

Abstract
The craniofacial region is assembled through the active migration of cells and the rearrangement and sculpting of facial prominences and pharyngeal arches, which consequently make it particularly susceptible to a large number of birth defects. Genetic, molecular, and cellular processes must be temporally and spatially regulated to culminate in the three-dimension structures of the face. The starting constituent for the majority of skeletal and connective tissues in the face is a pluripotent population of cells, the cranial neural crest cells (NCCs). In this review we discuss the newest scientific findings in the development of the craniofacial complex as related to NCCs. Furthermore, we present recent findings on NCC diseases called neurocristopathies and, in doing so, provide clinicians with new tools for understanding a growing number of craniofacial genetic disorders.
Keywords: neural crest cells, craniofacial development, neurocristopathies
Introduction
Malformations involving the craniofacial regions are observed in three-fourths of human birth defects (see Centers for Disease Control, Birth Defects Research and Prevention at http://www.cdc.gov/ncbddd/bd/centers.htm). In part, this may be attributable to the intricate means by which the craniofacial region is assembled during embryonic development. Tissues of the craniofacial complex are primarily derived from neural crest cells (NCCs), a population of transiently migratory cells that originate from the dorsal aspect of the neural tube during embryogenesis (Fig 1), then migrate to populate the frontonasal process (FNP) and the first, second, third and fourth pharyngeal arches [Lièvre and Douarin, 1975; Hunt et al., 1991a; Hunt et al., 1991b; Lumsden et al., 1991]. NCCs contribute to neural, skeletal, dermal, and mesenchymal structures. Because of this pluripotency they have sometimes been referred to as the fourth germ layer [Hall, 2000].
FIGURE 1.

Coronal section of murine embryo revealing neural crest cells migrating (blue, red arrows) from the dorsal neural tube (blue arrow).
A tightly controlled spatial and temporal signaling network is required for the induction, migration, proliferation and differentiation of NCCs that give rise to craniofacial structures. During migration, and even after arrival at their final destination, interactions between NCCs and the adjacent surface ectoderm, neuroectoderm and endoderm are necessary for normal development of the craniofacial region [Cordero et al., 2004; Creuzet et al., 2005; Creuzet et al., 2006; Sandell and Trainor, 2006]. Aberrations in any of the multi-step processes involved in regulating NCC behavior can result in craniofacial malformations. Clinicians who evaluate patients with such malformations will find that a working knowledge of NCC biology is beneficial for understanding the etiologies of these diseases.
Derivatives of the Neural Crest
NCCs contribute to multiple cell types and tissues throughout the body (enteric nervous system, glia, neurons, melanocytes, as well as connective tissues, chondrocytes, and myofibroblasts lining the blood vessels). In the face, however, they are the prime contributor. Specifically, the facial skeleton and the vast majority of facial connective tissues are derived exclusively from cranial NCCs. As a consequence, genetic diseases that affect aspects of NCC generation, migration, proliferation, or differentiation are more likely to manifest themselves in the craniofacial region. Therefore, we turn our attention first to the initiation of NCC development.
The Emerging Neural Crest
There are currently two theories regarding the initiation of NCCs specification. One theory that has been put forth is that NCCs are specified during gastrulation, and is based upon detailed studies in the avian model system [Basch et al., 2006]. In this study it was shown that a restricted region of epiblast, expressing Pax7, contributes to neural folds and migrating NCCs [Basch et al., 2006]. Inhibition of Pax7 protein production prevented the expression of several NCC markers like Slug (Snail2), Sox9, Sox10 and HNK-1 [Basch and Bronner-Fraser, 2006] independent of mesoderm or neural plate. A second group also showed that specification of NCC occurs during gastrulation but is dependent upon signals from dorso-lateral mesoderm, requiring both Wnt activation and BMP inhibition and a second step at neurulation, which requires both Wnt and BMP activation in adjacent tissues [Steventon et al., 2009]. This is in contrast to the classic theory that suggests that NCCs are specified around or at the time of neural tube closure.
In the second or “classical” model of NCC induction, planar interactions between the neural ectoderm and non-neural ectoderm, as well as signaling from underlying paraxial mesoderm, are required for NCC formation [Noden and Trainor, 2005; Selleck and Stern, 1991]. A number of signaling pathways have been shown to be necessary for NCC generation and/or survival including two Bone Morphogenetic Protein (BMP) antagonists, Chordin and Noggin [Anderson et al., 2006], Fibroblast Growth Factors (Fgfs) [Mayor et al., 1997], and Wnt signals [Garcia-Castro et al., 2002]. With the key molecular pathways identified, the present challenge is to understand how they are integrated in such a way as to induce the formation of this unique set of cells. Part of this integration relies on tightly regulated spatiotemporal expression of pathway activators. For example, the position of presumptive neural crest domain correlates broadly with the BMP4 expression domain [Ezin et al., 2009] and with the anterior-most extent of Wnt pathway activity [Li et al., 2009]. However, the integration of Wnt and BMP signaling pathways to produce NCCs is poorly understood. In some experimental models, BMP signals induce cells at the neural plate border to become NCCs, but only when Wnt signaling is inhibited [Patthey et al., 2009]. In other experimental models, Wnt signaling is sufficient for the generation of NCCs. For example, Wnt signaling induces the transcription factor Gbx2, which in turn is necessary for neural crest induction [Byrd and Meyers, 2005; Li et al., 2009].
Although both theories propose a different timing of specification, both agree that cranial NCCs are generated from the border between neural and non-neural ectoderm [Crane and Trainor, 2006; Knecht and Bronner-Fraser, 2002; Ruffins and Bronner-Fraser, 2000]. In addition the data from various model systems must be carefully compared [Steventon et al., 2005] and their extrapolation to human development must be critically assessed.
Neural Crest Migration
Independent of the timing of NCC specification, contact-mediated signaling between tissues in the dorsal neural tube results in neural ectoderm cells at the neuroectoderm and non-neuroectoderm border undergoing epithelial-mesenchymal transition (EMT), which is required for NCC migration. Contact-mediated signaling between tissues in the dorsal neural tube stimulates the cells at the neural/non-neural border to undergo a transition from an epithelial to a mesenchymal phenotype [Noden and Trainor, 2005]. This transition into a highly invasive phenotype is a hallmark of NCCs and one they share with metastatic cells [Radisky and LaBarge, 2008].
Epithelial-to-mesenchymal transition (EMT) is a multi-step process: For NCC migration to occur, NCCs must lose their apico-basal polarity, and simultaneously disassemble intercellular adhesion complexes that are required for epithelial formation [Acloque et al., 2009; Thiery and Sleeman, 2006]. The disassembly of intercellular adhesion complexes involves members of the Cadherin family. The expression of Cadherins are associated with the formation of epithelial and/or stable cell phenotypes and must be down-regulated by transcriptional repressors to complete the transition from a sedentary epithelium to motile mesenchyme [Obrink, 1986]. Furthermore, tight junctions, structures involved in maintaining cell-cell contacts and paracellular permeability and cell polarity [Lal-Nag and Morin, 2009] must also be eliminated in order to allow for cell migration. In terms of neural crest development and migration, Cadherins play an essential role. The neural plate can be delineated from the non-neural ectoderm by the expression of N-cadherin expression and the loss of E-cadherin expression. Further specification of the neural plate is acquired by induction of cadherin-6B expression in the dorsal neural folds, defining the domain that gives rise to premigratory neural crest cells. As neural crest cells undergo EMT in preparation of migration, cadherin-6B and N-cadherin are down-regulated, and cadherin-7 expression is induced in migratory neural crest cells [Taneyhill, 2008]. This process is initiated by expression of genes such as zinc-finger-like transcription factors like Snail1 and Slug (Snail 2) [Nieto, 2008; Nieto et al., 1994]. For example, over-expression of Snail leads to repression of E-cadherin gene transcription [Cano et al., 2000; Peinado et al., 2004a]. When this dynamic concert of cadherin gene expression fails to occur in the proper manner, NCC migration is disrupted and the result is a perturbation in craniofacial development.
SIP1 and SNAIL (SNAIL1 and SNAIL2 (SLUG) are known transcriptional repressors that control Cadherin activation [Comijn et al., 2001]. Micro-deletions or mutations in SIP1 transcriptional repressor have been identified in ∼200 patients with Mowat-Wilson syndrome (OMIM 235730) [Dastot-Le Moal et al., 2007; Saunders et al., 2009] an autosomal dominant disorder characterized by distinct facial [Saunders et al., 2009] dysmorphisms, microcephaly, mental retardation, and epilepsy [Mowat et al., 1998; Mowat et al., 2003; Horn et al., 2004; Zweier et al., 2005; Garavelli et al., 2009]. SIP1 normally represses E-cadherin expression [Comijn et al., 2001] and in mice carrying null mutations in Sip1, E-Cadherin levels stay abnormally high [Van de Putte et al., 2007]. Consequently, cells at the neural plate boundary cannot undergo an EMT and instead remain tethered to the epithelium. The end result is that NCC migration is delayed and affected mice exhibit abnormal morphology of the neural plate [Van de Putte et al., 2007].
The transcription factor Snail also represses E-cadherin gene transcription, through histone deacetylase-mediated chromatin remodeling [Peinado et al., 2004a; Peinado et al., 2004b]. Over-expression of Snail1 (which should down regulate E-cadherin function) is sufficient to induce EMT [Ikenouchi et al., 2003]. Snail1-null mice are early embryonic lethals. Conditional loss of Snail1 in murine neural crest cells has shown that Snail1 is not required for neural crest cell delamination and migration [Murray and Gridley, 2006a; Murray and Gridley, 2006b]. Redundant functions of Snail1 and Slug in cranial neural crest cells most likely account for a lack of phenotypic presentation in Snail1 conditional knock-out mice [Murray et al., 2006]. To date, no mutations in SNAIL have been associated with congenital malformations although the role of this gene in promoting epithelial-to-mesenchymal transitions in metastatic cancers is an area of active research [Herfs et al., 2010; Larriba et al., 2010; Wu and Zhou, 2010].
Slug, a marker of neural crest cells in Xenopus, zebrafish and chick embryos, is also involved in NCC migration. Slug functions via its up-regulation of rhoB [del Barrio and Nieto, 2002], a member of the Rho family of GTPases, which regulates actin organization and membrane trafficking [Prendergast, 2001]. RhoB expression pattern correlates with regions of NCC delamination, suggesting that NCCs that have already gone through the EMT and are ready to migrate express RhoB [Del Barrio and Nieto, 2004]. Furthermore, inactivation of RhoB impairs neural crest cell delamination [Liu and Jessell, 1998].
Homozygous deletions in Slug/SMAI2 that lead to the absence of the Slug protein have been identified in two unrelated patients with Waardenburg syndrome, type 2D (WS2D) (OMIM 608890) [Sanchez-Martin et al., 2002]. WS2D is a disorder primarily affecting the neural crest or neural crest derivatives, and is characterized by abnormal pigmentation, sensorineural hearing loss, and subtle differences in facial features [Read and Newton, 1997].
It is not surprising, then, that transcriptional misregulation of sets of genes involved in epithelial to mesenchymal transition of NCC like Snail and Slug may result in craniofacial defects. Such a mechanism is hypothesized to contribute to pathogenesis of CHARGE syndrome (OMIM 214800), a sporadic autosomal dominant disorder with cardinal features consisting of ocular coloboma, choanal atresia, heart malformations, growth restriction, genital and ear abnormalities (external and abnormal semicircular canals) [Sanlaville and Verloes, 2007; Verloes, 2005]. The face may also be square-shaped with a narrow bifrontal diameter, broad nasal bridge, malar flattening, cleft lip and palate bridge and a small mouth [Felix et al., 2006; Hall, 1979; Hittner et al., 1979; Jongmans et al., 2006; Oley et al., 1988; Sanlaville and Verloes, 2007]. Human mutations in the chromodomain helicase DNA-binding domain-7 member (CHD7) result in CHARGE syndrome [Johnson et al., 2006; Jongmans et al., 2006; Sanlaville et al., 2006; Vissers et al., 2004; Wincent et al., 2009] and account for 2/3 of the patients with this disorder [Sanlaville and Verloes, 2007]. Studies of CHD7 suggests that through enhancer elements it regulates transcription of genes involved in proliferation, differentiation and migration of cells that give rise to CHARGE-affected tissues [Zentner et al., 2010]. During human embryonic development CHD7 is expressed in the central nervous system and neural crest mesenchyme of the pharyngeal arches [Sanlaville et al., 2006]. It is felt that the underlying mechanism of this disorder involves abnormalities in NCCs [Siebert et al., 1985]. Knockdown studies in Xenopus laevis embryos using morpholino oligonucleotides targeting CHD7 resulted in abnormal migration of NCCs into the pharyngeal arches and a phenotype consistent with CHARGE syndrome [Bajpai et al., 2010]. Expression analysis in these embryos did not reveal down-regulation of genes (Pax3, Msx1, Zic1) involved in induction and formation of neural plate border but did show a down-regulation of genes (Slug/Snail, Twist, Sox9) that are involved in migration of NCC from the border. These data suggest that CHD7 is involved in the regulation of gene expression programs necessary for the formation of multipotent, migratory neural crest population but not in inductive events for specification of the neural plate border [Bajpai et al., 2010]. Human mutations in CHD7 may result in the same changes in NCC gene expression programs leading to the CHARGE phenotype. Mutations in SEMA3E which may be involved in NCC guidance have also been identified in CHARGE syndrome patients [Lalani et al., 2004].
Guidance Cues for Neural Crest Migration
NCCs follow well-delineated paths from the dorsal neural tube into the craniofacial region, and achieve this feat largely by communicating with the surrounding neural, facial, and pharyngeal epithelia, and cephalic mesoderm [Noden and Trainor, 2005]. NCCs utilize both repulsive and attractive factors as migratory guidance cues but the identities of most of these guidance cues have not been elucidated. There are, however, some notable exceptions: FGF-2 and FGF-8 both function in a chemotactic manner for NCCs [Kubota and Ito, 2000]. Semaphorins and Ephrin molecules also function as NCC guidance factors.
Perhaps the best studied of these guidance cues are the Ephrins, a family of ligands for the Eph receptors. Ephs constitute a subfamily of receptor tyrosine kinases with multiple functions [Nakamoto, 2000] including the modulation of the adhesion proteins such as members of the integrin family [Davy and Soriano, 2007; Murai and Pasquale, 2003; Peinado et al., 2004b]. Semaphorin proteins are involved in cellular processes such as axon guidance and cell migration. Mutations in semaphorin proteins are hypothesized to cause aberrant NCC migration and hence play an important role in pathogenesis of the CHARGE syndrome [Lalani et al., 2004].
Neural Crest Proliferation and Differentiation
Once NCCs arrive at their final destination, how do they know what to do? Two main theories have been proposed. The first theory suggests that NCCs are intrinsically programmed [Noden, 1983] with a facial patterning “blueprint”, and that they carry this molecular patterning information with them upon departure from the neural tube. The second theory suggests that NCCs acquire facial patterning information from the environment in which they eventually find themselves [del Barrio and Nieto, 2002]. This debate has been experimentally addressed in a number of ways, most recently by using quail and duck chimaeras. Quail NCCs transplanted into duck embryos produce ducks with a quail-like face (the “quck”), whereas duck NCCs put into a quail embryo produce quails with a duck-like bill (the “duail”) [Schneider and Helms, 2003]. These data argue that NCCs contain some sort of molecular blueprint for the structures they will eventually form. Other data argue the opposite point: that the fate of NCCs is only determined once they arrive in the facial prominences, and that fate can be altered by simply adjusting the molecular signals that the NCCs see [Hu et al., 2003]. So are NCCs pre-patterned or plastic? The prevailing opinion is that NCCs retain their multipotency even into late embryonic stages of development, which may explain why teratogens can exert their untoward effects even late in human gestation. On the other hand, even malformed faces retain definitive, species-specific characteristics and these appear to be immutable to a large degree.
Once in the prominences, NCCs proliferate and the facial structures begin to take shape. Many factors induce and regulate this proliferation. Abnormal regulation of proliferation contributes to Treacher Collins syndrome (TCS) (OMIM 154500), which manifests with severe craniofacial hypoplasia and dysplasia [Sakai and Trainor, 2009]. This autosomal dominant disorder is caused by mutations in TCOF1, which encodes for the Treacle protein. Treacle is a nuclear protein involved in ribosomal DNA gene transcription. Elegant experiments in mice have shown that haploinsufficiency (TCOF1 +/-) produces a significant decrease in the number of NCCs [Dixon et al., 2006]. This decrease in NCC is secondary to extensive neuroepithelial apoptosis and an accompanying decrease in NCC migration and proliferation [Dixon et al., 2006]. Apoptosis in TCS is caused by the activation of the p53 tumor suppressor pathway. It is hypothesized that mutation in the Treacle protein leads to deficient ribosome biogenesis and causes nuclear stress activation, which stabilizes p53. This stabilization results in activation of pro-apoptic genes and leads to the high degree of apoptosis observed in TCS. Inhibition of p53 provides a possible therapeutic approach in preventing the craniofacial birth defects seen in TCS or other neurocristopathies [Jones et al., 2008].
Other important factors involved in regulation of proliferation of NCC include the secreted proteins Sonic Hedgehog [Jeong et al., 2004] and Wnts [Brugmann et al., 2010]. Disruptions to these two pathways have profound effects on craniofacial morphogenesis. For example, Hedgehog signal transduction requires functional primary cilia; consequently, mutations in components of the primary cilia apparatus result in abnormal Hedgehog signaling, aberrant proliferation of neural crest cells and craniofacial malformations [Brugmann et al., 2010]. Based on these and other data, modulation of Hedgehog signaling may constitute a therapeutic approach to rescuing some types of facial dysmorphologies [Han et al., 2009].
Neural Crest Derivatives in the Face
NCCs make an integral contribution to the elaborate program of craniofacial development; consequently, we will devote the following paragraphs to outlining how the face develops, and the interactions between NCCs and adjacent cell populations that are required for normal craniofacial morphogenesis. The embryonic vertebrate face is made up of seven outgrowths or prominences: the singular frontonasal (FNP), and the paired lateral nasal, maxillary and mandibular prominences. We will consider each prominence separately because NCCs that populate each arch have distinct molecular signatures and interact with different epithelia during the development.
The Facial Prominences
The FNP gives rise to the forehead, middle of the nose, upper lip, philtrum, and primary palate [Helms et al., 2005]. Proper development of the FNP requires interactions between NCC and two epithelia, the forebrain and facial ectoderm [Hu and Helms, 1999]. Normally, the lateral region of the FNP fuses with the lateral nasal prominences (LNP) and medial region of the maxillary prominences (see below) to create the alae and columellae of the nose. The maxillary and mandibular prominences are both derived from the first pharyngeal arch. The maxillary prominences (MXP) [Helms et al., 2005] produce the upper jaw while the mandibular prominences (MNP) produce the lower jaw. The maxilla and mandible are both first arch derivatives and both require interactions between surface ectoderm, NCC, mesoderm and pharyngeal endoderm for their proper development [Couly et al., 2002; Ruhin et al., 2003].
Interactions between Neural Crest Cells and Cephalic Mesoderm
Although NCCs occupy the pharyngeal arches, they also have at their center a mass of mesodermal cells. These mesoderm-derived cells are surrounded by NCCs and externally covered by ectoderm and internally by endoderm [Graham, 2003] (Figure 2). This elaborate arrangement has great significance for craniofacial development because disruptions in the interactions between mesoderm, NCC, and the epithelia have profound effects on craniofacial development. A functional mouth, for example, depends upon the coordinated development of the facial skeleton, which is derived from NCCs, and its associated musculature, which is derived from mesoderm. New data demonstrate that the NCCs act as the conductor for this coordinated morphogenetic program: signals emanating from NCCs instruct and inform mesodermal cells to differentiate into myoblast precursors, and then to organize themselves around the developing skeletal elements [Grenier et al., 2009; Rinon et al., 2007].
FIGURE 2.
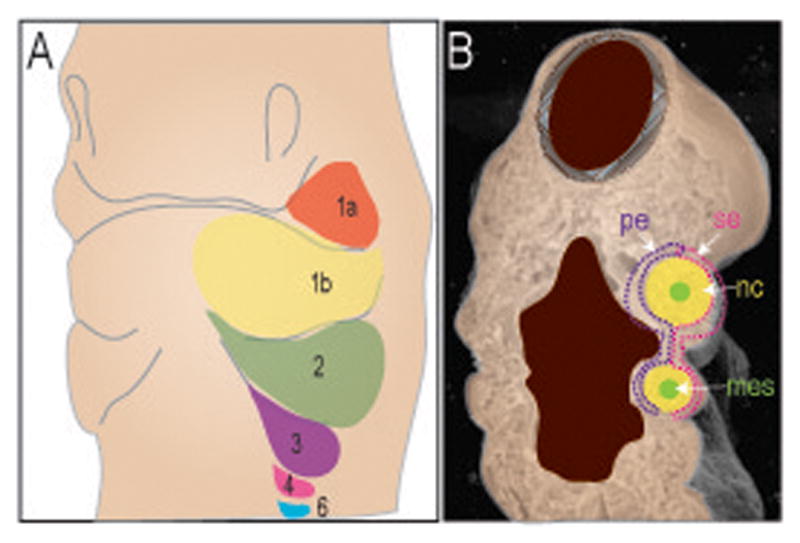
Schematic diagram of the pharyngeal arches. (A) The arches are indicated by different colors. The first arch is divided into maxillary (1a, orange) and mandibular (1b, yellow) component. The second arch (2, green); third arch (3, purple); forth arch (4, pink) and the residuals sixth arch (6, blue). B. Coronal cut through the embryo reveals the tissue contribution to each arch. The arches (1,2,3,4) are composed of a core of neural crest (nc, yellow) and mesoderm (mes, green) surrounded by both surface ectoderm (se, pink) and pharyngeal endoderm (pe, purple).
Much of what we now know about these tissues interactions originates from studies conducted in zebrafish that outline some of the functions of Endothelin (Edn) genes in craniofacial patterning. Edn1 encodes a secreted peptide Endothelin 1 (Edn 1) which signals from facial epithelia and mesoderm to the intervening NCCs that will later form the PA1 (pharyngeal arch 1) derived skeleton [Kimmel et al., 2001; Miller et al., 2000]. These zebrafish studies laid the foundation for understanding how Edn signaling regulated mammalian craniofacial development: for example, mice with a homozygous mutation in Edn1 have severely malformed bones in the jaw and throat [Kurihara et al., 1994] and defective musculature as well [Kimmel et al., 2003]. Data are now emerging from genetic studies in humans, which indicate that mutations in the Endothelin pathway contribute to Waardenburg-Shah syndrome (WS4A) (OMIM #277580; see reference [Edery et al., 1996]) and Hirschsprung disease (also known as aganglionic megacolon, OMIM #142623; see reference [Brooks et al., 2005]). Both Waardenburg and Hirschsprung disease are considered neurocristopathies; i.e., diseases whose etiologies are attributable to defects in NCC behavior. In addition to other NCC-related defects, patients with WS4A may exhibit heterochromia, ocular ptosis and hypertelorism.
The craniofacial features of the velo-cardio-facial / DiGeorge syndrome (DGS) (OMIM #188400) appear to result as a consequence of perturbation of crosstalk between the pharyngeal arch components. In addition to the spectrum of craniofacial dysmorphisms that includes cleft palate, neonatal hypocalcemia secondary to parathyroid hypoplasia, T cell deficiency resulting from hypoplasia or aplasia of the thymus gland, and a number of cardiac malformations that include the outflow tract [Ryan et al., 1997] are observed. The majority of cases involve a 1.5 to 3.0 Mb hemizygous interstitial deletion of chromosome 22q11.2 although terminal deletions and translocations that include 22q11.2 occur. This microdeletion includes between 30-50 genes. TBX1 a Tbox family of binding domain transcription factors is within this 22q11.2 critical region [Scambler, 2010]. In the developing pharyngeal arch Tbx1 is expressed in the endoderm and mesoderm as well as the epithelium of the palatal shelves and frontonasal process [Zoupa et al., 2006]. Decreased Tbx1 expression in the endoderm during pharyngeal arch and pouch development and epithelia of the palatal shelves [Zoupa et al., 2006] is believed to result in secondary NCC abnormalities (extrinsic to NCCs) [Walker and Trainor, 2006]. Supporting this idea, Tbx1 knockout mice show pharyngeal arch and pouch defects, which underlie the mouse phenotype and recapitulate the human phenotype [Walker and Trainor, 2006]. The cononical Wnt-beta-catenin signaling pathway has recently been shown to regulate Tbx1 expression. In mice beta-catenin inactivation results in malformations within the spectrum of DGS phenotypes and may be involved modifying the severity of DGS [Huh and Ornitz, 2010].
Neural Crest Cells and other Craniofacial Malformations
Identifying a molecular basis for one of the most common craniofacial malformations, clefting, has also been pursued heavily. Secondary palate is a product of MXPs while the primary palate is derived from the FNP. The palatal shelves themselves consist of NCC mesenchyme covered by surface and oral epithelia. The palatal shelves must perform an elaborate outgrowth in order for fusion to occur: first, the MXPs must expand and then must undergo a rotation from their initial vertical position to a horizontal position dorsal to the tongue, and finally fuse. If the embryonic tongue is too large, or if it fails to descend because it is ankylosed, this can impede the elevation of the palatal shelves and consequently cause secondary palatal clefting.
Secondary palatal clefting caused by TBX22 mutations
In the last decade, investigators have determined that a X-linked condition where cleft palate is accompanied by ankyloglossia (CPX) (OMIM # 303400) may be caused by mutations in a member of T-box transcription factor, TBX22 [Braybrook et al., 2001]. Unlike other types of clefting that have been attributed to environmental factors, CPX is recognized as an X-linked semi-dominant condition. Members of T-box family play an important role in vertebrate development, especially in mesoderm specification [Papaioannou and Silver, 1998]. Precisely how TBX22 regulates mesoderm-NCC interactions has yet to be determined.
Secondary palatal clefting associated with defects in Wnt signaling
There are other causes for secondary palatal clefting, attributable to defects in MXP outgrowth. For example, in mice with mutation in Wnt 9b, in particular, the basis for the clefting phenotype appears to be insufficient growth of the maxillary prominences [Lan et al., 2006]. Consequently, these prominences, from which the palatal shelves derive, fail to approximate and the result is palatal clefting [Juriloff et al., 2006]. In mammals, Wnt signaling is critical for the proliferation of NCCs within the MXPs [Brugmann et al., 2007; Brugmann et al., 2006] and disruptions in Wnt signaling specifically and profoundly influence the ability of NCC-derived facial prominences to fully develop [Brugmann et al., 2007]. As a consequence of down-regulated Wnt signaling, the FNP and MXPs are truncated and the result is palatal (and facial) clefting.
Palatal clefting and FOXE1 defects
FOXE1 (Forkhead box protein E1) is a member of a transcription factor family that is involved in embryonic pattern formation. Through positional cloning, candidate gene sequencing, and developmental gene expression analyses, a strong correlation has been discovered in between mutations in FOXE1 and the occurrence of cleft lip and palate in humans [Moreno et al., 2009]. Foxe1 is expressed in the secondary palate epithelium of mice [Dathan et al., 2002] and human embryos [Trueba et al., 2005]. Furthermore, mice with a null mutation in Titf2/Foxe1 have cleft palate and thyroid anomalies [De Felice et al., 1998]. The Foxe1 is expressed at the point of fusion between the MXPs and the FNP, further supporting its role in palatogenesis [Moreno et al., 2009].
Palatal clefting and Irf6
Another gene associated with facial clefting is the IRF6 (Interferon regulatory factor) gene, which is part of a larger family of transcription factors that bind to specific DNA sequences and regulate gene expression. In mice, disruption in Irf6 function results in clefting phenotypes [Ingraham et al., 2006; Rahimov et al., 2008]. In humans, mutations in IRF6 cause Van Der Woude syndrome and popliteal pterygium syndrome, two clefting disorders [Little et al., 2009]. There is also an increase risk of isolated cleft lip and palate in humans with variations in IRF6 [Blanton et al., 2005]. The occurrence of clefting in Irf6 mutant mice is hypothesized to be caused by a defect in elevation of palatine shelves, secondary to inappropriate adhesions between the palatal shelves and oral epithelium [Ingraham et al., 2006].
Palatal clefting and EGF receptor mutations
Once the palatal shelves have approximated, they must fuse. For this to occur, the medial edge epithelium must be removed in order for the NCC-derived mesenchyme to become confluent. This typically happens when the epithelial cells covering the palatal mesenchyme undergo programmed cell death and/or cell migration. Transforming growth factor-alpha (TGFα), an epidermal growth factor receptor (EGFR) ligand that is expressed in facial epithelia, plays a role in this fusion process. For example, Egfr-/- mice have midline defects that produce an elongated primary palate, micrognathia, and a high incidence of cleft palate [Miettinen et al., 1999]. In vitro experiments suggest that a delay in epithelial degeneration occurs in the absence of EGFR signaling. The molecular basis for this delay in epithelial degeneration and clefting seen in Egfr-/- embryos is associated with the loss of function of matrix metalloproteinases (MMPs), which are endopeptidases that cleave the extracellular matrix. These experiments illustrate the delicate balance between matrix remodeling and epithelial seam removal that is required for proper craniofacial fusions.
In conclusion, disruptions in the rate, the timing, or the extent of outgrowth of the facial prominences can all result in facial clefting and often these disruptions are attributable to defects in NCC behavior. Clefting malformations occur in approximately 1 out of 700 births, making it one of the most prevalent craniofacial birth defect; and the role of NCCs in this process cannot be overemphasized.
Pluripotency and Multipotency of the Neural Crest
NCCs, like hematopoietic stem cells, exhibit a hierarchical progression from being initially pluripotent to becoming progressively more restricted in their developmental potential. By means of in vitro serial subcloning, Le Douarin and colleagues identified both multipotent and oligopotent NCC progenitors that differed in their developmental repertoire, including their ability to self-renew [Trentin et al., 2004]. Whether NCCs actually retain their pluripotency into adulthood, however, is not entirely clear.
One approach to addressing this question is to follow the fate of NCCs in the adult animal. NCCs give rise to the embryonic facial skeleton, and they are also responsible for maintaining facial skeletal elements into adulthood [Leucht et al., 2008]. When NCC-derived skeletal stem cells are transplanted, they retain the ability to differentiate into chondrocytes and osteoblasts [Leucht et al., 2008]. These studies indicate that adult NCC-derived skeletal stem cells retain a bi-potential fate but they fall short of demonstrating that adult NCCs retain their self-renewing capacity.
The Future of Craniofacial Development
Much of our present knowledge of normal craniofacial development has come from genetic mutations in humans, genetically engineered mice and the results of embryonic exposure to teratogens in both humans and animals. Future advancements in our understanding of craniofacial development will come from utilizing new technologies and exploiting the knowledge of gene regulation provided by other disciplines, in particular the field of epigenetics. Understanding the importance of epigenetic regulation during neural crest cell development has already made an impact on understanding the etiology of craniofacial syndromes [Bajpai et al., 2010]. Furthermore, techniques such as next-generation sequencing can serve to identify patient-specific mutations in sporadic as well as congenital cases of craniofacial disorders, whereas recent advances in neural crest cell culture provide model systems of both indefinitely self-renewing primary human neural crest cultures [Thomas et al., 2008] and lineage-specific differentiation of pluripotent human embryonic stem cells into neural crest cells [Bajpai et al., 2010]. The exploitation of these techniques along with continued careful studies in craniofacial biology will hopefully provide an avenue for the accurate diagnosis and possible treatment of craniofacial disorders.
Acknowledgments
The authors would like to note the valuable insights and suggestions from Alan Shanske, Danny Huylebroeck, Paul Trainor and James Fraser. The manuscript described was supported by Award Number K12HD001255 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development and the Eleanor and Miles Shore Scholars in Medicine, Harvard Medical School to DC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health.
References
- Acloque H, Adams MS, Fishwick K, Bronner-Fraser M, Nieto MA. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest. 2009;119:1438–1449. doi: 10.1172/JCI38019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RM, Stottmann RW, Choi M, Klingensmith J. Endogenous bone morphogenetic protein antagonists regulate mammalian neural crest generation and survival. Dev Dyn. 2006;235:2507–2520. doi: 10.1002/dvdy.20891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang CP, Zhao Y, Swigut T, Wysocka J. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–962. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basch ML, Bronner-Fraser M. Neural crest inducing signals. Adv Exp Med Biol. 2006;589:24–31. doi: 10.1007/978-0-387-46954-6_2. [DOI] [PubMed] [Google Scholar]
- Basch ML, Bronner-Fraser M, Garcia-Castro MI. Specification of the neural crest occurs during gastrulation and requires Pax7. Nature. 2006;441:218–222. doi: 10.1038/nature04684. [DOI] [PubMed] [Google Scholar]
- Blanton SH, Cortez A, Stal S, Mulliken JB, Finnell RH, Hecht JT. Variation in IRF6 contributes to nonsyndromic cleft lip and palate. Am J Med Genet A. 2005;137A:259–262. doi: 10.1002/ajmg.a.30887. [DOI] [PubMed] [Google Scholar]
- Braybrook C, Warry G, Howell G, Mandryko V, Arnason A, Bjornsson A, Ross MT, Moore GE, Stanier P. Physical and transcriptional mapping of the X-linked cleft palate and ankyloglossia (CPX) critical region. Hum Genet. 2001;108:537–545. doi: 10.1007/s004390100518. [DOI] [PubMed] [Google Scholar]
- Brooks AS, Oostra BA, Hofstra RM. Studying the genetics of Hirschsprung's disease: unraveling an oligogenic disorder. Clin Genet. 2005;67:6–14. doi: 10.1111/j.1399-0004.2004.00319.x. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Allen NC, James AW, Mekonnen Z, Madan E, Helms JA. A primary cilia-dependent etiology for midline facial disorders. Hum Mol Genet. 2010;19:1577–1592. doi: 10.1093/hmg/ddq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugmann SA, Goodnough LH, Gregorieff A, Leucht P, ten Berge D, Fuerer C, Clevers H, Nusse R, Helms JA. Wnt signaling mediates regional specification in the vertebrate face. Development. 2007;134:3283–3295. doi: 10.1242/dev.005132. [DOI] [PubMed] [Google Scholar]
- Brugmann SA, Tapadia MD, Helms JA. The molecular origins of species-specific facial pattern. Curr Top Dev Biol. 2006;73:1–42. doi: 10.1016/S0070-2153(05)73001-5. [DOI] [PubMed] [Google Scholar]
- Byrd NA, Meyers EN. Loss of Gbx2 results in neural crest cell patterning and pharyngeal arch artery defects in the mouse embryo. Dev Biol. 2005;284:233–245. doi: 10.1016/j.ydbio.2005.05.023. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D, van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- Cordero D, Marcucio R, Hu D, Gaffield W, Tapadia M, Helms JA. Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J Clin Invest. 2004;114:485–494. doi: 10.1172/JCI19596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couly G, Creuzet S, Bennaceur S, Vincent C, Le Douarin NM. Interactions between Hox-negative cephalic neural crest cells and the foregut endoderm in patterning the facial skeleton in the vertebrate head. Development. 2002;129:1061–1073. doi: 10.1242/dev.129.4.1061. [DOI] [PubMed] [Google Scholar]
- Crane JF, Trainor PA. Neural crest stem and progenitor cells. Annu Rev Cell Dev Biol. 2006;22:267–286. doi: 10.1146/annurev.cellbio.22.010305.103814. [DOI] [PubMed] [Google Scholar]
- Creuzet S, Couly G, Le Douarin NM. Patterning the neural crest derivatives during development of the vertebrate head: insights from avian studies. J Anat. 2005;207:447–459. doi: 10.1111/j.1469-7580.2005.00485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creuzet SE, Martinez S, Le Douarin NM. The cephalic neural crest exerts a critical effect on forebrain and midbrain development. Proc Natl Acad Sci U S A. 2006;103:14033–14038. doi: 10.1073/pnas.0605899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dastot-Le Moal F, Wilson M, Mowat D, Collot N, Niel F, Goossens M. ZFHX1B mutations in patients with Mowat-Wilson syndrome. Hum Mutat. 2007;28:313–321. doi: 10.1002/humu.20452. [DOI] [PubMed] [Google Scholar]
- Dathan N, Parlato R, Rosica A, De Felice M, Di Lauro R. Distribution of the titf2/foxe1 gene product is consistent with an important role in the development of foregut endoderm, palate, and hair. Dev Dyn. 2002;224:450–456. doi: 10.1002/dvdy.10118. [DOI] [PubMed] [Google Scholar]
- Davy A, Soriano P. Ephrin-B2 forward signaling regulates somite patterning and neural crest cell development. Dev Biol. 2007;304:182–193. doi: 10.1016/j.ydbio.2006.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice M, Ovitt C, Biffali E, Rodriguez-Mallon A, Arra C, Anastassiadis K, Macchia PE, Mattei MG, Mariano A, Scholer H, Macchia V, Di Lauro R. A mouse model for hereditary thyroid dysgenesis and cleft palate. Nat Genet. 1998;19:395–398. doi: 10.1038/1289. [DOI] [PubMed] [Google Scholar]
- del Barrio MG, Nieto MA. Overexpression of Snail family members highlights their ability to promote chick neural crest formation. Development. 2002;129:1583–1593. doi: 10.1242/dev.129.7.1583. [DOI] [PubMed] [Google Scholar]
- Del Barrio MG, Nieto MA. Relative expression of Slug, RhoB, and HNK-1 in the cranial neural crest of the early chicken embryo. Dev Dyn. 2004;229:136–139. doi: 10.1002/dvdy.10456. [DOI] [PubMed] [Google Scholar]
- Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci U S A. 2006;103:13403–13408. doi: 10.1073/pnas.0603730103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edery P, Attie T, Amiel J, Pelet A, Eng C, Hofstra RM, Martelli H, Bidaud C, Munnich A, Lyonnet S. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome) Nat Genet. 1996;12(4):442–4. doi: 10.1038/ng0496-442. [DOI] [PubMed] [Google Scholar]
- Ezin AM, Fraser SE, Bronner-Fraser M. Fate map and morphogenesis of presumptive neural crest and dorsal neural tube. Dev Biol. 2009;330(2):221–36. doi: 10.1016/j.ydbio.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix TM, Hanshaw BC, Mueller R, Bitoun P, Murray JC. CHD7 gene and non-syndromic cleft lip and palate. Am J Med Genet A. 2006;140(19):2110–4. doi: 10.1002/ajmg.a.31308. [DOI] [PubMed] [Google Scholar]
- Garavelli L, Zollino M, Mainardi PC, Gurrieri F, Rivieri F, Soli F, Verri R, Albertini E, Favaron E, Zignani M, Orteschi D, Bianchi P, Faravelli F, Forzano F, Seri M, Wischmeijer A, Turchetti D, Pompilii E, Gnoli M, Cocchi G, Mazzanti L, Bergamaschi R, De Brasi D, Sperandeo MP, Mari F, Uliana V, Mostardini R, Cecconi M, Grasso M, Sassi S, Sebastio G, Renieri A, Silengo M, Bernasconi S, Wakamatsu N, Neri G. Mowat-Wilson syndrome: facial phenotype changing with age: study of 19 Italian patients and review of the literature. Am J Med Genet A. 2009;149A(3):417–26. doi: 10.1002/ajmg.a.32693. [DOI] [PubMed] [Google Scholar]
- Garcia-Castro MI, Marcelle C, Bronner-Fraser M. Ectodermal Wnt function as a neural crest inducer. Science. 2002;297(5582):848–51. doi: 10.1126/science.1070824. [DOI] [PubMed] [Google Scholar]
- Graham A. Development of the pharyngeal arches. Am J Med Genet A. 2003;119A(3):251–6. doi: 10.1002/ajmg.a.10980. [DOI] [PubMed] [Google Scholar]
- Grenier J, Teillet MA, Grifone R, Kelly RG, Duprez D. Relationship between neural crest cells and cranial mesoderm during head muscle development. PLoS One. 2009;4(2):e4381. doi: 10.1371/journal.pone.0004381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr. 1979;95(3):395–8. doi: 10.1016/s0022-3476(79)80513-2. [DOI] [PubMed] [Google Scholar]
- Hall BK. The neural crest as a fourth germ layer and vertebrates as quadroblastic not triploblastic. Evol Dev. 2000;2(1):3–5. doi: 10.1046/j.1525-142x.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- Han J, Mayo J, Xu X, Li J, Bringas P, Jr, Maas RL, Rubenstein JL, Chai Y. Indirect modulation of Shh signaling by Dlx5 affects the oral-nasal patterning of palate and rescues cleft palate in Msx1-null mice. Development. 2009;136(24):4225–33. doi: 10.1242/dev.036723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms JA, Cordero D, Tapadia MD. New insights into craniofacial morphogenesis. Development. 2005;132(5):851–61. doi: 10.1242/dev.01705. [DOI] [PubMed] [Google Scholar]
- Herfs M, Hubert P, Suarez-Carmona M, Reschner A, Saussez S, Berx G, Savagner P, Boniver J, Delvenne P. Regulation of p63 Isoforms by Snail and Slug Transcription Factors in Human Squamous Cell Carcinoma. Am J Pathol. 2010;176(4):1941–9. doi: 10.2353/ajpath.2010.090804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation—a syndrome. J Pediatr Ophthalmol Strabismus. 1979;16(2):122–8. doi: 10.3928/0191-3913-19790301-10. [DOI] [PubMed] [Google Scholar]
- Horn D, Weschke B, Zweier C, Rauch A. Facial phenotype allows diagnosis of Mowat-Wilson syndrome in the absence of Hirschsprung disease. Am J Med Genet A. 2004;124A(1):102–4. doi: 10.1002/ajmg.a.20298. [DOI] [PubMed] [Google Scholar]
- Hu D, Helms JA. The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126(21):4873–84. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- Hu D, Marcucio RS, Helms JA. A zone of frontonasal ectoderm regulates patterning and growth in the face. Development. 2003;130(9):1749–58. doi: 10.1242/dev.00397. [DOI] [PubMed] [Google Scholar]
- Huh SH, Ornitz DM. Beta-catenin deficiency causes DiGeorge syndrome-like phenotypes through regulation of Tbx1. Development. 2010;137(7):1137–47. doi: 10.1242/dev.045534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt P, Gulisano M, Cook M, Sham MH, Faiella A, Wilkinson D, Boncinelli E, Krumlauf R. A distinct Hox code for the branchial region of the vertebrate head. Nature. 1991a;353(6347):861–4. doi: 10.1038/353861a0. [DOI] [PubMed] [Google Scholar]
- Hunt P, Whiting J, Muchamore I, Marshall H, Krumlauf R. Homeobox genes and models for patterning the hindbrain and branchial arches. Dev Suppl. 1991b;1:187–96. [PubMed] [Google Scholar]
- Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003;116(Pt 10):1959–67. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- Ingraham CR, Kinoshita A, Kondo S, Yang B, Sajan S, Trout KJ, Malik MI, Dunnwald M, Goudy SL, Lovett M, Murray JC, Schutte BC. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat Genet. 2006;38(11):1335–40. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18(8):937–51. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D, Morrison N, Grant L, Turner T, Fantes J, Connor JM, Murday V. Confirmation of CHD7 as a cause of CHARGE association identified by mapping a balanced chromosome translocation in affected monozygotic twins. J Med Genet. 2006;43(3):280–4. doi: 10.1136/jmg.2005.032946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, Dixon MJ, Trainor P. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008;14(2):125–33. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, van Hagen JM, Donnai D, de Ravel TJ, Veltman JA, Geurts van Kessel A, De Vries BBA, Brunner HG, Hoefsloot LH, van Ravenswaaij CMA. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43(4):306–14. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ, McMahon AP, Carroll TJ, Lidral AC. Wnt9b is the mutated gene involved in multifactorial nonsyndromic cleft lip with or without cleft palate in A/WySn mice, as confirmed by a genetic complementation test. Birth Defects Res A Clin Mol Teratol. 2006;76(8):574–9. doi: 10.1002/bdra.20302. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Miller CT, Moens CB. Specification and morphogenesis of the zebrafish larval head skeleton. Dev Biol. 2001;233(2):239–57. doi: 10.1006/dbio.2001.0201. [DOI] [PubMed] [Google Scholar]
- Kimmel CB, Ullmann B, Walker M, Miller CT, Crump JG. Endothelin 1-mediated regulation of pharyngeal bone development in zebrafish. Development. 2003;130(7):1339–51. doi: 10.1242/dev.00338. [DOI] [PubMed] [Google Scholar]
- Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nat Rev Genet. 2002;3(6):453–61. doi: 10.1038/nrg819. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Ito K. Chemotactic migration of mesencephalic neural crest cells in the mouse. Dev Dyn. 2000;217(2):170–9. doi: 10.1002/(SICI)1097-0177(200002)217:2<170::AID-DVDY4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, Oda H, Kuwaki T, Cao WH, Kamada N, Jishage K, Ouchi Y, Azuma S, Toyoda Y, Ishikawa T, Kumada M, Yazaki Y. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368(6473):703–10. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- Lal-Nag M, Morin PJ. The claudins. Genome Biol. 2009;10(8):235. doi: 10.1186/gb-2009-10-8-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani SR, Safiullah AM, Molinari LM, Fernbach SD, Martin DM, Belmont JW. SEMA3E mutation in a patient with CHARGE syndrome. J Med Genet. 2004;41(7):e94. doi: 10.1136/jmg.2003.017640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan Y, Ryan RC, Zhang Z, Bullard SA, Bush JO, Maltby KM, Lidral AC, Jiang R. Expression of Wnt9b and activation of canonical Wnt signaling during midfacial morphogenesis in mice. Dev Dyn. 2006;235(5):1448–54. doi: 10.1002/dvdy.20723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larriba MJ, Bonilla F, Munoz A. The transcription factors Snail1 and Snail2 repress vitamin D receptor during colon cancer progression. J Steroid Biochem Mol Biol. 2010;121(1-2):106–9. doi: 10.1016/j.jsbmb.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Leucht P, Kim JB, Amasha R, James AW, Girod S, Helms JA. Embryonic origin and Hox status determine progenitor cell fate during adult bone regeneration. Development. 2008;135(17):2845–54. doi: 10.1242/dev.023788. [DOI] [PubMed] [Google Scholar]
- Li B, Kuriyama S, Moreno M, Mayor R. The posteriorizing gene Gbx2 is a direct target of Wnt signalling and the earliest factor in neural crest induction. Development. 2009;136(19):3267–78. doi: 10.1242/dev.036954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lièvre CSL, Douarin NML. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. Journal of Embryology and Experimental Morphology. 1975;34(1):125–154. [PubMed] [Google Scholar]
- Little HJ, Rorick NK, Su LI, Baldock C, Malhotra S, Jowitt T, Gakhar L, Subramanian R, Schutte BC, Dixon MJ, Shore P. Missense mutations that cause Van der Woude syndrome and popliteal pterygium syndrome affect the DNA-binding and transcriptional activation functions of IRF6. Hum Mol Genet. 2009;18(3):535–45. doi: 10.1093/hmg/ddn381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JP, Jessell TM. A role for rhoB in the delamination of neural crest cells from the dorsal neural tube. Development. 1998;125(24):5055–67. doi: 10.1242/dev.125.24.5055. [DOI] [PubMed] [Google Scholar]
- Lumsden A, Sprawson N, Graham A. Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development. 1991;113(4):1281–91. doi: 10.1242/dev.113.4.1281. [DOI] [PubMed] [Google Scholar]
- Mayor R, Guerrero N, Martinez C. Role of FGF and noggin in neural crest induction. Dev Biol. 1997;189(1):1–12. doi: 10.1006/dbio.1997.8634. [DOI] [PubMed] [Google Scholar]
- Miettinen PJ, Chin JR, Shum L, Slavkin HC, Shuler CF, Derynck R, Werb Z. Epidermal growth factor receptor function is necessary for normal craniofacial development and palate closure. Nat Genet. 1999;22(1):69–73. doi: 10.1038/8773. [DOI] [PubMed] [Google Scholar]
- Miller CT, Schilling TF, Lee K, Parker J, Kimmel CB. sucker encodes a zebrafish Endothelin-1 required for ventral pharyngeal arch development. Development. 2000;127(17):3815–28. doi: 10.1242/dev.127.17.3815. [DOI] [PubMed] [Google Scholar]
- Moreno LM, Mansilla MA, Bullard SA, Cooper ME, Busch TD, Machida J, Johnson MK, Brauer D, Krahn K, Daack-Hirsch S, L'Heureux J, Valencia-Ramirez C, Rivera D, López AM, Moreno MA, Hing A, Lammer EJ, Jones M, Christensen K, Lie RT, Jugessur A, Wilcox AJ, Chines P, Pugh E, Doheny K, Arcor-Borgos M, Marazita ML, Murray JC, Lidral AC. FOXE1 association with both isolated cleft lip with or without cleft palate, and isolated cleft palate. Hum Mol Genet. 2009;18(24):4879–96. doi: 10.1093/hmg/ddp444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat DR, Croaker GD, Cass DT, Kerr BA, Chaitow J, Ades LC, Chia NL, Wilson MJ. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet. 1998;35(8):617–23. doi: 10.1136/jmg.35.8.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat DR, Wilson MJ, Goossens M. Mowat-Wilson syndrome. J Med Genet. 2003;40(5):305–10. doi: 10.1136/jmg.40.5.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai KK, Pasquale EB. ‘Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci. 2003;116(Pt 14):2823–32. doi: 10.1242/jcs.00625. [DOI] [PubMed] [Google Scholar]
- Murray SA, Carver EA, Gridley T. Generation of a Snail1 (Snai1) conditional null allele. Genesis. 2006;44(1):7–11. doi: 10.1002/gene.20178. [DOI] [PubMed] [Google Scholar]
- Murray SA, Gridley T. Snail1 gene function during early embryo patterning in mice. Cell Cycle. 2006a;5(22):2566–70. doi: 10.4161/cc.5.22.3502. [DOI] [PubMed] [Google Scholar]
- Murray SA, Gridley T. Snail family genes are required for left-right asymmetry determination, but not neural crest formation, in mice. Proc Natl Acad Sci U S A. 2006b;103(27):10300–4. doi: 10.1073/pnas.0602234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamoto M. Eph receptors and ephrins. Int J Biochem Cell Biol. 2000;32(1):7–12. doi: 10.1016/s1357-2725(99)00096-5. [DOI] [PubMed] [Google Scholar]
- Nieto MA. Epithelial-Mesenchymal Transitions in development and disease: old views and new perspectives. Int J Dev Biol. 2008;53(8-10):1541–7. doi: 10.1387/ijdb.072410mn. [DOI] [PubMed] [Google Scholar]
- Nieto MA, Sargent MG, Wilkinson DG, Cooke J. Control of cell behavior during vertebrate development by Slug, a zinc finger gene. Science. 1994;264(5160):835–9. doi: 10.1126/science.7513443. [DOI] [PubMed] [Google Scholar]
- Noden DM. The role of the neural crest in patterning of avian cranial skeletal, connective and muscle tissues. Dev Biol. 1983;96:144–165. doi: 10.1016/0012-1606(83)90318-4. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207(5):575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrink B. Epithelial cell adhesion molecules. Exp Cell Res. 1986;163(1):1–21. doi: 10.1016/0014-4827(86)90554-9. [DOI] [PubMed] [Google Scholar]
- Oley CA, Baraitser M, Grant DB. A reappraisal of the CHARGE association. J Med Genet. 1988;25(3):147–56. doi: 10.1136/jmg.25.3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaioannou VE, Silver LM. The T-box gene family. Bioessays. 1998;20(1):9–19. doi: 10.1002/(SICI)1521-1878(199801)20:1<9::AID-BIES4>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Patthey C, Edlund T, Gunhaga L. Wnt-regulated temporal control of BMP exposure directs the choice between neural plate border and epidermal fate. Development. 2009;136(1):73–83. doi: 10.1242/dev.025890. [DOI] [PubMed] [Google Scholar]
- Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004a;24(1):306–19. doi: 10.1128/MCB.24.1.306-319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Marin F, Cubillo E, Stark HJ, Fusenig N, Nieto MA, Cano A. Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. J Cell Sci. 2004b;117(Pt 13):2827–39. doi: 10.1242/jcs.01145. [DOI] [PubMed] [Google Scholar]
- Prendergast GC. Actin' up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001;1(2):162–8. doi: 10.1038/35101096. [DOI] [PubMed] [Google Scholar]
- Radisky DC, LaBarge MA. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell. 2008;2(6):511–2. doi: 10.1016/j.stem.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, Domann FE, Govil M, Christensen K, Bille C, Melbye M, Jugessur A, Lie RT, Wilcox AJ, Fitzpatric DR, Green ED, Mossey PA, Little J, Steegers-Theunissen RP, Pennacchio LA. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet. 2008;40(11):1341–7. doi: 10.1038/ng.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34(8):656–65. doi: 10.1136/jmg.34.8.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinon A, Lazar S, Marshall H, Buchmann-Moller S, Neufeld A, Elhanany-Tamir H, Taketo MM, Sommer L, Krumlauf R, Tzahor E. Cranial neural crest cells regulate head muscle patterning and differentiation during vertebrate embryogenesis. Development. 2007;134(17):3065–75. doi: 10.1242/dev.002501. [DOI] [PubMed] [Google Scholar]
- Ruffins S, Bronner-Fraser M. A critical period for conversion of ectodermal cells to a neural crest fate. Dev Biol. 2000;218(1):13–20. doi: 10.1006/dbio.1999.9555. [DOI] [PubMed] [Google Scholar]
- Ruhin B, Creuzet S, Vincent C, Benouaiche L, Le Douarin NM, Couly G. Patterning of the hyoid cartilage depends upon signals arising from the ventral foregut endoderm. Dev Dyn. 2003;228(2):239–46. doi: 10.1002/dvdy.10380. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brøndum-Nielsen K, Scambler PJ. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34(10):798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai D, Trainor PA. Treacher Collins syndrome: unmasking the role of Tcof1/treacle. Int J Biochem Cell Biol. 2009;41(6):1229–32. doi: 10.1016/j.biocel.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Martin M, Rodriguez-Garcia A, Perez-Losada J, Sagrera A, Read AP, Sanchez-Garcia I. SLUG (SNAI2) deletions in patients with Waardenburg disease. Hum Mol Genet. 2002;11(25):3231–6. doi: 10.1093/hmg/11.25.3231. [DOI] [PubMed] [Google Scholar]
- Sandell LL, Trainor PA. Neural crest cell plasticity. size matters. Adv Exp Med Biol. 2006;589:78–95. doi: 10.1007/978-0-387-46954-6_5. [DOI] [PubMed] [Google Scholar]
- Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clément-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attié-Bitach T. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43(3):211–217. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007;15(4):389–99. doi: 10.1038/sj.ejhg.5201778. [DOI] [PubMed] [Google Scholar]
- Saunders CJ, Zhao W, Ardinger HH. Comprehensive ZEB2 gene analysis for Mowat-Wilson syndrome in a North American cohort: a suggested approach to molecular diagnostics. Am J Med Genet A. 2009;149A(11):2527–31. doi: 10.1002/ajmg.a.33067. [DOI] [PubMed] [Google Scholar]
- Scambler PJ. 22q11 Deletion Syndrome: A Role for TBX1 in Pharyngeal and Cardiovascular Development. Pediatr Cardiol. 2010;31(3):378–90. doi: 10.1007/s00246-009-9613-0. [DOI] [PubMed] [Google Scholar]
- Schneider RA, Helms JA. The cellular and molecular origins of beak morphology. Science. 2003;299(5606):565–8. doi: 10.1126/science.1077827. [DOI] [PubMed] [Google Scholar]
- Selleck MAJ, Stern CD. Fate mapping and cell lineage analysis of Hensen's node in the chick embryo. Development. 1991;112:615–626. doi: 10.1242/dev.112.2.615. [DOI] [PubMed] [Google Scholar]
- Siebert JR, Graham JM, Jr, MacDonald C. Pathologic features of the CHARGE association: support for involvement of the neural crest. Teratology. 1985;31(3):331–6. doi: 10.1002/tera.1420310303. [DOI] [PubMed] [Google Scholar]
- Steventon B, Araya C, Linker C, Kuriyama S, Mayor R. Differential requirements of BMP and Wnt signalling during gastrulation and neurulation define two steps in neural crest induction. Development. 2009;136(5):771–9. doi: 10.1242/dev.029017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steventon B, Carmona-Fontaine C, Mayor R. Genetic network during neural crest induction: from cell specification to cell survival. Semin Cell Dev Biol. 2005;16(6):647–54. doi: 10.1016/j.semcdb.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Taneyhill LA. To adhere or not to adhere: the role of Cadherins in neural crest development. Cell Adh Migr. 2008;2(4):223–30. doi: 10.4161/cam.2.4.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- Thomas S, Thomas M, Wincker P, Babarit C, Xu P, Speer MC, Munnich A, Lyonnet S, Vekemans M, Etchevers HC. Human neural crest cells display molecular and phenotypic hallmarks of stem cells. Hum Mol Genet. 2008;17(21):3411–25. doi: 10.1093/hmg/ddn235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trentin A, Glavieux-Pardanaud C, Le Douarin NM, Dupin E. Self-renewal capacity is a widespread property of various types of neural crest precursor cells. Proc Natl Acad Sci U S A. 2004;101(13):4495–500. doi: 10.1073/pnas.0400629101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueba SS, Auge J, Mattei G, Etchevers H, Martinovic J, Czernichow P, Vekemans M, Polak M, Attie-Bitach T. PAX8, TITF1, and FOXE1 gene expression patterns during human development: new insights into human thyroid development and thyroid dysgenesis-associated malformations. J Clin Endocrinol Metab. 2005;90(1):455–62. doi: 10.1210/jc.2004-1358. [DOI] [PubMed] [Google Scholar]
- Van de Putte T, Francis A, Nelles L, van Grunsven LA, Huylebroeck D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum Mol Genet. 2007;16(12):1423–36. doi: 10.1093/hmg/ddm093. [DOI] [PubMed] [Google Scholar]
- Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005;133A(3):306–8. doi: 10.1002/ajmg.a.30559. [DOI] [PubMed] [Google Scholar]
- Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, van der Vliet WA, Huys EH, de Jong PJ, Hamel BCJ, Schoenmakers EFPM, Brunner HG, Veltman JA, Geurts van Kessel A. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat Genet. 2004;36(9):955–7. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- Walker MB, Trainor PA. Craniofacial malformations: intrinsic vs extrinsic neural crest cell defects in Treacher Collins and 22q11 deletion syndromes. Clin Genet. 2006;69(6):471–9. doi: 10.1111/j.0009-9163.2006.00615.x. [DOI] [PubMed] [Google Scholar]
- Wincent J, Schulze A, Schoumans J. Detection of CHD7 deletions by MLPA in CHARGE syndrome patients with a less typical phenotype. Eur J Med Genet. 2009;52(4):271–2. doi: 10.1016/j.ejmg.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhou BP. Snail: More than EMT. Cell Adh Migr. 2010;4(2):199–203. doi: 10.4161/cam.4.2.10943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010;152A(3):674–86. doi: 10.1002/ajmg.a.33323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoupa M, Seppala M, Mitsiadis T, Cobourne MT. Tbx1 is expressed at multiple sites of epithelial-mesenchymal interaction during early development of the facial complex. Int J Dev Biol. 2006;50(5):504–10. doi: 10.1387/ijdb.052116mz. [DOI] [PubMed] [Google Scholar]
- Zweier C, Thiel CT, Dufke A, Crow YJ, Meinecke P, Suri M, Ala-Mello S, Beemer F, Bernasconi S, Bianchi P, Bier A, Devriendt K, Dimitrov B, Firth H, Gallagher RC, Garavelli L, Gillessen-Kaesbach G, Hudgins L, Kääriäinen H, Karstens S, Krantz I, Mannhardt A, Medne L, Mücke J, Kibaek M, Krogh LN, Peippo M, Rittinger O, Schulz S, Schelley SL, Temple IK, Dennis NR, Van der Knaap MS, Wheeler P, Yerushalmi B, Zenker M, Seidel H, Lachmeijer A, Prescott T, Kraus C, Lowry RB, Rauch A. Clinical and mutational spectrum of Mowat-Wilson syndrome. Eur J Med Genet. 2005;48(2):97–111. doi: 10.1016/j.ejmg.2005.01.003. [DOI] [PubMed] [Google Scholar]