


Abstract
Human c-sis/PDGF-B proto-oncogene has been shown to be overexpressed in a large percentage of human tumor cells establishing a growth-promoting, autocrine growth circuit. Triplex forming oligonucleotides (TFOs) can recognize and bind sequences in duplex DNA, and have received considerable attention because of their potential for targeting specific genomic sites. The c-sis/PDGF-B promoter contains a unique homopurine/homopyrimidine sequence (SIS proximal element, SPE), which is crucial for binding nuclear factors that provoke transcription. In order to develop specific transcriptional inhibitors of the human c-sis/PDGF-B proto-oncogene, 20 potential TFOs targeting part or all of the SPE were screened by gel mobility analysis. DNase I footprinting shows that the TFOs we designed can form a sequence-specific triplex with the target. Protein binding assays demonstrate that triplex formation inhibits nuclear factors binding the c-sis/PDGF-B promoter. Both transient and stable transfection experiments demonstrate that the transcriptional activity of the promoter is considerably inhibited by the TFOs. We propose that TFOs represent a therapeutic potential to specifically diminish the expression of c-sis/PDGF-B proto-oncogene in various pathologic settings where constitutive expression of this gene has been observed.
INTRODUCTION
Platelet-derived growth factor (PDGF) is a ubiquitous, potent mitogen and chemotactic factor for many connective tissue cells, which occurs as a three-disulfide-linked dimer composed of two homologous chains, A and B (1,2). The biological function of PDGF is mediated through binding to two cell surface proteins, PDGF receptors α and β (3–5). Binding of PDGF to the extracellular part of either receptor type leads to dimerization of receptor molecules, followed by activation of the receptor protein-tyrosine kinase (6) and generation of phosphorylation-mediated signals that initiate the biological response (7,8). Ever since the discovery, in 1983, that the v-sis oncogene of the simian sarcoma virus is a retroviral homolog of the cellular gene encoding the B chain of PDGF (9,10), much attention has been focused on the relationship between PDGF-B expression and malignant transformation. The hypothesis that unscheduled production of PDGF may contribute to the growth of spontaneous tumors is supported by the finding that PDGF is frequently produced by cell lines from human tumors such as glioblastoma and fibrosarcoma (11), melanoma (12), breast carcinoma (13), lung carcinoma (14), glioma (15), esophageal carcinoma (16) and Kaposi’s sarcoma (17). Gene transfer experiments have shown that overexpression of the normal human PDGF-B gene (c-sis, proto-oncogene) can cause the generation of fibrosarcoma (18), vascular connective tissue stroma with no necrosis (19) and tumorigenic and metastatic effects (20–22). Furthermore, PDGF has been implicated in the pathogenesis of several non-malignant proliferative diseases including atherosclerosis (23–24), fibrosis (25), restenosis following vascular angioplasty (26), giant cell arteritis (27), aseptic loosening (28) and bronchiolitis obliterans syndrome (29), etc.
The accumulating evidence for the involvement of PDGF in a variety of human proliferative disorders has led to a search for specific inhibition of PDGF expression. Oligonucleotides offer enormous potential for manipulating gene function by binding selectively to DNA [antigene or triplex forming oligonucleotides (TFOs)], to RNA (antisense and ribozyme oligonucleotides) and to proteins (aptamers). By now, antisense oligonucleotides (30), ribozymes (31) and aptamers (32) have been reported to specifically inhibit PDGF expression, yet, to our knowledge, no TFOs have ever been used to inhibit c-sis/PDGF-B proto-oncogene expression on a transcription level.
Oligonucleotide-directed triplex formation involves pyrimidine-rich (pyrimidine motif; T:A-T and C:G-C triplets) or purine-rich (purine motif; T:A-T and G:G-C triplets) oligonucleotides binding to a homopyrimidine/homopurine sequence in the major groove of duplex DNA. This method allows highly sequence-specific DNA recognition (33). Interest in triplexes was revived with the finding of Cooney et al. in 1988 that a purine oligonucleotide motif formed a triplex with a region of the c-myc P1 promoter and inhibited the in vitro transcription of the c-myc gene (34). A large number of genes have since been regulated through triplex formation, including HIV (35,36), HER2/neu (37), tumor necrosis factor (38), the interleukin-2 receptor (39), dihydrofolate reductase (40), Ha-ras (41), aldehyde dehydrogenase (42), granulocyte–macrophage colony-stimulating factor (43), DNA polymerase α (44), rat α1(I) collagen (45), insulin-like growth factor type I receptor (46,47), rhodopsin (48) and bcl-1 (49), etc. In this study, we investigated the capacity of TFOs to reduce c-sis/PDGF-B proto-oncogene transcription. Twenty potential purine motif TFOs of different sequence or length were designed against a dominant regulatory element within the PDGF-B promoter. Here we present data showing that 10 of them were able to form sequence-specific triplexes with the target site, and three of them effectively reduced the transcription level of the PDGF-B proto-oncogene in PMA-treated human K562 cell line.
MATERIALS AND METHODS
Oligonucleotides
All oligonucleotides were synthesized on a Beckman-Oligo1000 DNA synthesizer and purified by preparative 15–20% polyacrylamide gel electrophoresis. Duplex oligonucleotides were prepared by mixing equal amounts of complementary single strands in the presence of 0.25 M NaCl. The mixtures were annealed and purified on a 12% polyacrylamide gel, eluted and concentrated by ethanol precipitation.
Vectors
pUC18SPE was constructed by inserting the synthetic 38 bp double-stranded oligonucleotides between the SacI and SmaI sites of pUC18 containing both SIS proximal element (SPE) and purine rich region (PRR) as shown in Figure 1. Plasmid pGL3promoter was constructed as follows: a synthetic PDGF-B promoter (–252 to +3) was constructed by a technique of oligonucleotide overlap extension, and amplified by PCR. The resulting 255 bp fragment was cloned between the SacI and SmaI sites of pUC18 to give pUC18promoter, sequenced then subcloned into the SacI–HindIII site of the pGL3basic vector (Promega). To create pneorGL3promoter and pneorGL3control, pneorBSK(+) (constructed by our group) was digested with KpnI and SacI to release the 1.5 kb neor gene fragment, which was in turn subcloned into the KpnI–SacI site of pGL3promoter and pGL3control (Promega). Plasmid pRLSV40 was obtained from Promega.
Figure 1.

Schematic diagram of the human c-sis/PDGF-B gene promoter showing the triplex target sequence: SPE (–58 to –39 bp ) and PRR (–38 to –24); TATA, the TATA box (–24 to –20); TNS, TATA neighboring sequence (–18 to –9).
Cells
Human K562 erythroleukemia cells were obtained from American Type Culture Collection (CCL243, Rockville, MD) and maintained in complete RPMI (RPMI 1640 supplemented with 10% fetal bovine serum, 1 mM l-glutamine, 10 mM HEPES, 100 U/ml penicillin and 50 µg/ml streptomycin) in a 37°C/5% CO2 incubator.
Gel mobility shift analysis of triplex formation
The synthetic coding strand of PRR (15 bp) or SPE–PRR (35 bp) triplex target sequence was labeled with [γ-32P]ATP by T4 kinase and annealed to its oligonucleotide complement. The resulting duplex was purified on a 12% polyacrylamide gel, eluted and concentrated by ethanol precipitation. Potential TFOs were heated at 95°C for 3 min to avoid self-aggregation of the G-rich oligonucleotides, then cooled on ice before adding to the labeled 15 or 35 bp targets in 20 mM sodium cacodylate–HCl, pH 7.4, 10 mM MgCl2, 150 mM KCl and incubated at 37°C for 2 h. Samples were analyzed by electrophoresis on 10–15% native polyacrylamide gels at 4°C 100 V for 4 h. Gels were dried and autoradiographed at –70°C. Both gel and running buffer contained 90 mM Tris–borate, pH 8.0, 10 mM MgCl2.
DNase I footprinting
A 262 bp fragment with HindIII(399) and PvuII(628) sites containing the SPE (439–444) was isolated from pUC18SPE. This fragment was labeled at the HindIII site with [α-32P]dATP using the Klenow fragment of Escherichia coli DNA polymerase I. After incubation at 55°C for 5 min then cooling on ice, the labeled 262 bp fragment was incubated with increasing concentrations of TFO1, 17 and 19 that had been heated at 95°C for 3 min and cooled immediately, in 20 mM sodium cacodylate–HCl, pH 7.4, 10 mM MgCl2, 150 mM KCl at 37°C for 2 h. Samples were precooled on ice, then digested with DNase I for 40 s on ice. Digestion was terminated by the addition of 20 mM EDTA in 98% formamide followed by heating at 95°C for 5 min to inactivate DNase I. Samples were quickly cooled on ice, then analyzed by electrophoresis on an 8 M urea, 9% polyacrylamide sequencing gel at 50 W. The gel was dried and autoradiographed at –70°C.
Protein binding assays
Nuclear extracts from PMA-treated K562 cells were prepared according to the method of Dignam et al. (50). A 255 bp fragment of the human PDGF-B gene promoter (–252 to +3) with EcoRI and SmaI ends isolated from pUC18promoter was labeled with [α-32P]dATP at the EcoRI site using the Klenow fragment of E.coli DNA polymerase I. The probe was incubated with increasing concentrations of TFOs in 3 µl 20 mM sodium cacodylate–HCl, pH 7.4, 10 mM MgCl2, 150 mM KCl. After preincubation at 37°C for 2 h, 6 µg PMA-treated K562 cells nuclear extracts were added, resulting in a final volume of 10 µl containing 20 mM Tris–HCl, pH 8.0, 5 mM MgCl2, 5 mM CaCl2, 0.1 mM EDTA, 0.1 mM DTT, 0.5 µg BSA, 100 mM KCl, 3% glycerol and 1 µg poly(dI-dC). The DNA–protein mixtures were kept at room temperature for 15 min, followed by electrophoresis in a 5% polyacrylamide gel in 0.25× TBE (22.5 mM Tris–borate, 0.5 mM EDTA) at 10 V/cm at room temperature for 2 h. Gels were then dried and autoradiographed at –70°C.
Transient transfection experiments
Aliquots of 40 µg pGL3promoter or pGL3control were incubated with increasing concentrations of TFOs in 20 mM sodium cacodylate–HCl, pH 7.4, 10 mM MgCl2, 150 mM KCl at 37°C for 2 h before transfection. Exponentially growing K562 cells were counted, harvested by centrifugation and resuspended in RPMI 1640 to a concentration of 1.25 × 107 cells/ml. For each transfection, 6.25 × 106 cells (0.5 ml) were mixed on ice with plasmid–TFO mixtures and 0.5 ng pRLSV40 plasmid (Promega). The cell–DNA mixtures were pulsed at 300 V/975 µF capacitance using a Bio-Rad Gene Pulser and immediately resuspended in complete RPMI. Approximately 24 h after electroporation, the cell culture volumes were treated with phorbol 12-myristate 13-acetate (PMA; final concentration 2 ng/ml; Sigma Chemical Co.). Cultures were harvested ∼48 h after electroporation by centrifugation, washed three times with cold PBS and resolved in 500 µl lysis buffer (Luciferase Assay System, Promega). After 1 h at room temperature, the lysate was collected. A 30 µl volume of the lysate was added to 30 µl of luciferase assay Reagent II (Promega) in a clear polystyrene 12 × 75 mm tube, which was immediately placed in a luminometer (Lumat LB 9507, EG&G Berthold) and light production was measured for 10 s. The first measurement was firefly luciferase activity from pGL3promoter plasmid, then 30 µl of Stop & Glu reagent (Promega) was added immediately to measure the Renilla luciferase activity from pRLSV40 plasmid. Relative firefly luciferase activity was indicated by the proportions of the first and second measurements.
Stable transfection experiments
To establish the K562 luciferase or control cell line, 20 µg pneorGL3promoter or pneorGL3control was mixed with 6.25 × 106 cells in 0.5 ml RPMI 1640 on ice and electroporated as described above. Approximately 48 h after transfection, 0.5 mg neomycin analog G418 (Gibco BRL) was added to culture medium per ml. The transfectants were replenished with fresh selection medium every 2–3 days. Resistant colonies were pooled into a 96-well plate after 2 weeks of selection and propagated in the presence of 0.5 mg/ml of G418. 6.25 × 106 cells K562 luciferase and control cells were electroporated as described above in 0.5 ml RPMI 1640 containing 50 µM TFOs. Twelve hours after electroporation, the cells were treated with PMA as mentioned above. Twenty-four hours after electroporation, cells were harvested and counted. The firefly luciferase activity of equal numbers of living cells was measured as above.
RESULTS
Selection of target sites and design of TFOs
The human c-sis/PDGF-B promoter contains a 20 bp SPE, which is the binding site for the Sp family of transcription factors. Ablation of this region results in a complete loss of promoter activity (51,52). The SPE sequence has a run of 20 pyrimidines interrupted by three T-As and one C-G, while the 15 bp PRR downstream is strictly homopurine/homopyrimidine run (Fig. 1). A series of purine motif TFOs of different sequence or length were designed (Table 1): a 16mer TFO1 was designed to target the perfect 15 bp PRR. G was placed in the oligonucleotide in apposition to each G-C pair in the target (G:GC triplet) while A was placed in apposition to each A-T pair (A:AT triplet). A 13mer TFO2 was the same as TFO1 except without three As at the 3′-end. Twelve 22mers, TFO3–14, were designed to target part of the SPE and the entire PRR. At each inversion site, C, G, A or T was aligned with the C-G (C:CG, G:CG, A:CG or T:CG triplets) and T, A or G was aligned with the T-A (T:TA, A:TA or G:TA triplets). Another two 33mers, TFO15–16, were designed to target the entire SPE and PRR, with T at the C-G and A or G at the T-A. TFOs17–20 were the same as TFOs13–16, respectively, except for the addition of three As at the 3′-end. TFOs1–20 were all antiparallel in orientation to the target purine strand, while another TFO-p, with the same sequence as TFO1, was in parallel orientation.
Table 1. Oligonucleotide sequences and their alignment with the duplex target.
| Target | 5′ T C T C C A C C C A C C T C T C G C A C T C T C C C T T C T C C T T T 3′ | Kd (µM) |
| |
3′ A
G A G G T G G G T G G A G A G C G T G A G A G G G A A G A G G A A A 5′ |
|
| TFO-p | 3′ G A G A G G G A A G A G G A A A -5′ | >50 |
| TFO1 | 5′ G A G A G G G A A G A G G A A A -3′ | 0.8 |
| TFO2 | 5′ G A G A G G G A A G A G G 3′ | 0.2 |
| TFO3 | 5′ G G A G A G C G T G A G A G G G A A G A G G 3′ | >50 |
| TFO4 | 5′ G G A G A G C G A G A G A G G G A A G A G G 3′ | >50 |
| TFO5 | 5′ G G A G A G C G G G A G A G G G A A G A G G 3′ | >50 |
| TFO6 | 5′ G G A G A G G G T G A G A G G G A A G A G G 3′ | >50 |
| TFO7 | 5′ G G A G A G G G A G A G A G G G A A G A G G 3′ | >50 |
| TFO8 | 5′ G G A G A G G G G G A G A G G G A A G A G G 3′ | >50 |
| TFO9 | 5′ G G A G A G A G T G A G A G G G A A G A G G 3′ | >50 |
| TFO10 | 5′ G G A G A G A G A G A G A G G G A A G A G G 3′ | >50 |
| TFO11 | 5′ G G A G A G A G G G A G A G G G A A G A G G 3′ | >50 |
| TFO12 | 5′ G G A G A G T G T G A G A G G G A A G A G G 3′ | 6 |
| TFO13 | 5′ G G A G A G T G A G A G A G G G A A G A G G 3′ | 1.5 |
| TFO14 | 5′ G G A G A G T G G G A G A G G G A A G A G G 3′ | 0.6 |
| TFO15 | 5′ A G A G G A G G G A G G A G A G T G A G A G A G G G A A G A G G 3′ | 0.2 |
| TFO16 | 5′ A G A G G A G G G A G G A G A G T G G G A G A G G G A A G A G G 3′ | 0.1 |
| TFO17 | 5′ G G A G A G T G A G A G A G G G A A G A G G A A A 3′ | 5 |
| TFO18 | 5′ G G A G A G T G G G A G A G G G A A G A G G A A A 3′ | 2 |
| TFO19 | 5′ A G A G G A G G G A G G A G A G T G A G A G A G G G A A G A G G A A A 3′ | 0.6 |
| TFO20 | 5′ A G A G G A G G G A G G A G A G T G G G A G A G G G A A G A G G A A A 3′ | 0.3 |
The equilibrium dissociation constant (Kd) for third strand binding was estimated as the concentration of TFO at which apparent binding was half maximal, as judged by gel mobility shift assay in Figure 2.
Triplex formation with the c-sis/PDGF-B promoter
Triplex formation was demonstrated by gel mobility shift analysis and DNase I footprinting.
In gel mobility shift assays, triplex DNA migrates more slowly than duplex DNA because of its decreased charge density. First, the 16mer TFO1 and the 13mer TFO2 in antiparallel orientation, together with the 16mer TFO-p in parallel orientation, were used to bind PRR. The results are shown in Figure 2(a–c). As expected, increasing concentrations of TFOs1 and 2 shifted the 16 bp duplex target (D) to a distinct, more slowly migrating band (T). The equilibrium dissociation constant (Kd) for third strand binding was estimated as the concentration of TFO at which apparent binding was half maximal (53). The results from darkness scanning showed that the Kd of TFO1 was at 0.8 µM, while that of TFO2 was at 0.2 µM. This 4-fold difference between TFOs1 and 2 is similar to the results obtained by Arimondo et al. (54), indicating the negative effect of the three As at the 3′-end of the TFOs on their binding affinity to the target. However, both TFOs showed complete formation of triplex DNA at 3 µM. In contrast, TFO-p with an identical sequence to TFO1 but in a parallel orientation to the target purine strand, did not show any triplex formation under the same conditions. Then, 12 22mers, TFO3–14, without the three As at the 3′-end were screened for the best choice of bases at each inversion site in the target. As shown in Figure 2(d–f), TFOs12–14 shifted the 35 bp duplex to triplex in a concentration-dependent manner. At 3 µM concentration, TFO12 shifted ∼25%, TFO13 ∼80% and TFO14 100% duplex to triplex. In contrast, TFOs3–11 did not have any effect on the target at a concentration as high as 50 µM (Fig. 2i). According to Table 1, at the C-G conversion site C, G or A were placed in TFOs3–11, while T was placed in TFOs12–14. TFO12 had a T, TFO13 an A and TFO14 a G against the T-A conversion site. These results indicate that a single base change in the TFO sequence at the mismatched site will dramatically reduce its ability to bind the duplex target, which further shows the sequence specificity of TFOs. Based on these results, another two 33mers, TFO15 and 16, were designed with a T at the C-G conversion site, an A at the first two T-A conversion sites and A or G at the third T-A conversion. As shown in Figure 2 (g and h), increasing concentrations of TFOs15 and 16 can shift the target duplex to triplex. At 3 µM concentration, both TFOs shifted 100% duplex to triplex. To explore the effects of the three As at the 3′-end of TFO, TFOs17–20 were designed with the same sequences as TFOs13–16, respectively, with the addition of three As at the 3′-end. Gel mobility shift analysis was done and the Kd was estimated as mentioned above. Triplex formation by TFOs17–20 occurred in a similar concentration-dependent manner to TFOs13–16 (data not shown). The Kd data shown in Table 1 indicates that the absence of the three As at the 3′-end provided a 4-fold increase in triplex stability, however, as shown in Figure 2j, at 20 µM, TFOs17–20 shifted 100% duplex to triplex, as did TFOs1 and 2 and TFOs13–16. Therefore, at higher concentrations, both types of TFO (with or without three As at the 3′-end) were able to form triplex quite well.
Figure 2.
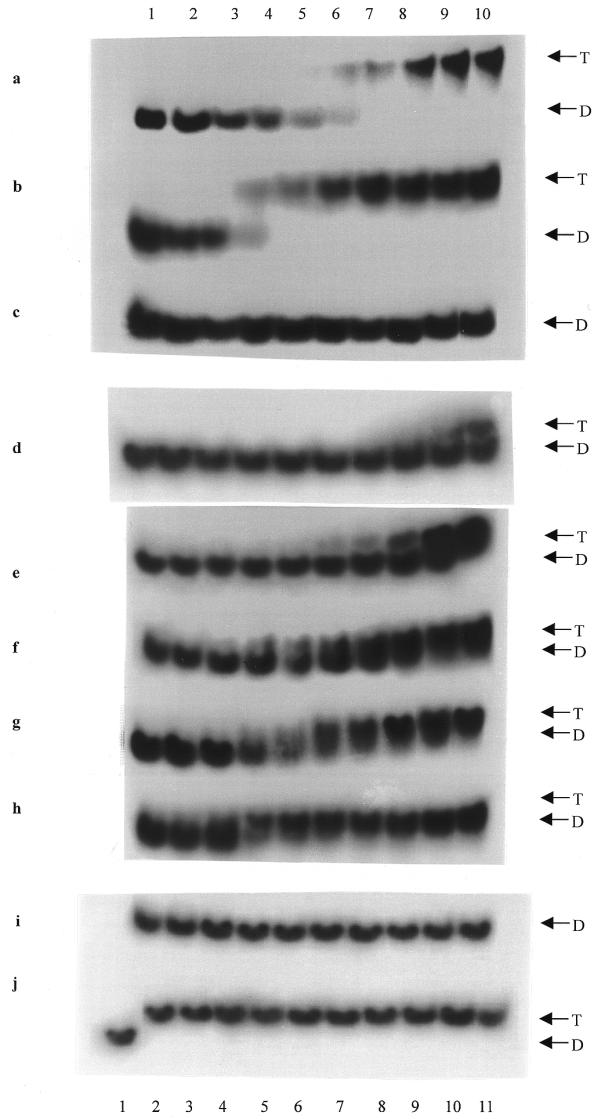
Gel mobility shift assays. (a) TFO1, (b) TFO2 and (c) TFO-p triplex formation with the synthetic 15 bp PRR duplex. Lanes 1–10, 0, 0.05, 0.1, 0.2, 0.4, 0.8, 1.5, 3, 6.0 and 12 µM TFO1, TFO2 or TFO-p were added to 30 nM 32P-labeled 15 bp PRR duplex. (d) TFO12, (e) TFO13, (f) TFO14, (g) TFO15 and (h) TFO16 triplex formation with the synthetic 35 bp SPE–PRR duplex. Lanes 1–10, 0, 0.001, 0.025, 0.05, 0.1, 0.2, 0.4, 0.8, 1.5 and 3.0 µM TFOs were added to 30 nM 32P-labeled 35 bp SPE–PRR duplex. (i) Triplex formation by equal amount of TFOs3–11 with the synthetic 35 bp SPE–PRR duplex. Lane 1, duplex control without TFOs. Lanes 2–10, 50 µM TFOs3–11 were added to 30 nM 32P-labeled 35 bp SPE–PRR duplex, respectively. (j) Triplex formation by equal amounts of TFOs1, 2 and 13–20 with the synthetic 35 bp SPE–PRR duplex. Lanes 2–11, 20 µM TFO1, 2 and 13–20 were added to 30 nM 32P-labeled 35 bp SPE–PRR duplex, respectively. Lane 1, duplex control without TFOs. T, triplex; D, duplex.
DNase I footprinting experiments further confirmed that triplex formation occurred in a sequence-specific manner. As shown in Figure 3, protection of the target sequences by TFO1, 17 and 19 were concentration-dependent in a manner consistent with the gel mobility shift analysis. At 20 µM, the antiparallel TFO1, 17 and 19 yielded similar DNase I protection patterns documenting complete protection of the 16, 25 and 35 bp target sequence, respectively. These data further suggest that triplex formation is completely sequence-specific.
Figure 3.
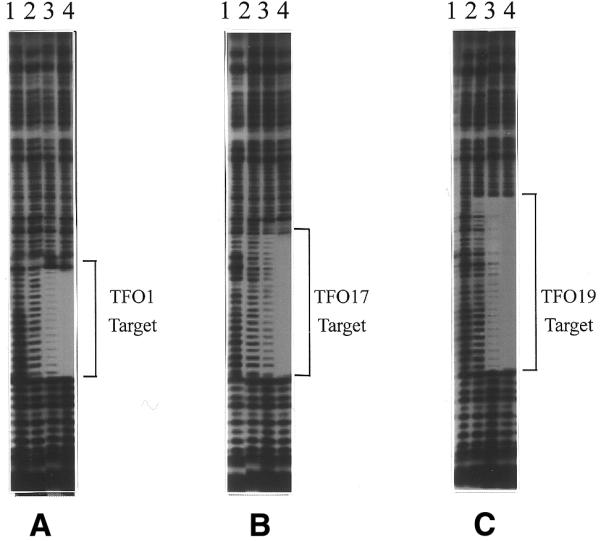
DNase I footprinting analysis demonstrating sequence-specific binding of TFO1 (A), TFO17 (B) and TFO19 (C) to the 262 bp target duplex containing SPE and PRR. Lane 1, no TFOs; lanes 2–4, 1, 10 and 20 µM relevant TFO were incubated with 30 nM 262 bp 32P-labeled fragment followed by limited DNase I digestion.
Effect of triplex formation on nuclear factor binding
The effect of oligonucleotide-directed triplex formation on nuclear factors binding to the 255 bp promoter fragment was determined by protein binding assays (Fig. 4). This fragment, lying immediately 5′ to the SIS/PDGF-B mRNA initiation site (+1), contains SPE between –58 and –39, which is the binding site for the Sp family of transcription factors including Sp1 and Sp3 (at least). In Figure 4, lane 2 contains the labeled 255 bp c-sis/PDGF-B promoter alone, while lane 1 shows that the labeled 255 bp target is bound by nuclear factors as evidenced by retardation of labeled duplex target following incubation with nuclear extracts of PMA-treated K562 cells. In lane 3, 20-fold unlabeled 255 bp target competed with the labeled target and bound most of the nuclear factors. Increasing amounts of TFO (1, 5 and 20 µM) successfully eliminated the formation of protein–DNA complexes partly (TFO1, lanes 5–7) and completely (TFO18, lanes 11–13; TFO20, lanes 14–16). In contrast, lane 4 shows that 20 µM TFO3 has little effect on the binding between nuclear factors and duplex target. Compared with TFO1, TFO2 has a rather slight effect on protein binding, as indicated in lanes 8–10.
Figure 4.

Electrophoretic mobility shift analysis of the effect of TFOs on the binding of nuclear factors present in PMA-treated K562 cells to the target region of the c-sis/PDGF-B promoter. Radiolabeled duplex target, the 255 bp promoter fragment isolated from the pUC18promoter, (30 nM) was preincubated with increasing concentration (1, 5 or 20 µM) of single-stranded TFO1 (lanes 5–7), TFO2 (lanes 8–10), TFO18 (lanes 11–13), TFO20 (lanes 14–16 ), 20 µM TFO3 (lane 4) and no TFOs (lanes 1-3). Samples were then incubated with the nuclear extracts from PMA-treated K562 cells except in lane 2, which contained labeled duplex only. In lane 3, 20-fold more unlabeled duplex target was added. a and b, protein–DNA complexes; c, free duplex probe.
Effect of triplex formation on c-sis/PDGF-B transcription
A reporter plasmid pGL3promoter carrying a firefly luciferase gene driven by the 255 bp c-sis/PDGF promoter was constructed to measure the in vivo effects of TFOs on c-sis/PDGF-B transcription. Triplex was formed in vitro by incubating supercoil pGL3promoter with increasing concentrations of TFO (1, 5 and 20 µM), and then the entire DNA complex was transfected into K562 cells. A dose-dependent decrease of luciferase activity was observed in all the three kind of TFOs. At 20 µM, TFO1 caused a 56% decrease in luciferase activity, while TFO18 caused a 68% and TFO20 an 87% decrease. As a non-triplex-forming control, TFO3 had little effect on luciferase activity. At the same time, all TFOs have no effect on the expression of firefly luciferase driven by SV40 promoter in pGL3control plasmid under the same conditions (Fig. 5A). For stable transfection experiments, another two K562 cell lines were created. One was the K562 luciferase cell line, which was stably transfected with pneorGL3promoter, the other was K562 control cell line, which was stably transfected with pneorGL3control. Twenty-four hours after electroporating both cell lines with 50 µM TFOs, a marked decrease of luciferase activity was observed only in the former cell line. The inhibition of luciferase activity was as high as 51% by TFO1, 69% by TFO18 and 82% by TFO20. Again, as expected, all TFOs have no effect on the K562 control cell line, neither did TFO3 have any effect on the K562 luciferase cell line at 50 µM (Fig. 5B). These results suggest that inhibition of PDGF-B transcription by TFOs is due to sequence-specific triplex formation within the PDGF-B promoter.
Figure 5.

Luciferase assay demonstrating specific inhibition of c-sis/PDGF transcription. (A) Transient transfection. (B) Stable transfection.
DISCUSSION
The intracellular, autocrine growth circuit of growth factors is extremely important to the maintenance of tumor cell lines. These interactions are usually inaccessible to therapeutic strategies such as extracellularly-added antibodies or receptor antagonists. In a variety of neoplastic conditions where PDGF-B is implicated, the activation of the endogenous PDGF-B gene remains an important step within the PDGF-B autocrine loop, a step that could potentially be targeted to achieve suppression of the growth-promoting circuit.
Compared with antisense or ribozyme techniques, the triplex strategy has a potential stoichiometric advantage, because there are generally one to two targets per cell as compared with the hundreds to thousands of mRNA targets for antisense or ribozyme oligonucleotides, thus offering the potential for low-dose long-acting therapeutics. The major limitation of the application of oligonucleotide-directed triplex formation to naturally occurring sequences is the requirement for predominantly homopurine/homopyrimidine tracts. Fortunately, these tracts occur relatively often in the promoters of eukaryotic genes and mostly take an active part in the control of gene transcription (55). Previous studies on TFOs targeting the coding tract of a gene suggest a non-permanent barrier to chromosomal replication where the DNA replication/recombination complex could efficiently remove the block (56). Accordingly, inhibition of transcription through intermolecular triplex formation with the promoter is very attractive.
Two previous studies on the regulatory elements of the human PDGF-B (c-sis, proto-oncogene) have shed considerable light on the cis-acting elements, trans-acting factors and their functional properties. The 20 bp SPE region within the promoter immediately upstream of the transcription start site is the binding site for the Sp family of transcription factors including Sp1 and Sp3. This region is essential for transcriptional activity, and thus provides an attractive target for the design of a sequence-specific DNA binding agent that may inhibit transcription of this biologically important gene.
Since a single interruption in the duplex homopurine/homopyrimidine tract may significantly destabilize the triplex, and SPE contains four purine interruptions in a run of 20 pyrimidines, we developed a new strategy by making use of the 15 bp PRR downstream. A series of GA-rich potential TFOs of different sequence or length were designed. We did not use pyrimidine third strand, as pH constrains the formation of pyrimidine-directed triple-helical complexes, which require cytosine protonation for stabilization. Each TFO we designed contains a run of 15 nt purine sequence at its 3′-end targeting PRR. TFOs of different lengths were designed to target part of or the entire of SPE. Data from gel mobility shift analysis demonstrates that, as expected, TFOs1 and 2, which target the perfect 15 bp PRR, showed high affinity for the target. In contrast, TFOs3–11 (25 nt), containing A, C or G opposite a single C-G base pair, bind the duplex target poorly, while TFOs12–14, which have a T placed at the position of C-G inversion, showed higher affinity to the target. Previous research (57) has shown that G in a pyrimidine motif TFO can recognize T-A conversion in the target, while here we show that in a purine motif TFO, T is the best choice to recognize the C-G inversion. According to the results from gel shift, TFO13 is more easily able to form triplex than TFO12, and TFO14 is the best of the three, indicating an order of T<A<G in affinity to the T-A inversion. However, in the following designed 33mer TFOs15 and16, we did not choose G opposite the first two T-A inversions since it would result in a G tract at least 6–9 nt long, which would be undesirable due to the propensity to form self-aggregates such as G-tetrads. Instead, A was used at the first two T-A inversions, G at the third and T at the C-G inversion. The results from gel shift assays demonstrated that both TFOs could form triplexes perfectly. The effect of the three As at the 3′-end of TFO was further investigated by designing another four 35mer TFO17–20. Kd data showed that three As had a small negative effect on triplex formation by decreasing nearly 4-fold stability. The following DNase I footprinting experiments confirmed the sequence-specificity of triple helix formation by TFO1, 17 and 19. Based on these facts, we conclude that although G is a little better than A at T-A inversion site, A is still a good choice when it is undesirable to use G in a long tract of G surroundings.
In order to evaluate the effect of TFO on the binding of nuclear factors contained in nuclear extracts, we employed protein binding assays using the nuclear extracts from PMA-treated K562 cells, as PMA can promote PDGF-B gene expression at the transcription level (58). As shown in Figure 4, the formation of protein–DNA complexes was partly inhibited by increasing amounts of TFO1, completely inhibited by TFO18 and TFO20, and slightly inhibited by TFO2. These results are as expected, since TFO1, which is targeted to the 15 nt PRR downstream SPE, did not directly interfere with the binding site of the Sp family of transcription factors. The part inhibition may be due to the bending or stiffening of the target duplex by triplex formation, thus affecting the formation of the transcription initiation complex. Since TFO2 has three As absent at the 3′ end, in spite of the higher affinity to the target, the region it covers is rather too small to effectively alter the flexibility of the target duplex. The negative effect of three As at the 3′-end became less important when weighed against the possible benefits of covering a larger region on the target, the higher stability of TFO itself in vivo and more sequence specificity in triplex formation. The longer TFOs18 and 20 interfere with the SPE partly or entirely, therefore their ability to form triplex within the SPE contributes to the occlusion of the transcriptional factors. TFO1, 18 and 20 were used in further transfection experiments. Results from both the transient and stable transfections parallel the findings of the protein binding assays. The TFOs appear to specifically interact with the c-sis/PDGF-B gene promoter and repress PDGF-B transcription, since the K562 cell lines transiently and stably transfected with the luciferase gene driven by the same promoter showed dramatically decreased activity. Although we cannot directly demonstrate that triplexes formed inside cells, we propose that these unusual DNA structures formed by TFO1, 18 and 20 may be responsible for the reduction in PDGF-B expression in PMA-treated K562 cells. This inhibitory effect was found only in cells treated with TFO1, 18 and 20, and not in cells treated with TFO3 sharing nearly the same TFO composition with only one base difference.
Many modifications have been employed to improve the stability of TFO in vivo and its affinity to the target such as phosphorothioate (59), N3′→P5′ phosphoramidate (np) linkages (60) and psoralen-conjugated TFOs (61), etc. Meanwhile, several specific ligands that can intercalate into triplexes and stabilize them have also been developed (62). In this report, unmodified phosphodiester oligonucleotides were used. Therefore, there still remains a promising future for modified TFOs on the regulation of PDGF-B expression in vivo. Further experiments to explore the therapeutic applications of these TFOs in suppression of tumor growth and in animal model systems are in progress.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Dr Chen Wang, Li Chen, Feng Xu, Ge Jiang, Xing-Jun Fan, Yu-Ping Gong, Wei-Guo Han, Quan-Sheng Liu, Jie Jia and Xiang-Long Chen for their helpful advice and technical support, Jun-Ru Bao for her kind help in obtaining reagents, and Pei-Juan Gong and Jian Chen for their assistance with automated DNA synthesis. This work was supported by Grant No. G1998051103 from State Key Programs Basic Research of China and Grant No. 863-102-08-8 from National High Technology Programs of China.
References
- 1.Raines E.W., Bowen-Pope,D.F. and Ross,R. (1990) In Sporn,M.B. and Roberts,A.B. (eds), Handbook in Experimental Pharmacology. Peptide Growth Factors and Their Receptors. Springer, Heidelberg, pp 173–262.
- 2.Heldin C.-H. (1992) Structural and functional studies on platelet-derived growth factor. EMBO J., 11, 4251–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yarden Y., Escobendo,J.A., Kuang,W.-J., Yang-Feng,T.L., Daniel,T.O., Tremble,P.M., Chen,E.Y., Ando,M.E., Harkins,R.N., Francke,U., Friend,V.A., Ullrich,A. and Williams,L.T. (1986) Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature, 323, 2326–2327. [DOI] [PubMed] [Google Scholar]
- 4.Matsui T., Heidaran,M., Miki,T., Popescu,N., La Rochelle,W., Kraus,M., Pierce,J. and Aaronson,S.A. (1988) Isolation of a novel receptor cDNA establishes the existence of two PDGF receptor genes. Science, 243, 800–804. [DOI] [PubMed] [Google Scholar]
- 5.Claesson-Welsh L., Eriksson,A., Westermark,B. and Heldin,C.-H. (1989) cDNA cloning and expression of the human A-type platelet-derived growth factor (PDGF) receptor establishes structural similarity to the B-type PDGF receptor. Proc. Natl Acad. Sci. USA, 86, 4917–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heldin C.-H., Ernlund,A., Rorsman,C. and Ronnstrand,L. (1989) Dimerization of B-type platelet-derived growth factor receptors occurs after ligand binding and is closely associated with receptor kinase activation. J. Biol. Chem., 264, 8905–8912. [PubMed] [Google Scholar]
- 7.Kelly J.D., Haldeman,B.A., Grant,F.J., Murray,M.J., Seifert,R.A., Bowen Pope,D.F., Cooper,J.A. and Kazlauskas,A. (1991) Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem., 266, 8987–8992. [PubMed] [Google Scholar]
- 8.Escobedo J.A., Barr,P.J. and Williams,E.T. (1988) Role of tyrosine kinase and membrane-spanning domains in signal transduction by the platelet-derived growth factor receptor. Mol. Cell. Biol., 8, 5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waterfield M.D., Scrace,G.T., Whittle,N., Stroobant,P., Johnsson,A., Wasteson,A., Westermark,B., Heldin,C.H., Huang,J.S. and Deuel,T. (1983) Platelet-derived growth factor is structurally related to the putative trans-forming protein p28sis of simian sarcoma virus. Nature, 304, 35–39. [DOI] [PubMed] [Google Scholar]
- 10.Doolittle R.F., Hunkappiller,M.W., Hood,L.E., Devare,S.G., Robbins,K.C., Aaronson,S.A. and Antoniades,H.N. (1983) Simian sarcoma virus oncogene, v-sis is derived from the gene (or genes) encoding platelet-derived growth factor. Science, 221, 275–277. [DOI] [PubMed] [Google Scholar]
- 11.Pantazis P., Pelicci,P.G., Dalla-Favera,R. and Antoniades,H.N. (1985) Synthesis and secretion of proteins resembling platelet-derived growth factor by human glioblastoma and fibrosarcoma cells in culture. Proc. Natl Acad. Sci. USA, 82, 2404–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barnhill R.L., Xiao,M., Graves,D. and Antoniades,H.N. (1996) Expression of platelet-derived growth factor (PDGF)-A, PDGF-B and the PDGF-alpha receptor, but not the PDGF-beta receptor, in human malignant melanoma in vivo. Br. J. Dermatol., 135, 898–904. [DOI] [PubMed] [Google Scholar]
- 13.Bronzert D.A., Pantazis,P., Antoniades,H.N., Kasid,A., Davidson,N., Dicknson,R.B. and Lippman,M.E. (1987) Synthesis and secretion of platelet-derived growth factor by human breast cancer cell lines. Proc. Natl Acad. Sci. USA, 84, 5763–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antoniades H.N., Galanopoulos,T., Neville-Golden,J. and O’Hara,C.J. (1991) Malignant epithelial cells in primary human lung carcinomas coexpress in vivo platelet-derived growth factor (PDGF) and PDGF receptor mRNA and their protein products. Proc. Natl Acad. Sci. USA, 89, 3942–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nister M., Claesson-Welsh,L., Eriksson,A.C., Heldin,C.-H. and Westermark,B. (1991) Differential expression of platelet derived growth factor receptors in human malignant glioma cell lines. J. Biol. Chem., 266, 16755–16763. [PubMed] [Google Scholar]
- 16.Liu Y.C., Chen,S.C., Chang,C., Leu,C.M. and Hu,C.P. (1996) PDGF is an autocrine stimulator for the growth and survival of human esophageal carcinoma cell lines. Exp. Cell Res., 228, 206–211. [DOI] [PubMed] [Google Scholar]
- 17.Ralf K., Blatt,L.M., Streubert,M., Zietz,C., Hermeking,H., Brysch,W. and Stürzl,M. (1996) Consensus-interferon and platelet-derived growth factor adversely regulate proliferation and migration of Kaposi’s sarcoma cells by control of c-myc expression. Am. J. Pathol., 149, 1871–1885. [PMC free article] [PubMed] [Google Scholar]
- 18.Pech M., Gazit,A., Arnstein,P. and Aaronson,S.A. (1989) Generation of fibrosarcomas in vivo by a retrovirus that expresses the normal B chain of platelet-derived growth factor and mimics the alternative splice pattern of the v-sis oncogene. Proc. Natl Acad. Sci. USA, 86, 2693–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsberg K., Valyi-Nagy,I., Heldin,C.-H. and Meenhard,H. (1993) Platelet-derived growth factor (PDGF) in oncogenesis: development of a vascular connective tissue stroma in xenotransplanted human melanoma producing PDGF-BB. Proc. Natl Acad. Sci. USA, 90, 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J., Coltrera,M.D. and Gown,A.M. (1994) Cell proliferation in human soft tissue tumors correlates with platelet-derived growth factor B chain expresson: an immunohistochemical and in situ hybridization study. Cancer Res., 54, 560–564. [PubMed] [Google Scholar]
- 21.Potapova O., Fakhrai,H., Baird,S. and Mercola,D. (1996) Platelet-derived growth factor-B/v-sis confers a tumorigenic and metastatic phenotype to human T98G glioblastoma cells. Cancer Res., 56, 280–286. [PubMed] [Google Scholar]
- 22.Skobe M. and Fusenig,N.E. (1998) Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc. Natl Acad. Sci. USA, 95, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross R., Masuda,J., Raines,E.W., Gown,A.M., Katsuda,S., Sasahara,M., Malden,L.T., Masuko,H. and Sato,H. (1990) Localization of PDGF-B protein in macrophages in all phases of atherogenesis. Science, 248, 1009–1012. [DOI] [PubMed] [Google Scholar]
- 24.Uchida K., Sasahara,M., Morigami,N., Hazama,F. and Kinoshita-Muchida,K. (1996) Expression of PDGF B-chain in neointimal smooth muscle cells of balloon injured rabbit femoral arteries. Atherosclerosis, 124, 9–23. [DOI] [PubMed] [Google Scholar]
- 25.Wangoo A., Taylor,I.K., Haynes,A.R. and Shaw,R.J. (1993) Up-regulation of alveolar macrophage PDGf-B mRNA by interferon-α from Mycobacterium tuberculosis antigen (PPD)–stimulated lymphocytes. Clin. Exp. Immunol., 94, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson D.E., Hinohara,T., Selmon,M.R., Braden,L.J. and Simpson,J.B. (1990) Primary peripheral arterial stenoses and restenoses excised by transluminal atherectomy: a histopathologic study. J. Am. Coll. Cardiol., 15, 419–425. [DOI] [PubMed] [Google Scholar]
- 27.Kaiser M., Weyand,C.M., Bjornsson,J. and Goronzy,J.J. (1998) PDGF, intimal hyperplasia and ischemic complications in giant cell arteritis. Arthritis. Rheum., 41, 623–633. [DOI] [PubMed] [Google Scholar]
- 28.Xu J.W., Konttinen,Y.T., Li,T.F., Waris,V., Lassus,J., Matucci-Cerinic,M., Sorsa,T. and Santavirta,T.S. (1998) Production of PDGF in aseptic loosening of total hip replacement. Rheumatol. Int., 17, 215–221. [DOI] [PubMed] [Google Scholar]
- 29.Bergmann M., Tiroke,A., Schafer,H., Barth,J. and Haverich,A. (1998) Gene expression of profibrotic mediators in bronchiolitis obliterans syndrome after lung transplantation. Scand. Cardiovasc. J., 32, 97–103. [DOI] [PubMed] [Google Scholar]
- 30.Nitta T. and Sato,K. (1994) Specific inhibition of c-sis protein synthesis and cell proliferation with antisense oligodeoxynucleotides in human glioma cells. Neurosurgery, 34, 309–315. [DOI] [PubMed] [Google Scholar]
- 31.Dorai T., Kobayashi,H., Holland,J.F. and Ohnuma,T. (1994) Modulation of platelet-derived growth factor-beta mRNA expression and cell growth in a human mesothelioma cell line by a hammerhead ribozyme. Mol. Pharmacol., 46, 437–444. [PubMed] [Google Scholar]
- 32.Green L.S., Jellinek,D., Jenison,R., Ostman,A., Heldin,C.H. and Janjie,N. (1996) Inhibitory DNA ligands to platelet-derived growth factor B-chain. Biochemistry, 35, 14413–14424. [DOI] [PubMed] [Google Scholar]
- 33.Moser H.E. and Dervan,P.B.(1987) Sequence-specific cleavage of double helical DNA by triple helix formation. Science, 238, 645–650. [DOI] [PubMed] [Google Scholar]
- 34.Cooney M., Czernuszewicz,G., Postel,E.H., Flint,S.J. and Hogan,M.E. (1988) Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science, 241, 456–459. [DOI] [PubMed] [Google Scholar]
- 35.McShan W.M., Rossen,R.D., Laughter,A.H., Trial,J., Kessler,D.J., Zendegui,J.G., Hogan,M.E. and Orson,F.M. (1992) Inhibition of transcription of HIV-1 in infected human cells by oligodeoxynucleotides designed to form DNA triple helices. J. Biol. Chem., 267, 5712–5721. [PubMed] [Google Scholar]
- 36.Giovannangeli C., Diviacco,S., Labrousse,V., Gryaznov,S., Charneau,P. and Helene,C. (1997) Accessibility of nuclear DNA to triplex-forming oligonucleotides: the integrated HIV-1 provirus as a target. Proc. Natl Acad. Sci. USA, 94, 79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Porumb H., Gousset,H., Letellier,R., Salle,V., Briane,D., Vassy,J., Amor-Gueret,M., Israel,L. and Taillandier,E. (1996) Temporary ex vivo inhibition of the expression of the human oncogene HER2 (NEU) by a triple helix-forming oligonucleotide. Cancer Res., 56, 515–522. [PubMed] [Google Scholar]
- 38.Aggarwal B.B., Schwarz,L., Hogan,M.E. and Rando,R.F. (1996) Triple helix-forming oligodeoxyribonucleotides targeted to the human tumor necrosis factor (TNF) gene inhibit TNF production and block the TNF-dependent growth of human glioblastoma tumor cells. Cancer Res., 56, 5156–5164. [PubMed] [Google Scholar]
- 39.Grigoriev M., Praseuth,D., Robin,P., Hemar,A., Saison-Behmoaras,T., Dautry-Varsat,A., Thuong,N.T., Helene,C. and Harel-Bellan,A. (1992) A triple helix-forming oligonucleotide–intercalator conjugate acts as a transcriptional repressor via inhibition of NF kappa B binding to interleukin-2 receptor alpha-regulatory sequence. J. Biol. Chem., 267, 3389–3395. [PubMed] [Google Scholar]
- 40.Gee J.E., Blume,S., Snyder,R.C., Ray,R. and Miller,D.M. (1992) Triplex formation prevents Sp1 binding to the dihydrofolate reductase promoter. J. Biol. Chem., 267, 11163–11167. [PubMed] [Google Scholar]
- 41.Mayfield C., Ebbinghaus,S., Gee,J., Jones,D., Rodu,B., Squibb,M. and Miller,D. (1994) Triplex formation by the human Ha-ras promoter inhibits Sp1 binding and in vitro transcription. J. Biol. Chem., 269, 18232–18238. [PubMed] [Google Scholar]
- 42.Tu G.C., Cao,Q.N. and Israel,Y. (1995) Inhibition of gene expression by triple helix formation in hepatoma cells. J. Biol. Chem., 270, 28402–28407. [DOI] [PubMed] [Google Scholar]
- 43.Kochetkova M. and Shannon,M.F. (1996) DNA triplex formation selectively inhibits granulocyte-macrophage colony-stimulating factor gene expression in human T cells. J. Biol. Chem., 271, 14438–14444. [DOI] [PubMed] [Google Scholar]
- 44.Alama A., Barbieri,F., Cagnoli,M., Mazzei,M., Grandi,T. and Nicolin,A. (1996) Inhibition of DNA polymerase alpha expression and cell growth, a possible triple helix mechanism. Antisense Nucleic Acid Drug Dev., 6, 95–101. [DOI] [PubMed] [Google Scholar]
- 45.Kovacs A., Kandala,J.C., Weber,K.T. and Guntaka,R.V. (1996) Triple helix-forming oligonucleotide corresponding to the polypyrimidine sequence in the rat alpha 1(I) collagen promoter specifically inhibits factor binding and transcription. J. Biol. Chem., 271, 1805–1812. [DOI] [PubMed] [Google Scholar]
- 46.Rininsland F., Johnson,T.R., Chernicky,C.L., Schulze,E., Burfeind,P. and Ilan,J. (1997) Suppression of insulin-like growth factor type I receptor by a triple-helix strategy inhibits IGF-I transcription and tumorigenic potential of rat C6 glioblastoma cells. Proc. Natl Acad. Sci. USA, 94, 5854–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alexander S., Burfeind,P., Schulze,S., Rininsland,F., Johnson,T.R., Trojan,J., Chernicky,C.L., Hlne,C. and Ilan,J. (1997) Potential triple helix-mediated inhibition of IGF-I gene expression significantly reduces tumorigenicity of glioblastoma in an animal model. Cancer Gene Ther., 4, 105–112. [PubMed] [Google Scholar]
- 48.Perkins B.D., Wilson,J.H., Wensel,T.G. and Vasquez,K.M. (1998) Triplex targets in the human rhodopsin gene. Biochemistry, 37, 11315–11322. [DOI] [PubMed] [Google Scholar]
- 49.Kim H.G. and Miller,D.M. (1998) A novel triplex-forming oligonucleotide targeted to human cyclin D1 (bcl-1, proto-oncogene) promoter inhibits transcription in HeLa cells. Biochemistry, 37, 2666–2672. [DOI] [PubMed] [Google Scholar]
- 50.Dignam J.D., Lebovitz,R.M. and Roeder,R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 5, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liang Y., Robinson,D.F., Dennig,J., Suske,G. and Fahl,W.E. (1996) Transcriptional regulation of the SIS/PDGF-B gene in human osteosarcoma cells by the Sp family of transcription factors. J. Biol. Chem., 271, 11792–11797. [DOI] [PubMed] [Google Scholar]
- 52.Liang Y., Robinson,D.F., Kujoth,G.C. and Fahl,W.E. (1996) Functional analysis of the SIS proximal element and its activating factors: regulated transcription of the c-SIS/PDGF-B gene in human erythroleukemia cells. Oncogene, 13, 863–871. [PubMed] [Google Scholar]
- 53.Durland R.H., Kessler,D.J., Gunnell,S., Duvic,M., Pettitt,B.M. and Hogan,M.E. (1991) Binding of triple helix forming oligonucleotides to sites in gene promoters. Biochemistry, 30, 9246–9255. [DOI] [PubMed] [Google Scholar]
- 54.Arimondo P.B., Barcelo,F., Sun,J.S., Maurizot,J.C., Garestier,T. and Helene,C. (1998) Triple helix formation by (G, A)-containing oligonucleotides: asymmetric sequence effect. Biochemistry, 37, 16627–16635. [DOI] [PubMed] [Google Scholar]
- 55.Lu G. and Ferl,R.J. (1993) Homopurine/homopyrimidine sequences as potential regulatory elements in eukaryotic cells. Int. J. Biochem., 25, 1529–1537. [DOI] [PubMed] [Google Scholar]
- 56.Maine I.P. and Kodadek,T. (1994) Efficient unwinding of triplex DNA by a DNA helicase. Biochem. Biophys. Res. Commun., 204, 1119–1124. [DOI] [PubMed] [Google Scholar]
- 57.Griffin L.C. and Dervan,P.B. (1989) Recognition of thymine adenine base pairs by guanine in a pyrimidine triple helix motif. Science, 245, 967–971. [DOI] [PubMed] [Google Scholar]
- 58.Colamonici O.R., Trepel,J.B., Vidal,C.A. and Neckers,L.M. (1986) Phorbol ester induces c-sis gene transcription in stem cell line K-562. Mol. Cell. Biol., 6, 1847–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joseph J., Kandala,J.C., Veerapanane,D., Weber,K.T. and Guntaka,R.V. (1997) Antiparallel polypurine phosphorothioate oligonucleotides form stable triplexes with the rat α1(I) collagen gene promoter and inhibit transcription in cultured rat fibroblasts. Nucleic Acids Res., 25, 2182–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou-Sun B.W., Sun,J.S., Gryaznov,S.M., Liquier,J., Garestier,T., Helene,C. and Tailandier,E. (1997) A physico-chemical study of triple helix formation by an oligodeoxythymidylate with N3′→P5′ phosphoramidate linkages. Nucleic Acids Res., 25, 1782–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miller P.S., Bi,G., Kipp,S.A., Fok,V. and DeLong,R.K. (1996) Triplex formation by a psoralen-conjugated oligodeoxyribonucleotide containing the base analog 8-oxo-adenine. Nucleic Acids Res., 15, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Escudé C., Nguyen,C.H., Kukreti,S., Janin,Y., Sun,J.S., Bisagni,E., Garestier,T. and Hélène,C. (1998) Rational design of a triple helix-specific intercalating ligand. Proc. Natl Acad. Sci. USA, 95, 3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]