

Abstract
Recent years have seen a rapid increase in the mechanistic and structural information on enzymes that are involved in the biosynthesis and breakdown of naturally occurring phosphonates. This review focuses on these recent developments with an emphasis on those enzymes that have been characterized crystallographically in the past five years, including proteins involved in the biosynthesis of phosphinothricin, fosfomycin, and dehydrophos and proteins involved in resistance mechanisms.
Keywords: antibiotics, phosphonates, X-ray crystallography, methyl transferase, dioxygenase, epoxidase, resistance
Introduction
The recent surge in available genomic information for a wide range of bacteria has coincided with, and at the same time stimulated, a strong renewed interest in natural product biosynthesis. Many recent genome-aided studies have focused on the products of non-ribosomal peptide synthesis, polyketides, isoprenoids, and ribosomally synthesized natural products [1-3]. By comparison, relatively little was known until recently about the biosynthetic pathways leading to phosphonate natural products. Seminal pioneering studies by Seto and coworkers established that the P-C bond in most phosphonates is formed by the enzyme phosphoenolpyruvate (PEP) mutase [4], which converts PEP to phosphonopyruvate (PnPy, Figure 1), and that the biosynthetic pathways of this class of compounds contain many unusual chemical transformations [5, 6]. Recent years have seen the characterization of the first biosynthetic clusters for phosphonates, including phosphinothricin [7, 8], fosfomycin [9], FR900098 [10], dehydrophos [11], and rhizocticin [12] (Figure 1). The genetic information in turn has allowed the elucidation of the biosynthetic pathways and study of individual enzymes. In parallel, the use of phosphonates in the clinic has fueled studies of emerging resistance mechanisms. This review will focus on recent developments in phosphonate biosynthesis and resistance with emphasis on those enzymes that have been investigated mechanistically and characterized crystallographically.
Figure 1.
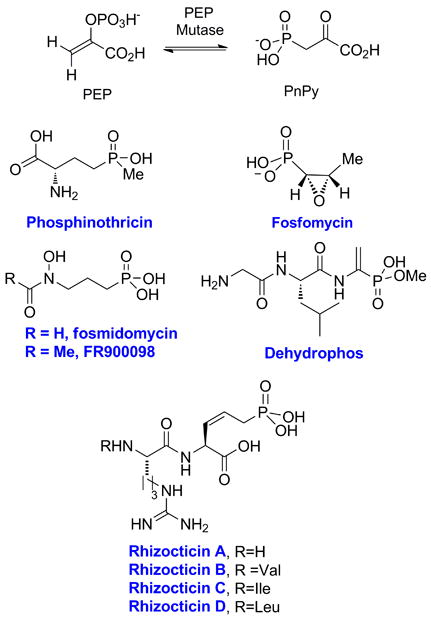
Phosphorus-carbon bond formation by PEP mutase and structures of the phosphonate natural products for which the gene clusters have been determined.
Phosphinothricin biosynthesis
Metcalf and coworkers recently revised the early steps in the biosynthesis of phosphinothricin [13], an unusual phosphininate containing two P-C bonds (Figure 1). Phosphinothricin is the active ingredient of commercial herbicides as Herbiace and BASTA, and is used in combination with LibertyLink genetically modified crops including corn, canola, soybean, and cotton. Phosphinothricin mimics the tetrahedral intermediate generated in Gln synthase [14, 15] by using a monoanionic phosphinate group containing two P-C bonds. One of the revised biosynthetic steps features the conversion of 2-hydroxyethyl phosphonate (2-HEP) to hydroxymethylphosphonate (HMP, Figure 2A). In vitro reconstitution of this transformation catalyzed by the enzyme encoded by the phpD gene demonstrated that the protein only requires ferrous ion and oxygen for activity, and that 2-HEP is converted into HMP and formate [16]. Experiments with 18O-labeled molecular oxygen showed that the label is incorporated into both HMP and formate, demonstrating the enzyme is a 2-hydroxyethylphosphonate dioxygenase (HEPD). However, about 40% of the hydroxyl oxygen in HMP was derived from solvent, requiring a solvent exchangeable intermediate during catalysis. Use of deuterium labeled substrates showed that the enzyme abstracts a hydrogen atom from C2, and that the hydrogens on C1 are retained in the HMP product.
Figure 2.
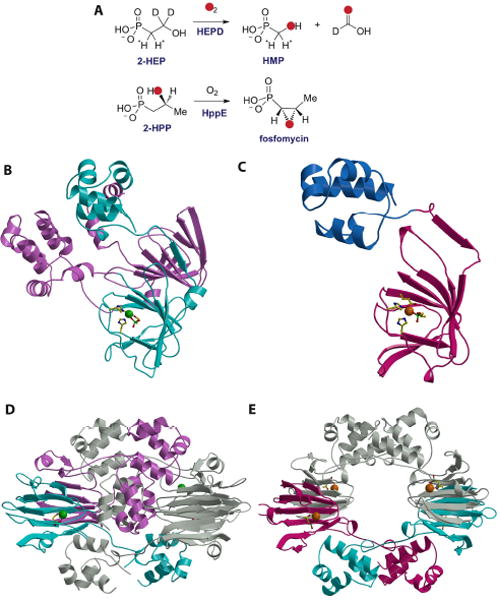
(A) Reactions catalyzed by HEPD and HppE. (B) Structure of the HEPD-HEP complex with the active metal-containing domain colored in cyan and the vestigial domain colored in pink. The bound Cd(II) is shown as a green sphere, with protein ligands and the substrate molecule shown as stick figures. (C) Structure of the HppE-fosfomycin complex with the cupin domain colored in cyan and the novel alpha domain colored in blue. The requisite Fe(II) is colored in orange with protein ligands and the substrate molecule shown as stick figures. (D) Structure of the HEPD dimer suggesting the relevance of the vestigial repeat in forming the composite active site. (E) Structure of the HppE tetramer shown in the same orientation as that for the HEPD dimer.
The X-ray crystal structure of HEPD, determined to a resolution of 1.8 Å (Figure 2B), demonstrates that the polypeptide is composed of two repeats of a cupin domain [16]. This structural homology is unexpected given that the enzyme does not show sequence similarities to other proteins that share the cupin architecture. Members of the cupin superfamily are non-heme mononuclear iron-dependent proteins that adopt a characteristic antiparallel β-barrel fold. The two cupin domains in HEPD are decorated with a helical region consisting of roughly five α-helices and this topology of a cupin domain fused to a helical region is unique to HEPD and HppE, an iron-dependent enzyme involved in the biosynthesis of fosfomycin (see below).
Although the structure of HEPD consists of two structurally intertwined repeats of this fold, only one of the two repeats contains a functional metal-binding site made up of the characteristic 2-His-1-Glu facial triad composed of residues His129, Glu176, and His182. As crystallization was conducted in the presence of a cadmium salt, a Cd(II) ion is positioned at the metal-binding site with three solvent molecules completing the six-coordinate metal center. The corresponding metal-binding site in the second repeat lacks this facial triad and is further occluded by residues Tyr359 and Lys404. Mutational studies document that residues within this vestigial metal site are not essential for catalytic activity. The crystal structure of Cd(II)-HEPD in complex with 2-HEP was solved to 1.9 Å resolution and reveals bi-dentate coordination of the substrate to the metal. Although substrate binding does not induce any major structural reorganization of the polypeptide, the side chain of Tyr98 undergoes a torsional rotation to form a hydrogen bond with one of the phosphonate oxygen atoms of the substrate, with Asn126 providing an additional hydrogen bond with the substrate.
On the basis of the mechanistic studies and crystallographic information, two mechanisms were proposed for the conversion of 2-HEP to HMP and formate [16] (Figure 3A). For both, the ferrous enzyme reacts with oxygen to form a formally ferric-superoxo species I that abstracts a hydrogen atom from the substrate. The resulting substrate radical can then either be hydroxylated or hydroperoxylated. In the former case, an acetal results that can undergo a retro-Claisen type reaction to provide formate and the stabilized anion in intermediate II, which may attack the electrophilic ferryl species resulting in HMP. In the case of hydroperoxylation, a Criegee rearrangement would afford ester III, which upon hydrolysis would also provide HMP and formate. For both mechanisms exchange of oxygen from O2 with solvent can be explained but by different mechanisms. For the hydroxylation pathway, the ferryl intermediate in II can undergo exchange as reported previously for other systems [17-19]. For the hydroperoxylation mechanism, exchange requires an unusual hydrolysis of the ester III via attack of hydroxide (which can exchange with solvent) on C1 rather than at the carbonyl carbon. Experiments with a series of substrate analogs provide direct support for hydroperoxylation. For instance, with 1-hydroxyethyl phosphonate, the direct product of the Criegee rearrangement step, 1-acetylphosphate, was produced (Figure 3B) [20].
Figure 3.

(A) Two proposed mechanisms for the HEPD reaction. (B) The substrate analog 1-HEP is converted to acetylphosphate by HEPD. (C) Two proposed mechanisms for the HppE reaction. The phosphonate groups are shown as dianionic but could be monoprotonated. L = undefined ligand, probably water or hydroxide.
Fosfomycin biosynthesis
Fosfomycin is a clinically used antibiotic that inhibits UDP-GlcNAc enolpyruvoyl transferase (MurZ), an essential enzyme in cell wall biosynthesis, via a covalent mechanism [21]. The biosynthetic gene cluster in Streptomyces wedmorensis has been characterized [22, 23], as has its biosynthetic pathway. The most interesting of the biosynthetic enzymes that have been studied thus far is an unusual epoxidase that converts (2S)-hydroxypropyl phosphonate (2-HPP) to fosfomycin (Figure 2A). During this transformation, the hydroxyl oxygen of 2-HPP ends up in the epoxide [24]. Liu and coworkers have extensively studied this process [25-30] and Drennan and coworkers solved the structure of the enzyme [31].
The crystal structure of the oxidative cyclase consists of a single β-barrel containing cupin domain augmented with a novel helical region composed of five α-helices (Figure 2C). The facial triad of HppE consists of His138, Glu142 and His180 and crystallographic studies of Fe(II)-HppE revealed a hexacoordinate metal with three solvent molecules completing the coordination sphere. Structural studies of catalytically inert aerobic Co(II)-HppE and active Fe(II)-HppE, obtained anerobically, showed two modes of binding for the substrate 2-HPP suggestive of a precatalytic two-step binding process. Within the two copies of the polypeptide in the crystallographic asymmetric unit, one molecule shows a monodentate coordination of a phosphonate oxygen to the metal and the second shows a bidentate interaction. Notably, bidentate coordination of the substrate is accompanied by a reorganization of an active site loop that results in the reorganization of active site residue Tyr105 to form a hydrogen bond with one of the oxygen atoms in the 2-HPP substrate. Interestingly, substrate binding in HEPD induced a torsional rotation in the side chain of the equivalent Tyr98 to similarly form a hydrogen bond interaction with an oxygen atom in the substrate 2-HEP.
A comparison of the crystal structure of HppE with that of HEPD provides several insights into the evolution of these mononuclear non-heme oxygenases. First, although the composition of the active site facial triad is similar in both polypeptides, the first two metal ligands in HEPD are separated by 48 amino acids, which is distinct from the 1-4 residue spacing that is typical for other facial triad enzymes such as HppE. Second, both enzymes employ similar mechanisms of substrate orientation that involves structural reorganization to position a catalytically requisite tyrosine residue within hydrogen bonding distance to the substrate. Lastly, a comparison of the quarternary structure of both proteins suggests a role for the vestigial cupin domain in HEPD. Specifically, in the active site of HppE, the substrate HPP is engaged by Tyr105 and Lys23, and mutational analysis established the necessity of both of these residues in catalysis [31]. In the crystal structure of the HppE monomer, Lys23 is distal to the active site. However, within the structure of the biological tetramer, a Lys23 from one subunit engages in intermolecular interactions with the active site of another subunit (Figure 2E). Similarly, in the structure of HEPD, a lysine residue from the vestigial domain of one subunit (Lys16) protrudes into the functional active site of the other subunit to stabilize the phosphonate (Figure 2D), recapitulating the stabilizing interactions that are observed in the structures of Fe(II)- and Co(II)-HppE-HPP. Thus, it is likely that the vestigial cupin of HEPD serves to position Lys16 for intermolecular interactions with substrate in a composite active site.
Despite the structural similarities of HppE and HEPD, the two enzymes catalyze remarkably different reactions on very similar substrates (Figure 2A). Bidentate binding of the substrate may explain how both enzymes can activate O2 to form a ferric-superoxo species (I in HEPD and IV in HppE), a step that is normally thermodynamically unfavorable. From there, one main difference between the two enzymes is that HppE requires the input of two additional electrons from a reductase. In principle, this allows cleavage of the O-O bond of molecular oxygen prior to substrate activation (Figure 3C, top pathway), an option not available to HEPD. As a result, HppE can access a ferryl intermediate V, a more reactive species than IV that may be required to abstract a hydrogen atom from C1 of 2-HPP, which is less activated than the hydrogen abstracted from C2 of 2-HEP by HEPD. The radical at C2 of 2-HPP can then undergo an unusual intramolecular rebound-like combination with the alkoxide group at C2 to provide fosfomycin [30]. Alternatively, the superoxo intermediate IV might abstract a hydrogen atom to generate a radical at C2 and ferric hydroperoxide VI (Figure 3C, bottom pathway). Electron transfer from the reductase may then cleave the O-O bond to generate ferryl intermediate VII that could undergo a similar rebound-like mechanism to provide fosfomycin and a ferric enzyme that needs to be reduced to the active ferrous HppE for another turnover [30]. Different variants of this latter mechanism can be drawn but they have in common hydrogen atom abstraction by a ferric-superoxo intermediate. Recent 18O kinetic isotope studies on the HppE reaction showed that the formation of an FeIII–OOH species is involved in the rate-limiting step of O2 activation. This step may either be hydrogen atom abstraction or proton-coupled electron transfer from the substrate or electron transfer from the reductase [32].
Biosynthesis of dehydrophos
Dehydrophos produced by Streptomyces luridus is unusual in that it is the only natural product phosphonate known to date in which the phosphonate group is esterified [33] (Figure 4A). Its biosynthetic gene cluster was recently sequenced revealing a candidate gene for the methyltransferase that installs the methyl ester [11]. Heterologous expression and purification allowed characterization of the enzyme. Evaluation of a series of synthetic potential substrates in the dehydrophos biosynthetic pathway demonstrated that the enzyme is rather promiscuous and can methylate a large range of phosphonates (Figure 4A) [34]. By far the most efficient substrates are tripeptides such as 1, suggesting that methylation takes place late in the biosynthesis of dehydrophos. This conclusion implies that methylation is not a self-defense mechanism to prevent toxicity of the biosynthetic intermediates for the producing strain but rather a potential role in its mechanism of action. Indeed, if dehydrophos is imported by a general peptide permease followed by proteolysis, as is observed for other peptides containing phosphonate groups [35, 36], then this process would release acetylphosphonate monomethyl ester, a known inhibitor of pyruvate dehydrogenase [31]. This model is supported by structure-activity relationship studies with synthetic analogs of dehydrophos [37]. Interestingly, the substrate promiscuity extends to other natural product phosphonates. For instance, fosmidomycin, fosfomycin, and L-AP4 (Figure 4A) were all methylated by the enzyme. Phospho-serine, the phosphate ester analog of L-AP4, was also a substrate indicating that DhpI does not strongly differentiate between a phosphonate and its corresponding phosphate ester [34].
Figure 4.
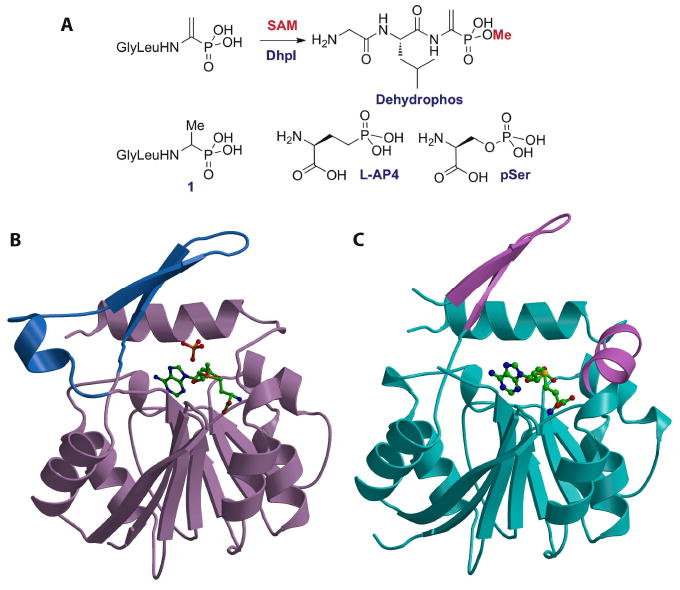
(A) Reaction catalyzed by DhpI and structures of several of the phosphonates that are also substrates for the enzyme. (B) Structure of the DhpI-SAM-sulfate complex showing the nucleotide binding Rossman fold in brown and the unusual insertions necessary for substrate binding in blue. The SAM co-factor and sulfate anion are shown as stick figures. (C) Structure of the DhpI-SAH complex with the nucleotide-binding domain colored in cyan and part of the novel insertion as well as a newly formed helix colored in pink. The helix of the insertion is disordered in the SAH complex.
The 2.3 Å resolution crystal structure of DhpI in complex with S-adenosylmethionine (SAM) revealed a core nucleotide binding Rossman fold domain, typical of SAM-dependent methyltransferases, augmented with additional structural elements that are important for substrate recognition [34]. The novel structural features include an amino terminal extension of thirty residues and an insertion of a short helix and two subsequent β-strands (Figure 4B). A sulfate molecule, originating fortuitously from the crystallization solution, was found adjacent to SAM, where the phosphonate substrate would bind. Biochemical analyses of variants of residues that engage the anion confirmed their respective roles in substrate recognition [34].
The structures of the DhpI-SAM-sulfate and DhpI-SAM-2-HEP complexes revealed that the enzyme forms a homodimer, with the helical insertion from one molecule crossing over into the active site of the other molecule. Residues within this helix that form inter-subunit interactions stabilize the substrate, and mutations at Tyr29, which is distal to the substrate binding site but participates in inter-subunit interactions, results in a 500-fold decrease in kcat/Km for substrate Gly-l-Leu-l-Ala-P, relative to the wild-type enzyme. DhpI is the first example of a methyltransferase harboring a composite active site composed of residues from multiple subunits. The composite active site contains an extended binding cleft in which a number of residues within the cross-over helical insert are poised to form hydrogen-bond interactions with extended substrates, such as the peptidyl chain of dehydrophos.
A comparison of the DhpI-SAM structure with the structure of DhpI in complex with product S-adenosylhomocysteine (SAH), determined to 1.5 Å resolution (Figure 4C), reveals significant rearrangements around the active site. Notably, conformational shifts in the secondary structural elements that harbor residues mediating inter-subunit interactions result in the disruption of the composite active site. As a consequence, residues within the helical insert are disordered further disrupting the organization of active site residues that engage the substrate. The concerted re-alignment of these active site residues in the DhpI-SAH structure may facilitate the release of methylphosphonate products following catalysis.
Commonalities in phosphonate recognition
Recent years have been a continuous influx of structural data on phosphonate biosynthetic enzymes, and the small, but notable, number of such available studies suggests some generalities with respect to phosphonate recognition. For example, despite the fact that HppE and HEPD share no discernable sequence identity, and carry out chemically distinct reactions, the overall architecture of their respective active sites and the determinants of phosphonate recognition are quite similar.
Both enzymes contain a 2His/1Glu facial triad that harbors a six-coordinate metal center, which engages their respective substrates through a bidentate interaction with the metal (Figure 5A & B). Binding of the substrate induces a significant conformational rearrangement in HppE, and a more modest side chain rotation in HEPD, which results in the formation of a hydrogen bonding interaction between the substrate and a tyrosine residue (Tyr105 in HppE, and Tyr98 in HEPD) that is essential for catalysis. Similarly, the active sites of both enzymes are composed of residues from different subunits in an oligomeric assembly. In HppE, Lys23 from one subunit of the biological tetramer interacts with HPP in the active site of an adjacent subunit (Figure 5A), and mutational studies have established that this residue is essential for activity [31]. In the structure of the HEPD dimer, Lys16 from the vestigial repeat of one subunit interacts with 2-HEP in the active site of a different subunit (Figure 5B). Mutational studies at Lys16 in HEPD have not yet been carried out.
Figure 5.
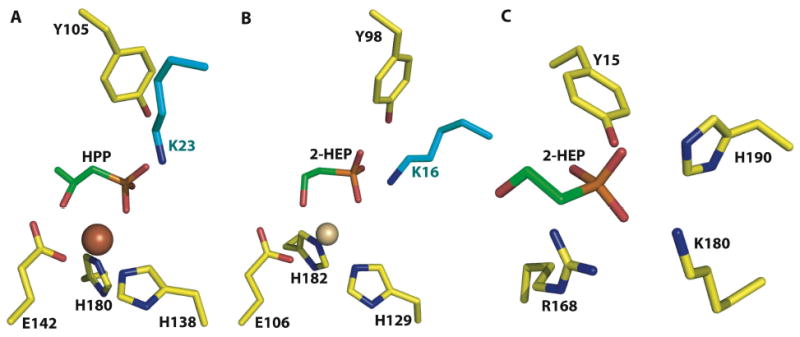
A close-up view of the actives sites of (A) HppE in complex with 2-HPP, (B) HEPD in complex with 2-HEP, and (C) DhpI in complex with 2-HEP. Despite the lack of any notable sequence similarities, both HEPD and HppE use a near identical constellation of active site residues to carry out their respective reactions. Notably, both polypeptides contain composite active sites with a catalytically essential lysine residue (colored in cyan) from a different subunit interacting with the active site. A comparison of HppE, HEPD, and DhpI co-crystal structures illustrates the chemical features that are used by functionally and structurally distinct enzymes to harbor phosphonate substrates.
A comparison of the HppE-2-HPP and HEPD-2-HEP co-crystal structures with that of the DhpI-2-HEP-SAM complex shows striking similarities in substrate recognition (Figures 5A-C). Despite the fact that DhpI is completely unrelated to the non-heme iron-dependent enzymes, recognition of 2-HEP is carried out using a similar ligand set consisting of Tyr15 and Lys180. The importance of these residues has been established by mutational analysis [34]. Of course, in HEPD the catalytically requisite metal provides an additional interaction with the phosphonate substrate and the lack of a stabilizing metal in DhpI is compensated by additional protein ligands including Arg168 and His190. The universality of the tyrosine/lysine pair as phosphonate ligands remains to be seen given the small number of co-crystal structures available, and additional structural data will be required on the growing number of phosphonate biosynthetic enzymes.
Phosphonate resistance determinants
Several different characterized resistance determinants target fosfomycin, each utilizing a unique mechanism for inactivation of the antibiotic. The most common examples of resistance against fosfomycin are the result of alterations in the transport or uptake of the antibiotic [38, 39] and mutation to the MurA target [40]. More recently, several enzymes that inactivate fosfomycin by covalent modification have been identified, and these include the phosphonate kinase FomA [9, 41] and the thiol transferases FosA and FosB, which catalyze the addition of glutathione or cysteine to the antibiotic (Figure 6A) [42, 43]. Lastly, genome mining studies identified a homolog of these thiol transferases called FosX that catalyzes the hydration of fosfomycin (Figure 6B) [44].
Figure 6.
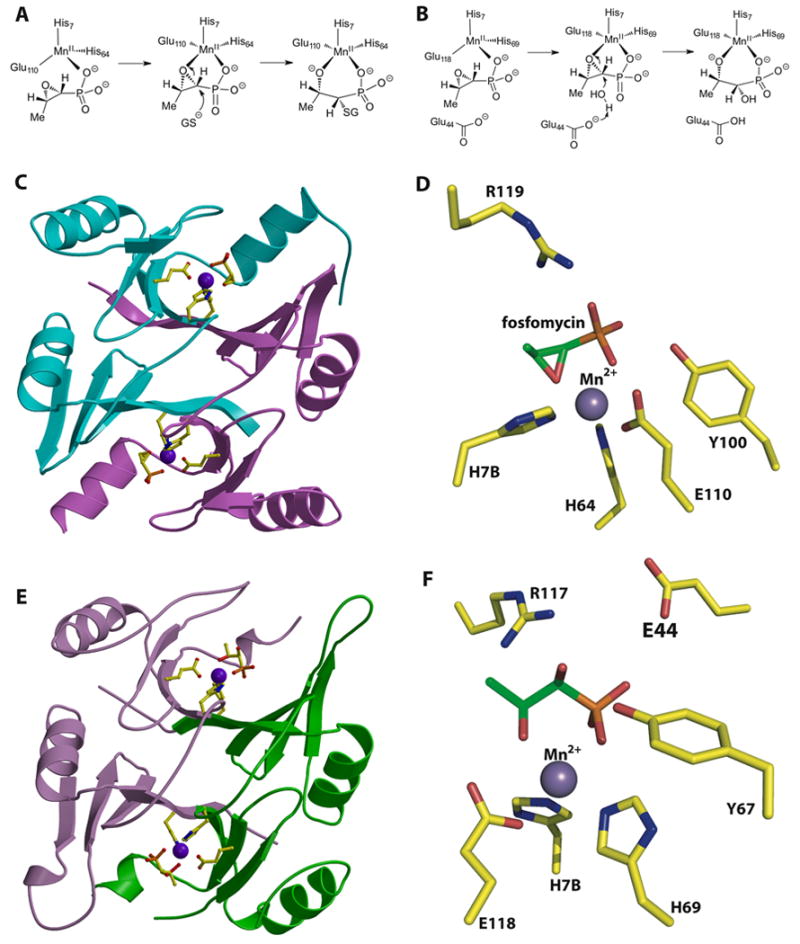
(A) Reaction catalyzed by the fosfomycin thiol transferase FosA. (B) Reaction catalyzed by the homologous FosX metalloenzyme that hydrates the antibiotic. (C) Structure of the fosfomycin inactivating enzyme FosA. (D) Close-up view of the active site of FosA. (E) Structure of the fosfomycin hydrolase FosX. (F) Close-up view of the active site of FosX revealing a glutamate residue (Glu44) that is proximal to the substrate and that is essential for catalysis. This residue is absent in FosA.
Biochemical and structural studies of FosA established that the enzyme catalyzes the Mn(II)-dependent addition of glutathione to the oxirane of fosfomycin [42]. The FosA and FosB thiol transferases belong to the vicinal oxygen chelate superfamily of enzymes that provide a cavity for metal-binding with open coordination sites to promote electrophilic catalysis by the metal ion. In the 1.19 Å resolution crystal structure of FosA, the metal sites are composed in a domain-swapped arrangement with binding motifs derived from different subunits (Figure 6C) [45]. The catalytically requisite manganese ion is liganded by residues His7, His64, and Glu110, resulting in an unexpected four-coordinate distorted tetrahedral geometry (Figure 6A). In the co-crystal structure of FosA in complex with fosfomycin, the Mn(II) shifts to a five-coordinate geometry with a phosphonate oxygen and an oxirane oxygen of the substrate completing the trigonal bipyramidal coordination sphere (Figure 6D). These studies suggest a role for the metal ion in promoting electrophilic catalysis, probably by stabilization of the alkoxide in the transition state complex.
Bioinformatic queries of bacterial genome sequences using the primary sequence of FosA and FosB identified a subfamily of enzymes, termed FosX, in a number of pathogenic organisms [44]. Although the active site metal triad of FosX is conserved to those found in FosA/B, unexpectedly the product of the reaction with fosfomycin was identified to be 1,2-dihydroxypropylphosphonic acid, a hydration product. The 1.83 Å resolution crystal structure of FosX revealed that the overall fold and several active site features are identical to those observed in crystal structures of FosA (Figure 6E). However, a glutamate side chain (Glu44) is novel to members of the FosX family and is poised in the vicinity of the active site, 5 Å away from C1 of the antibiotic. Mutational analysis establishes that Glu44 is essential for catalytic activity and single-turnover 18O exchange experiments demonstrate that the carboxylate acts as a general base to promote the attack of water onto the antibiotic and is not a nucleophile that opens the ring [44].
In addition to the FosA/B/X family of resistance genes, a second class of determinants encoded by the fomA and fomB loci have been characterized to confer high-level resistance against fosfomycin in the producer strains [41]. These genes encode polypeptides with low-level sequence identity to eukaryotic protein kinases, and biochemical studies with recombinant enzymes established that FomA catalyzes the initial phosphorylation of fosfomycin to fosfomycin monophosphate, and FomB the subsequent phosphorylation of this product to fosfomycin diphosphate. The 1.6 Å resolution co-crystal structure of the FomA-AMP-PNP-fosfomycin complex established FomA as a member of the amino acid kinase superfamily [46]. The substrate fosfomycin is bound by the amino-terminal lobe of the polypeptide and recognition of the phosphonate by the protein is effected by hydrogen bonding with active site residues. Interactions between the nucleotide and the substrate are indirect and occur largely through solvent-mediated hydrogen bonds. Based on structural similarities with N-acetyl-L-glutamate kinase, the phosphorylation reaction was suggested to occur through an associative in-line phosphoryl transfer.
Several microorganisms have been demonstrated to utilize phosphonates as a nutrient source [47]. Currently, five different enzyme activities cleave C-P bonds and these can be divided into two mechanistic classes. The first class encompasses enzymes that act on phosphonates that contain a β-carbonyl group, and these include phosphonoacetate or phosphonoacetaldehyde hydrolase, phosphonopyruvate hydrolase and phosphoenolpyruvate phosphomutase. For the latter, the P-C cleavage reaction is the reverse of the P-C bond forming reaction used for phosphonate biosynthesis (Figure 1). The second class consists of carbon-phosphorus lyase, a multi-enzyme system that can directly cleave C-P bonds without forming enolate-stabilized carbanions. Although the biochemical characterization of C-P lyase activity has not yet been established, radiolabeling studies using crude cell lysates demonstrate that the products of C-P lyase are the corresponding hydrocarbon and a phosphonate derivative [48].
Genetic analyses demonstrated that CP-lyase activity is encoded by the phn operon that contains fourteen different open reading frames that function in response to phosphate depletion [49]. Gene deletion studies showed that only a subset of the fourteen genes, namely phnGHIJKLM, are necessary for catalysis, with the other genes involved in regulation and/or transport. Biochemical characterization of these proteins has been minimal, as many have proven recalcitrant to in vitro reconstitution of activity [47]. For example, genetic analysis of phnH showed that this gene is essential for C-P lyase activity, and while a high-resolution crystal structure of PhnH is available, no function has yet been assigned for this protein [50]. Functional annotation has been established for PhnD, a periplasmic organophosphate binding protein [51], PhnN, an ATP-dependent kinase [52], and PhnO, an acetyl-CoA transferases that functions on several different phosphonates [53]. Lastly, the phnP locus was shown to encode a phosphodiesterase that shares structural similarities with tRNase Z endonucleases, and likely shares mechanistic similarities with these enzymes [54].
Outlook
The last decade has lead to a resurgence of investigations into the microbiology, biochemistry and structural biology of enzymes that catalyze the biosynthesis of phosphonate natural products. Genome mining experiments, based on using the PEP mutase genes as genetic markers for the identification of phosphonate encoding gene clusters, have yielded a wealth of information on the novel C-P containing products, and their underlying biosynthesis [55]. Continued biochemical and structural biological elucidation into the enzymology of these biosynthetic enzymes will likely provide more examples of unusual catalysts and shed additional light into the molecular determinants of phosphonate recognition.
Acknowledgments
This work has been supported by the National Institutes of Health (PO1 GM077596 to WAV and SKN). We thank our collaborator Bill Metcalf and his laboratory for their contributions to the work described in this review.
Abbreviations used
- 2-HEP
2-hydroxyethylphosphonate
- 2-HPP
(2S)-hydroxypropylphosphonate
- SAM
S-adenosylmethionine
- HEPD
2-hydroxyethylphosphonate dioxygenase
- HppE
(2S)-hydroxypropylphosphonate epoxidase
- DhpI
phosphonate methyltransferase
- FosA/B
fosfomycin thiol transferases
- FosX
fosfomycin hydrolase
- HMP
hydroxymethylphosphonate
- SAH
S-adenosylhomocysteine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bode HB, Muller R. Angew Chem Int Ed Engl. 2005;44:6828–6846. doi: 10.1002/anie.200501080. [DOI] [PubMed] [Google Scholar]
- 2.Oman TJ, van der Donk WA. Nat Chem Biol. 2010;6:9–18. doi: 10.1038/nchembio.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Challis GL. J Med Chem. 2008;51:2618–2628. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]
- 4.Seidel HM, Freeman S, Seto H, Knowles JR. Nature. 1988;335:457–458. doi: 10.1038/335457a0. [DOI] [PubMed] [Google Scholar]
- 5.Hidaka T, Seto H, Imai S. J Am Chem Soc. 1989;111:8012–8013. [Google Scholar]
- 6.Seto H, Kuzuyama T. Nat Prod Rep. 1999;16:589–596. doi: 10.1039/a809398i. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz D, Berger S, Heinzelmann E, Muschko K, Welzel K, Wohlleben W. Appl Environ Microbiol. 2004;70:7093–7102. doi: 10.1128/AEM.70.12.7093-7102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blodgett JA, Zhang JK, Metcalf WW. Antimicrob Agents Chemother. 2005;49:230–240. doi: 10.1128/AAC.49.1.230-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodyer RD, Shao Z, Metcalf WM, Thomas PM, Kelleher NL, van der Donk WA, Zhao H. Chem Biol. 2006;13:1171–1182. doi: 10.1016/j.chembiol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 10.Eliot AC, Griffin BM, Thomas PM, Johannes TW, Kelleher NL, Zhao H, Metcalf WW. Chem Biol. 2008;15:765–770. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Circello BT, Eliot AC, Lee J-L, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:402–411. doi: 10.1016/j.chembiol.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borisova SA, Circello BT, Zhang JK, van der Donk WA, Metcalf WW. Chem Biol. 2010;17:28–37. doi: 10.1016/j.chembiol.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blodgett JA, Thomas PM, Li G, Velasquez JE, van der Donk WA, Kelleher NL, Metcalf WW. Nat Chem Biol. 2007;3:480–485. doi: 10.1038/nchembio.2007.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leason M, Cunliffe D, Parkin D, Lea PJ, Miflin BJ. Phytochemistry. 1982;21:855–857. [Google Scholar]
- 15.Tachibana K, Watanabe T, Sekizawa Y, Takematsu T. Nippon Noyaku Gakkaishi; J Pestic Sci. 1986;11:27–31. [Google Scholar]
- 16.Cicchillo RM, Zhang H, Blodgett JAV, Whitteck JT, Li G, Nair SK, van der Donk WA, Metcalf WW. Nature. 2009;459:871–874. doi: 10.1038/nature07972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kikuchi Y, Suzuki Y, Tamiya N. Biochem J. 1983;213:507–512. doi: 10.1042/bj2130507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baldwin JE, Adlington RM, Crouch NP, Pereira IAC. Tetrahedron. 1993;49:7499–7518. [Google Scholar]
- 19.Pestovsky O, Bakac A. Inorg Chem. 2006;45:814–820. doi: 10.1021/ic051868z. [DOI] [PubMed] [Google Scholar]
- 20.Whitteck JT, Cicchillo RM, van der Donk WA. J Am Chem Soc. 2009;131:16225–16232. doi: 10.1021/ja906238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marquardt JL, Brown ED, Lane WS, Haley TM, Ichikawa Y, Wong CH, Walsh CT. Biochemistry. 1994;33:10646–10651. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- 22.Hidaka T, Goda M, Kuzuyama T, Takei N, Kidaka M, Seto H. Mol Gen Genet. 1995;249:274–280. doi: 10.1007/BF00290527. [DOI] [PubMed] [Google Scholar]
- 23.Kuzuyama T, Hidaka T, Imai S, Seto H. J Antibiot. 1993;46:1478–1480. doi: 10.7164/antibiotics.46.1478. [DOI] [PubMed] [Google Scholar]
- 24.Hammerschmidt F. J Chem Soc Perkin Trans. 1991;1:1993–1996. [Google Scholar]
- 25.Liu P, Murakami K, Seki T, He X, Yeung SM, Kuzuyama T, Seto H, Liu HW. J Am Chem Soc. 2001;123:4619–4620. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- 26.Zhao Z, Liu P, Murakami K, Kuzuyama T, Seto H, Liu HW. Angew Chem Int Ed Engl. 2002;41:4529–4532. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 27.Zhao ZB, Liu PH, Murakami K, Kuzuyama T, Seto H, Liu HW. Angew Chem Int Ed. 2002;41:4529–4532. doi: 10.1002/1521-3773(20021202)41:23<4529::AID-ANIE4529>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 28.Liu P, Liu A, Yan F, Wolfe MD, Lipscomb JD, Liu HW. Biochemistry. 2003;42:11577–11586. doi: 10.1021/bi030140w. [DOI] [PubMed] [Google Scholar]
- 29.Liu P, Mehn MP, Yan F, Zhao Z, Que L, Jr, Liu HW. J Am Chem Soc. 2004;126:10306–10312. doi: 10.1021/ja0475050. [DOI] [PubMed] [Google Scholar]
- 30.Yan F, Moon SJ, Liu P, Zhao Z, Lipscomb JD, Liu A, Liu HW. Biochemistry. 2007;46:12628–12638. doi: 10.1021/bi701370e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Higgins LJ, Yan F, Liu P, Liu HW, Drennan CL. Nature. 2005;437:838–844. doi: 10.1038/nature03924. [DOI] [PubMed] [Google Scholar]
- 32.Mirica LM, McCusker KP, Munos JW, Liu HW, Klinman JP. J Am Chem Soc. 2008;130:8122–8123. doi: 10.1021/ja800265s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whitteck JT, Ni W, Griffin BM, Eliot AC, Thomas PM, Kelleher NL, Metcalf WW, van der Donk WA. Angew Chem Int Ed Engl. 2007;46:9089–9092. doi: 10.1002/anie.200703810. [DOI] [PubMed] [Google Scholar]
- 34.Lee JH, Bae B, Kuemin M, Circello BT, Metcalf WW, Nair SK, Van der Donk WA. 2010 doi: 10.1073/pnas.1006848107. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen JG, Havas L, Leicht E, Lenox-Smith I, Nisbet LJ. Antimicrob Agents Chemother. 1979;16:306–313. doi: 10.1128/aac.16.3.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Atherton FR, Hall MJ, Hassall CH, Lambert RW, Lloyd WJ, Lord AV, Ringrose PS, Westmacott D. Antimicrob Agents Chemother. 1983;24:522–528. doi: 10.1128/aac.24.4.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuemin M, van der Donk WA. 2010 submitted for publication. [Google Scholar]
- 38.Kadner RJ, Winkler HH. J Bacteriol. 1973;113:895–900. doi: 10.1128/jb.113.2.895-900.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuruoka T, Yamada Y. J Antibiot. 1975;28:906–911. doi: 10.7164/antibiotics.28.906. [DOI] [PubMed] [Google Scholar]
- 40.Wu HC, Venkateswaran PS. Ann N Y Acad Sci. 1974;235:587–592. doi: 10.1111/j.1749-6632.1974.tb43292.x. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi S, Kuzuyama T, Seto H. Antimicrob Agents Chemother. 2000;44:647–650. doi: 10.1128/aac.44.3.647-650.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernat BA, Laughlin LT, Armstrong RN. Biochemistry. 1997;36:3050–3055. doi: 10.1021/bi963172a. [DOI] [PubMed] [Google Scholar]
- 43.Suarez JE, Mendoza MC. Antimicrob Agents Chemother. 1991;35:791–795. doi: 10.1128/aac.35.5.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fillgrove KL, Pakhomova S, Newcomer ME, Armstrong RN. J Am Chem Soc. 2003;125:15730–15731. doi: 10.1021/ja039307z. [DOI] [PubMed] [Google Scholar]
- 45.Rife CL, Pharris RE, Newcomer ME, Armstrong RN. J Am Chem Soc. 2002;124:11001–11003. doi: 10.1021/ja026879v. [DOI] [PubMed] [Google Scholar]
- 46.Pakhomova S, Bartlett SG, Augustus A, Kuzuyama T, Newcomer ME. J Biol Chem. 2008;283:28518–28526. doi: 10.1074/jbc.M803709200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Quinn JP, Kulakova AN, Cooley NA, McGrath JW. Environ Microbiol. 2007;9:2392–2400. doi: 10.1111/j.1462-2920.2007.01397.x. [DOI] [PubMed] [Google Scholar]
- 48.Avila LZ, Draths KM, Frost JW. Bioorg Med Chem Lett. 1991;1:51–54. [Google Scholar]
- 49.Yakovleva GM, Kim SK, Wanner BL. Appl Microbiol Biotechnol. 1998;49:573–578. doi: 10.1007/s002530051215. [DOI] [PubMed] [Google Scholar]
- 50.Adams MA, Luo Y, Hove-Jensen B, He SM, van Staalduinen LM, Zechel DL, Jia Z. J Bacteriol. 2008;190:1072–1083. doi: 10.1128/JB.01274-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rizk SS, Cuneo MJ, Hellinga HW. Protein Sci. 2006;15:1745–1751. doi: 10.1110/ps.062135206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hove-Jensen B, Rosenkrantz TJ, Haldimann A, Wanner BL. J Bacteriol. 2003;185:2793–2801. doi: 10.1128/JB.185.9.2793-2801.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Errey JC, Blanchard JS. Biochemistry. 2006;45:3033–3039. doi: 10.1021/bi052297p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Podzelinska K, He SM, Wathier M, Yakunin A, Proudfoot M, Hove-Jensen B, Zechel DL, Jia Z. J Biol Chem. 2009;284:17216–17226. doi: 10.1074/jbc.M808392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Metcalf WW, van der Donk WA. Annu Rev Biochem. 2009;78:65–94. doi: 10.1146/annurev.biochem.78.091707.100215. [DOI] [PMC free article] [PubMed] [Google Scholar]