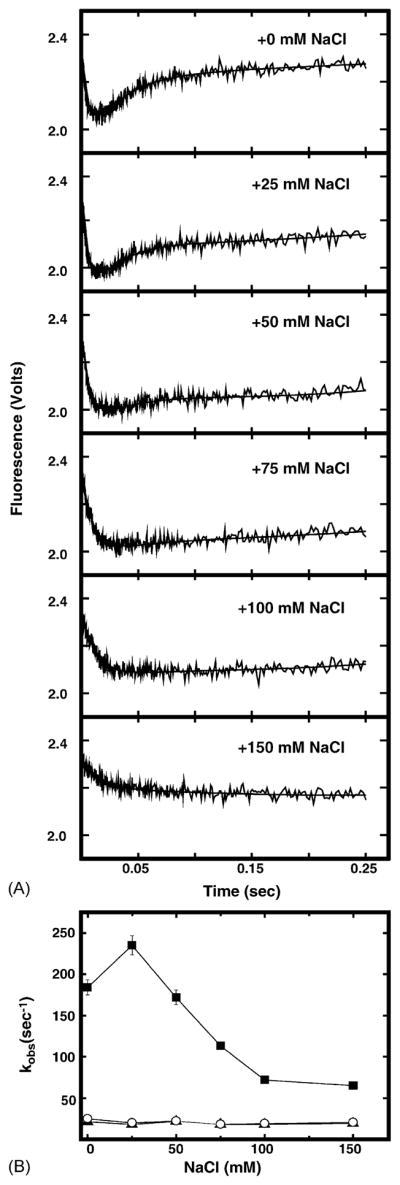
Effect of NaCl concentration on dsU·A-25-mer-induced UNG intrinsic fluorescence. (A) UNG (100 nM) was mixed at 25 °C with 1 μM dsU·A-25-mer containing 0, 25, 50, 75, 100, or 150 mM of NaCl in buffer A as indicated and the fluorescence intensity was monitored in a stopped-flow spectrometer. Each trace shown represents an average of 10 individual acquisitions. The solid lines represent the best-fit curves to Eq. (2) as described in Section 2. (B) The observed rate constants for fluorescence quenching (kQa, filled squares and kQb, filled triangles) and fluorescence recovery (kR, open circles) were obtained by fitting each kinetic trace to Eq. (2) as described in Section 2 and graphed as a function of NaCl concentration.
3.5. UNG fluorescence change associated with binding uracil in double-stranded DNA
To examine the effect of Arg276 mutations on the change in UNG intrinsic fluorescence associated with binding uracil-DNA, each Arg276 mutant protein was mixed with dsU·A-25-mer DNA and the change in protein fluorescence was monitored. As shown in Fig. 5, the change in intrinsic fluorescence of each mutant protein differed from that of wild-type UNG in several respects. First, the extent of mutant protein fluorescence quenching was significantly reduced relative to UNG (compare to Fig. 3B). Second, the fluorescence recovery phase was not detected for any mutant protein during the 250 ms time scale. A longer recovery period (2–4 s) was required before intrinsic fluorescence returned to initial intensity levels. One exception was the fluorescence of R276E, which remained essentially unchanged (Fig. 5, R276E). Third, the magnitude of fluorescence quenching of R276C, R276H, R276L, R276W, and R276Y mutant proteins was reduced, and resembled that of UNG at various concentrations of NaCl (Fig. 4A). Data analysis revealed that the kQa of each Arg276 mutant protein was significantly reduced (~28 to ~98%) compared to UNG (Fig. 7A, solid black bars); the kQa of R276E had the greatest reduction. However, both the kQb and kR values of each mutant were essentially the same as those of UNG (Fig. 7A, open and hatched bars, respectively).
Fig. 5.
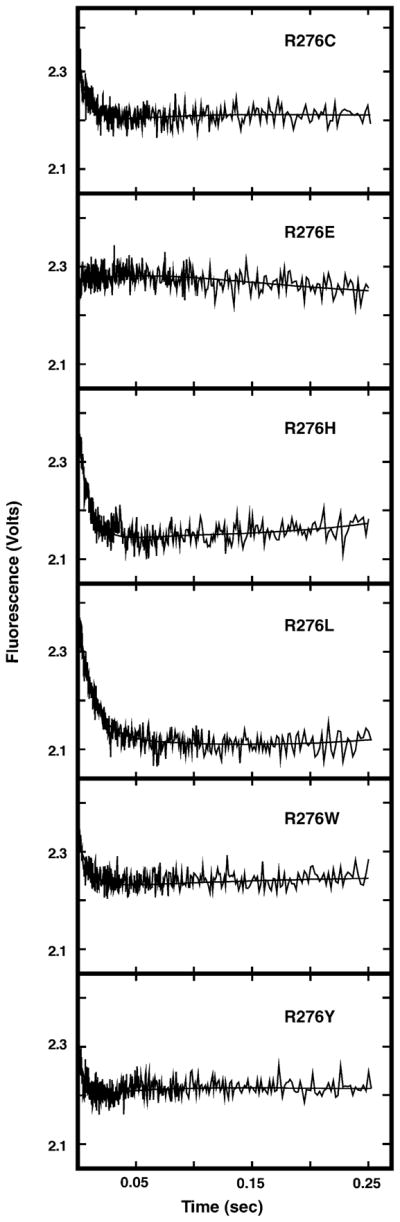
Stopped-flow time trace of Arg276 mutant protein intrinsic fluorescence change upon binding dsU·A-25-mer. Each Arg276 mutant protein (100 nM) was mixed with dsU·A-25-mer (1 μM) in buffer A at 25 °C and the intrinsic protein fluorescence monitored using a stopped-flow spectrometer. Each kinetic trace shown represents an average of 10 individual acquisitions; the identity of the Arg276 mutant protein is indicated in the upper right corner of the time trace. The solid lines represent the best-fit curves to Eq. (2) as described in Section 2.
Fig. 7.
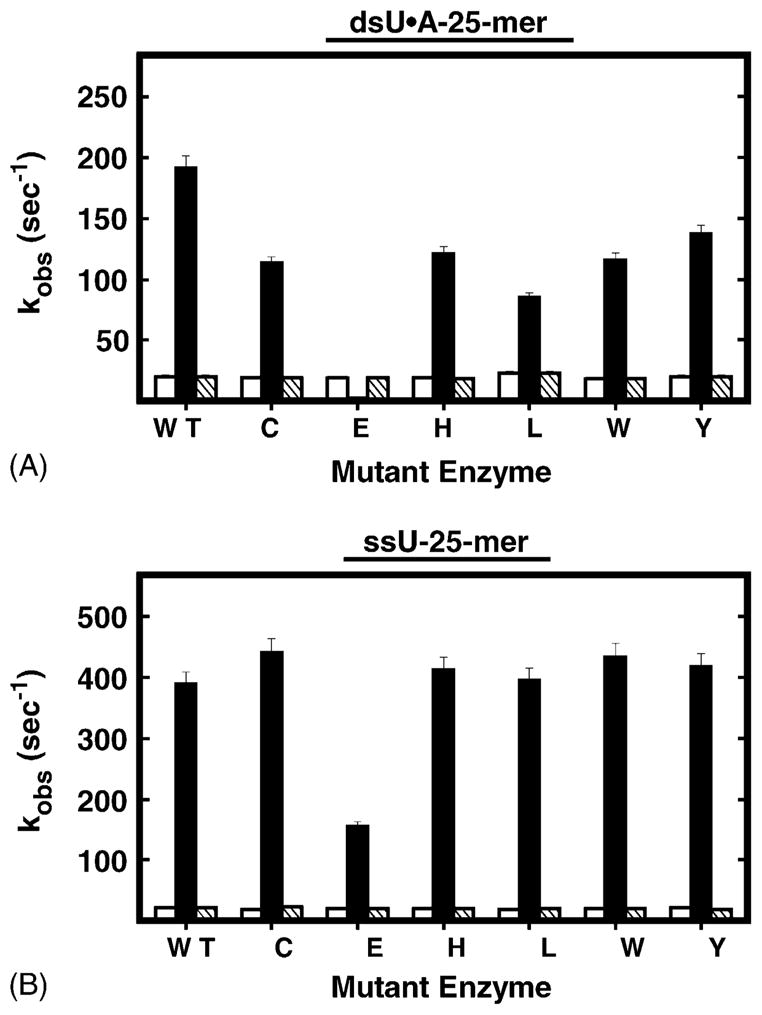
Observed rate constants for ssU-25-mer- and dsU·A-25-mer-induced intrinsic protein fluorescence change. (A) The observed rate constants associated with dsU·A-25-mer were obtained from Figs. 3B and 5. (B) The observed rate constants associated with ssU-25-mer were obtained from Fig. 6. The filled columns and opened columns represent the rates of rapid (kQa) and slow (kQb) fluorescence quenching, respectively. The hatched columns represent the rate of slow fluorescence recovery (kR). The Arg276 mutant proteins are identified by single-letter amino acid abbreviations; WT stands for UNG.
3.6. UNG fluorescence change associated with binding uracil in single-stranded DNA
Previously, we observed that the mutational effects of substitution at Arg276 on UV-crosslinking to ψU-DNA were less severe in single-stranded DNA compared to double-stranded DNA [12]. In addition, UNG had been reported to remove uracil more rapidly from single-stranded (ss) DNA than from double-stranded (ds) DNA [7]. Therefore, to further investigate the effect of Arg276 mutations on the change in UNG intrinsic fluorescence upon binding ssDNA, UNG was mixed with ssU-25-mer and monitored for changes in intrinsic fluorescence. As shown in Fig. 6, UNG (WT) exhibited rapid quenching of fluorescence in the first 15 ms, followed by a slow fluorescence recovery over ~150 ms. Notably, the fluorescence recovery from binding single-stranded U-DNA was faster than that observed for double-stranded U-DNA (Fig. 3B). Overall, the ssU-25-mer kinetic trace of UNG fluorescence change resembled that of UNG with dsU·A-25-mer (Fig. 6, WT panel, compared to Fig. 3B). Moreover, intrinsic fluorescence change was specific for binding uracil-DNA, since no fluorescence change was observed when UNG reacted with ssT-25-mer DNA (data not shown). Next, the R276C, R276E, R276H, R276L, R276W, and R276Y mutants were reacted with ssU-25-mer and monitored for intrinsic tryptophan fluorescent change. Analysis of the stopped-flow traces showed that the intrinsic fluorescence change of each mutant protein was analogous to that of wild-type UNG, exhibiting rapid fluorescence quenching in the first 15 ms, followed by slower fluorescence recovery over ~150 ms (Fig. 6). However, the recovery of R276E fluorescence to the starting level required more time (0.2–0.3 s) than did UNG (Fig. 6, panel R276E). Data analysis revealed that the kQa of each Arg276 mutant protein was essentially the same as that of UNG (Fig. 7B, solid black bars), except that the kQa of R276E was reduced by about 60%. Both the kQb and kR of each mutant were very similar to those of UNG (Fig. 7B, open and hatched bars, respectively).
Fig. 6.
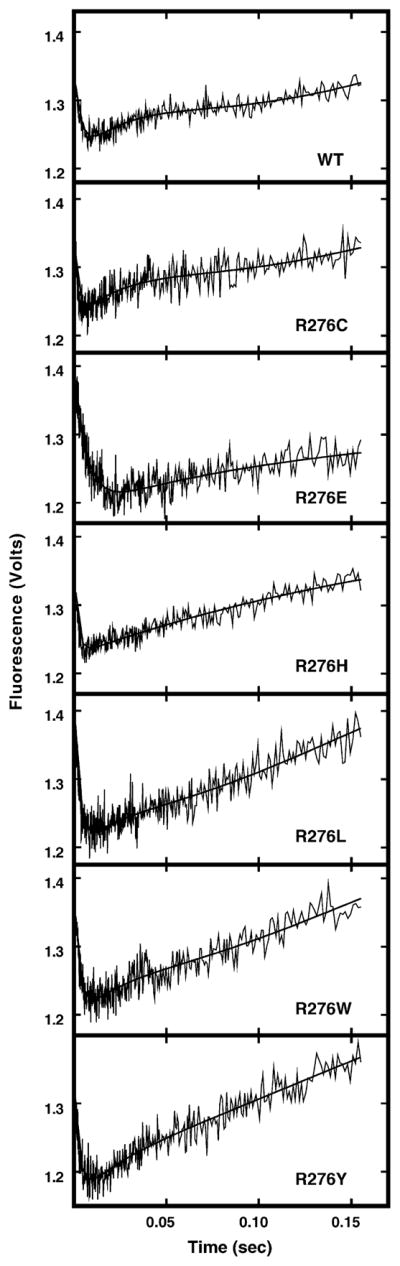
Effect of single-stranded U-25-mer DNA on the intrinsic protein fluorescence of UNG and Arg276 mutant proteins. Each Arg276 mutant protein (100 nM) was mixed with ssU-25-mer DNA (1 μM) in buffer A at 25 °C and the intrinsic protein fluorescence monitored using a stopped-flow spectrometer. Each kinetic trace shown represents an average of 10 individual acquisitions; the identity of the wild-type UNG and Arg276 mutant proteins is indicated in the lower right corner of the time trace. The solid lines represent the best-fit curves to Eq. (2) as described in Section 2.
3.7. Effect of mutations at Arg276 on uracil-excision activity
Since intrinsic protein fluorescence change correlated well with enzyme catalysis, and the effects of mutations at Arg276 on intrinsic protein fluorescence were relieved in reactions containing single-stranded uracil-DNA (Fig. 6), it was of interest to learn how these mutations would affect catalytic activity on single-stranded versus double-stranded uracil-containing DNA. In a preliminary experiment, UNG and the R276E mutant enzyme were titrated (0.22–80 nM) into excision reactions (Fig. 8) containing the same 5′-FAM-U-25-mer and 5′-FAM-U·A-25-mer DNA substrates that were utilized in the anisotropy binding assay (Table 1). Control reactions showed that >95% of the 5′-FAM-U·A-25-mer substrate was converted to 5′-FAM-10-mer product by E. coli Ung (Fig. 8A, lane C). The minor amount (<10%) of single-stranded 5′-FAM-ssU-25-mer DNA substrate that was apparently refractory to cleavage by E. coli Ung was also refractory to cleavage by UNG (Fig. 8B, lanes C and 1, respectively), and did not materially impact the experimental results. Titration of the 5′-FAM-dsU·A-25-mer cleavage reaction with increasing amounts of UNG (Fig. 8A, lanes 10–1) showed that approximately 50% of the substrate DNA was converted to product in reactions containing 8.75 fmol of UNG (Fig. 8A, lane 9). In contrast, when UNG was replaced by equal molar amounts of R276E and assayed using the 5′-FAM-dsU·A-25-mer substrate under the identical reaction conditions, little 5′-FAM-10-mer product was observed at any R276E amount less than 1.6 pmol (Fig. 8A, lanes 12–20). When UNG was reacted with the single-stranded 5′-FAM-ssU-25-mer DNA substrate, approximately 15 fmol of enzyme resulted in 50% product (Fig. 8B, lanes 7). When the single-stranded DNA uracil excision assay was repeated with R276E (Fig. 8B, lanes 11–20), the results were essentially indistinguishable from those obtained with wild-type UNG; that is, R276E appeared to be as active on single-stranded uracil-containing DNA as UNG.
Fig. 8.
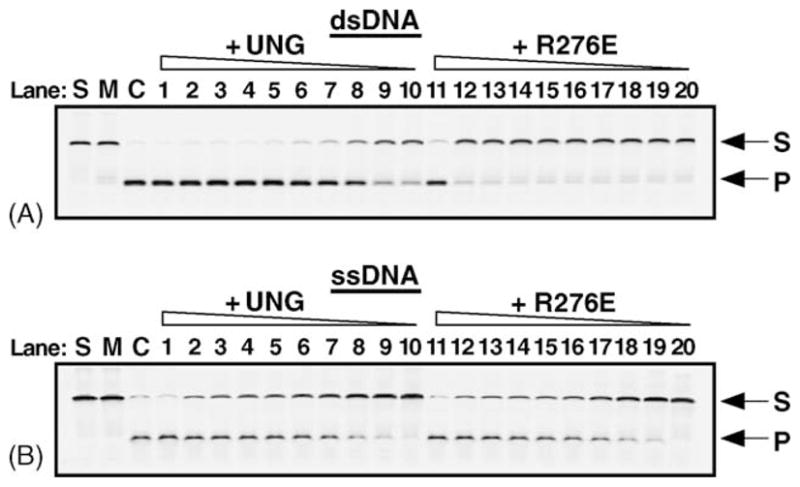
Uracil base excision activity of UNG and R276E on single- and double-stranded uracil-containing DNA. UNG and the R276E mutant enzyme (fraction IV) were assayed for uracil-DNA glycosylase activity using the 5′-end carboxyfluorescein (FAM)-labeled double- and single-stranded uracil-containing oligonucleotide substrates, 5′-FAM-dsU·A-25-mer (panel A) and 5′-FAM-ssU-25-mer (panel B). Control reactions consisted of: S, substrate alone; M, mock reaction mixtures containing substrate in reaction buffer but lacking enzyme; and C, reaction mixtures to which E. coli Ung (88 fmol) was added (lanes S, M, and C, respectively). Reactions contained 1.6 pmol, 80, 40, 30, 20, 17.5, 15, 10, 8.75, and 4.375 fmol of UNG or R276E, as indicated, in lanes 1–10, and 11–20, respectively. Following incubation at 30 °C for 15 min, reaction mixtures that contained duplex 5′-FAM-dsU·A-25-mer (A), were subjected to Endo IV (1 unit) treatment to cleave abasic (AP) sites generated by uracil excision, while the reaction mixtures containing the single-stranded substrate 5′-FAM-ssU-25-mer were subjected to hot alkaline treatment in order to cleave AP sites. Reaction products were analyzed by 12% denaturing polyacrylamide gel electrophoresis and the gels were scanned using a FMBioII fluorescence imaging system. Arrows indicate the locations of the 5′-FAM-ssU-25-mer oligonucleotide substrate (S) and 5′-FAM-10-mer uracil-excision product (P).
In order to determine the kcat of UNG and the R276 mutants, time course reactions were carried out using the 5′-FAM-U-25-mer and 5′-FAM-U·A-25-mer DNA substrates, and the reaction products were resolved by denaturing polyacrylamide and quantified by laser scanning as described in Section 2. To ascertain whether UNG and the R276 mutants exhibited differential sensitivity to NaCl, reactions contained 0–100 mM NaCl. The kcat of the R276 enzymes was determined by the linear dependence of product formation on time, and the results are reported in Table 2. Examination of these data shows that the activities of the R276 mutants on the single-stranded U-25-mer DNA substrate were comparable to UNG, but considerably reduced on the double-stranded U·A-25-mer substrate. Interestingly, the activities of UNG and the R276 mutants on single-stranded DNA were differentially affected by NaCl concentration. For example, the glutamic acid mutant (R276E) was quite active at 0 mM NaCl, but significantly inhibited at NaCl concentrations ≥20 mM. The activities of the H, L, W, and Y R276 mutants were stimulated at 20 mM NaCl compared to 0 mM NaCl, and dropped more slowly with increasing NaCl concentration compared to R276E. The R276C mutant was equally active at 0 and 20 mM NaCl, and the most active mutant enzyme on single-stranded DNA. The activity of UNG on single-stranded DNA was clearly stimulated by NaCl, and ranged from 238 min−1 at 0 mM NaCl to 1860 min−1 at 40 mM NaCl. In the absence of NaCl, UNG exhibited the slowest turnover on single-stranded DNA of the R276 enzymes; however, at 100 mM NaCl, UNG remained more than three-fold faster than any R276 mutant. When double-stranded DNA was used for substrate, the activities of the R276 mutants dropped sharply with increasing NaCl concentration. In contrast, the activity of UNG on double-stranded DNA was stimulated by NaCl up to 60 mM, and at 100 mM NaCl, the activity of UNG (kcat = 231 min−1) was still relatively robust, while that of the R276 mutants was extremely low (kcat ~ 1 min−1). Therefore, the kcat values of UNG and the R276 mutants for single- and double-stranded DNA correlated well with the rates of conformational change observed in the pre-steady-state kinetics experiments (Figs. 5–7) utilizing these DNAs.
Table 2.
| NaCl (mM) | UNG | C | E | H | L | W | Y |
|---|---|---|---|---|---|---|---|
| 5′-FAM-U-25-mer | |||||||
| 0 | 238 | 2456 | 1532 | 629 | 337 | 1004 | 461 |
| 20 | 1696 | 2539 | 387 | 1640 | 1542 | 1655 | 1667 |
| 40 | 1860 | 1363 | 601 | 1241 | 1124 | 925 | 1127 |
| 60 | 1582 | 564 | 44 | 822 | 567 | 340 | 595 |
| 100 | 822 | 155 | 17 | 175 | 260 | 82 | 171 |
| 5′-FAM-U·A-25-mer | |||||||
| 0 | 308 | 214 | 9.0 | 392 | 242 | 134 | 195 |
| 20 | 814 | 67 | 1.5 | 153 | 96 | 44 | 56 |
| 40 | 804 | 13 | 0.5 | 46 | 24 | 13 | 11 |
| 60 | 628 | 5 | 0.4 | 13 | 5 | 6 | 2 |
| 100 | 231 | 1 | 0.2 | 1 | nd | nd | |
Turnover per min.
Listed by single letter amino acid abbreviation.
4. Discussion
In this report, pre-steady-state kinetic analysis of intrinsic protein fluorescence was used to examine the effects of mutations at Arg276 on the UNG conformational change associated with binding uracil in double- and single-stranded DNA. In addition, the overall dissociation constant (Kd) of the mutant enzymes for dψU·2AP-25-mer, 5′-FAM-dψU-25-mer, 5′-FAM-U-25-mer, and 5′-FAM-U·A-25-mer was determined, and the uracil excision activity of UNG and the Arg276 mutant proteins was compared on double- and single-stranded uracil-containing DNA substrates. The results show that, without exception, mutations at Arg276 resulted in a significant reduction in the rate of enzyme conformational change associated with binding uracil in double-stranded DNA, reduced DNA binding affinity, and decreased catalytic activity on double-stranded uracil-containing DNA. However, these mutational effects were not observed when the Arg276 mutant proteins were reacted with uracil-containing single-stranded DNA.
Jiang and Stivers [20] studied the effect of replacing the key leucine-loop residue Leu191 in E. coli Ung with alanine. The extrahelical state of the uracil nucleoside analog 2′-β-fluoro-deoxyuridine opposite adenine was assessed by 2AP-fluorescence, and the enzyme conformational change by initial quenching of intrinsic protein fluorescence [20]. Their data indicated that flipping of the uracil nucleotide analog by the L191 mutant protein was unimpaired; however, the change of intrinsic protein fluorescence that typically occurs concomitantly with base flipping was not observed [20]. These results suggested that the L191A mutant protein did not effectively isomerize to the closed conformation required to lock the uracil residue in the active site [20]. The catalytic activity of the analogous UNG mutation, L272A, was less than 1% of wild-type [10]. In agreement with Stivers et al. [16], the data presented here suggest that the change in uracil-DNA glycosylase tryptophan fluorescence likely reflects the global enzyme conformational change associated with the leucine-loop insertion step. As demonstrated in Fig. 3C, the fact that the rate of change in UNG tryptophan fluorescence showed a hyperbolic dependence on uracil-DNA concentration suggested that UNG followed a multistep binding mechanism similar to that proposed for E. coli Ung [16]: formation of a weak non-specific enzyme-DNA complex, followed by reversible uracil-flipping and enzyme conformational change centered around the flipped-out base [20].
As proposed by Wong et al. [21], penetration of the leucine-loop into the DNA minor groove appears to act more as a “doorstop” to prevent the return of the flipped-out uracil residue than as a piston to push the uracil nucleotide out of the DNA helix. This hypothesis is consistent with the observation by Jiang et al. [22] that the E. coli L191 mutation could be rescued if a pyrene nucleotide were incorporated in the DNA strand opposite to the uracil base. The bulky pyrene moiety was thought to maintain the uracil nucleotide in an extrahelical state, since it filled most or all of the space in the DNA helix normally occupied by a dsU·A base pair. Extrapolating from their results, Jiang et al. [22] described the role of the Leu191 side chain as a “plug” to tilt the equilibrium of uracil flipping toward the enzyme active site pocket rather than the helical base stack. Based on the structural and kinetic work discussed above, it appears that the reduced kQa exhibited by the Arg276 mutant proteins for dsU·A-25-mer DNA was caused by defective leucine-loop intercalation, which allowed the flipped uracil nucleotide to return to the base stack. Thus, the Arg276 mutant proteins were less efficient at capturing the uracil base into the uracil-specificity pocket, and the rate of enzyme conformational change was reduced. Failure to lock in the flipped uracil and generate the active site through conformational change led to reduced uracil-excision activity on double-stranded DNA (Table 2).
The anisotropy of the 5′-FAM-U-25-mer probe increased when an R276 enzyme bound the DNA, thereby decreasing the rate of its rotational diffusion. The observed anisotropy was a population-weighted average of the anisotropy of the free and enzyme-bound fluorescent probe [23], and therefore, a direct measurement of binding affinity. The Kd of R276E for single-stranded DNA (1958 nM) was significantly elevated relative to the other R276 enzymes; however, in the absence of NaCl, R276E exhibited rapid turnover (kcat = 1532 min−1). Under the same reaction conditions, the wild-type enzyme UNG was approximately six-fold slower (kcat = 238 min−1). The addition of NaCl to the R276E activity reaction mixtures (Table 2) resulted in a precipitous decline in kcat; however, the activity of UNG was significantly stimulated by 40 mM NaCl. These observations are interesting because, given the kcat of the R276 enzymes and the enzyme concentrations used in the anisotropy binding assay, all of the uracil in the 5′-FAM-U-25-mer fluorescent probe would have been excised during the thermal equilibration period (3 min) prior to the anisotropy measurement. Therefore, the dissociation constants of the R276 enzymes were actually determined for AP-site DNA. We interpret the results of the kcat and Kd determinations to mean that tight binding of UNG to its AP-site reaction product inhibited enzymatic turnover. The introduction of NaCl to UNG reaction mixtures increased kcat most likely by increasing Kd. For those enzymes tested (UNG, R276L, and R276E), NaCl concentrations to 25 mM produced a linear decrease in anisotropy in binding reactions (data not shown). Since the Kd of R276E in the absence of NaCl was already elevated, binding to AP-site DNA was weaker and, hence, less inhibitory. Increasing NaCl concentrations reduced the activity of R276E most likely by reducing Ka. It is interesting to note the rate of conformational change, kQa, of UNG for ssDNA actually increased 34% in the presence of 25 mM NaCl and declined at higher concentrations (Fig. 4). Thus, the effect of NaCl on UNG kcat for single-stranded DNA reflected the sum of changes in kQa, Kd, and Ka. Since the rate of uracil capture, kQa, was rapid (~400 s−1) compared to kcat (238 min−1), dissociation from the AP-site DNA reaction product appeared to be rate limiting. The affinity of the R276 mutants for double-stranded AP-site DNA was 3–10-fold weaker than for single-stranded AP-site DNA, and the activity was reduced, primarily as the result of the inability of the mutants to effectively flip and bind the uracil residue (Fig. 5).
Parikh et al. [10] deduced from the UNG/U·G DNA co-crystal structure that the εN of Arg276 formed a water-mediated hydrogen bond with the Leu272 carbonyl and the N3 of the adenine (Ade6) nucleotide 3′ to the uracil nucleotide. These water-mediated interactions might not be necessary for the Leu272 side chain to facilitate docking of the uracil-nucleotide in the enzyme active site pocket. Uracil capture might be easier in ssDNA because base stacking and hydrogen bonding interactions are greatly reduced in ssDNA, and the sugar-phosphate backbone is more flexible than in dsDNA. The uracil nucleotide in ssDNA can be bound without generating torsional strain in the phosphate backbone; therefore, it has a better chance of docking in the uracil-specificity pocket, where it can make the high affinity interactions that result in enzyme conformational change. The fact that the kQa of UNG and Arg276 mutant proteins was higher for ssU-25-mer than for dsU·A-25-mer suggested that the binding affinity of UNG and the Arg276 mutant proteins for ssDNA was greater than for dsDNA, as confirmed by the anisotropy binding assays (Table 1). The slower rate of UNG fluorescence quenching, kQb, and the slow rate of fluorescence recovery, kR, may represent isomerization of the enzyme from the open to the closed, or from the closed to the open conformation, respectively. The magnitude of kQb and kR were almost identical, and both kQb and kR were independent of uracil-DNA concentration. The kQb and kR for ssDNA were almost the same as those for dsDNA. In addition, both kQb and kR were insensitive to NaCl. These results suggested that mutations at Arg276 did not affect the rate of enzyme isomerization once the uracil nucleotide was docked into the enzyme active site. Taken together, these observations show that mutations at Arg276 effectively transform UNG into a single-stranded DNA-specific uracil-DNA glycosylase.
Acknowledgments
This work is dedicated to the memory of Dr. Dale W. Mosbaugh, deceased February 17, 2004. We acknowledge Dr. Michael Schimerlik (Oregon State University) for assistance with fluorescence-based kinetic studies and critical reading of the manuscript. This work was supported by National Institutes of Health Grants GM66245 (to S.E.B.) and GM32823 (to D.W.M.), and the National Institute of Environmental Health Sciences Grant P30ES00210.
References
- 1.Mosbaugh DW, Bennett SE. Uracil-excision DNA repair. Prog Nucleic Acid Res Mol Biol. 1994;48:315–370. doi: 10.1016/s0079-6603(08)60859-4. [DOI] [PubMed] [Google Scholar]
- 2.Verri A, Mazzarello P, Biamonti G, Spadari S, Focher F. The specific binding of nuclear protein(s) to the cAMP responsive element (CRE) sequence (TGACGTCA) is reduced by the misincorporation of U and increased by the deamination of C. Nucleic Acids Res. 1990;18:5775–5780. doi: 10.1093/nar/18.19.5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duncan BK, Weiss B. Specific mutator effects of ung (uracil-DNA glycosylase) mutations in Escherichia coli. J Bacteriol. 1982;151:750–755. doi: 10.1128/jb.151.2.750-755.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B. DNA N-glycosidases. Properties of a uracil-DNA glycosidase from Escherichia coli. J Biol Chem. 1977;252:3286–3294. [PubMed] [Google Scholar]
- 5.Nilsen H, Otterlei M, Haug T, Solum K, Nagelhus TA, Skorpen F, Krokan HE. Nuclear and mitochondrial uracil-DNA glycosylases are generated by alternative splicing and transcription from different positions in the UNG gene. Nucleic Acids Res. 1997;25:750–755. doi: 10.1093/nar/25.4.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mol CD, Arvai AS, Sanderson RJ, Slupphaug G, Kavli B, Krokan HE, Mosbaugh DW, Tainer JA. Crystal structure of human uracil-DNA glycosylase in complex with a protein inhibitor: protein mimicry of DNA. Cell. 1995;82:701–708. doi: 10.1016/0092-8674(95)90467-0. [DOI] [PubMed] [Google Scholar]
- 7.Slupphaug G, Eftedal I, Kavli B, Bharati S, Helle NM, Haug T, Levine DW, Krokan HE. Properties of a recombinant human uracil-DNA glycosylase from the UNG gene and evidence that UNG encodes the major uracil-DNA glycosylase. Biochemistry. 1995;34:128–138. doi: 10.1021/bi00001a016. [DOI] [PubMed] [Google Scholar]
- 8.Kavli B, Slupphaug G, Mol CD, Arvai AS, Petersen SB, Tainer JA, Krokan HE. Excision of cytosine and thymine from DNA by mutants of human uracil-DNA glycosylase. EMBO J. 1996;15:3442–3447. [PMC free article] [PubMed] [Google Scholar]
- 9.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 10.Parikh SP, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc Natl Acad Sci USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CY, Mosbaugh DW, Bennett SE. Mutational analysis of Arginine 276 in the leucine-loop of human uracil-DNA glycosylase. J Biol Chem. 2004;279:48177–48188. doi: 10.1074/jbc.M407836200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Schimerlik MI, Peterson VJ, Hobbs PD, Dawson MI, Leid M. Kinetic and thermodynamic analysis of 9-cis-retinoic acid binding to retinoid X receptor alpha. Biochemistry. 1999;38:6732–6740. doi: 10.1021/bi9829478. [DOI] [PubMed] [Google Scholar]
- 15.Lakowicz JR. Principles of Fluorescence Spectroscopy. Plenum Press; New York: 1983. [Google Scholar]
- 16.Stivers JT, Pankeiwicz KW, Watanabe KA. Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry. 1999;38:952–963. doi: 10.1021/bi9818669. [DOI] [PubMed] [Google Scholar]
- 17.Dinner AR, Blackburn GM, Karplus M. Uracil-DNA glycosylase acts by substrate autocatalysis. Nature. 2001;413:752–755. doi: 10.1038/35099587. [DOI] [PubMed] [Google Scholar]
- 18.Jiang YL, Ichikawa Y, Song F, Stivers JT. Powering DNA repair through substrate electrostatic interactions. Biochemistry. 2003;42:1922–1929. doi: 10.1021/bi027014x. [DOI] [PubMed] [Google Scholar]
- 19.Bennett SE, Schimerlik MI, Mosbaugh DW. Kinetics of the uracil-DNA glycosylase/inhibitor protein association. J Biol Chem. 1993;268:26879–26885. [PubMed] [Google Scholar]
- 20.Jiang YL, Stivers JT. Mutational analysis of the base-flipping mechanism of uracil DNA glycosylase. Biochemistry. 2002;41:11236–11247. doi: 10.1021/bi026226r. [DOI] [PubMed] [Google Scholar]
- 21.Wong I, Lundquist AJ, Bernards AS, Mosbaugh DW. Presteady-state analysis of a single catalytic turnover by Escherichia coli uracil-DNA glycosylase reveals a “pinch–pull–push” mechanism. J Biol Chem. 2002;277:19424–19432. doi: 10.1074/jbc.M201198200. [DOI] [PubMed] [Google Scholar]
- 22.Jiang YL, Kwon K, Stivers JT. Turning on uracil-DNA glycosylase using a pyrene nucleotide switch. J Biol Chem. 2001;276:42347–42354. doi: 10.1074/jbc.M106594200. [DOI] [PubMed] [Google Scholar]
- 23.Shaw RW, Feller JA, Bloom LB. Contribution of a conserved phenylalanine residue to the activity of Escherichia coli uracil DNA glycosylase. DNA Repair. 2004;3:1273–1283. doi: 10.1016/j.dnarep.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Parikh SS, Slupphaug G, Korkan HE, Blackburn GM, Tainer JA. Crystal Structure of Human Uracil-DNA Glycosylase Bound to Uncleaved Substrate-Containing DNA. PDB 1EMH. 2000 [Google Scholar]