

Abstract
A fascinating feature of some bifunctional enzymes is the presence of an internal channel or tunnel to connect the multiple active sites. A channel can allow for a reaction intermediate generated at one active site to be used as a substrate at a second active site, without the need for the intermediate to leave the safety of the protein matrix. One such bifunctional enzyme is carbon monoxide dehydrogenase/acetyl-CoA synthase from Moorella thermoacetica (mtCODH/ACS). A key player in the global carbon cycle, CODH/ACS uses a Ni–Fe–S center called the C-cluster to reduce carbon dioxide to carbon monoxide and uses a second Ni–Fe–S center, called the A-cluster, to assemble acetyl-CoA from a methyl group, coenzyme A, and C-cluster-generated CO. mtCODH/ACS has been proposed to contain one of the longest enzyme channels (138 Å long) to allow for intermolecular CO transport. Here, we report a 2.5 Å resolution structure of xenon-pressurized mtCODH/ACS and examine the nature of gaseous cavities within this enzyme. We find that the cavity calculation program CAVENV accurately predicts the channels connecting the C- and A-clusters, with 17 of 19 xenon binding sites within the predicted regions. Using this X-ray data, we analyze the amino acid composition surrounding the 19 Xe sites and consider how the protein fold is utilized to carve out such an impressive interior passageway. Finally, structural comparisons of Xe-pressurized mtCODH/ACS with related enzyme structures allow us to study channel design principles, as well as consider the conformational flexibility of an enzyme that contains a cavity through its center.
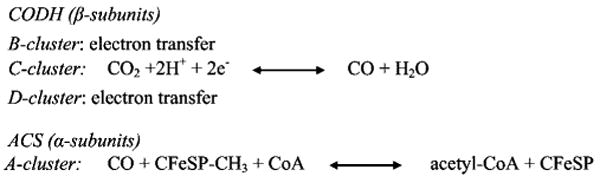
Carbon monoxide dehydrogenase/acetyl-CoA synthase (CODH/ACS)1 is a key component of various energy yielding pathways in microbes. In acetogens such as Moorella thermoacetica, CODH/ACS is a bifunctional enzyme that is responsible for the reduction of CO2 to CO and subsequent assembly of acetyl-CoA, as part of the Wood–Ljungdahl carbon fixation pathway. Methanogenic archea also use a CODH/ACS variant sometimes referred to as acetyl-CoA decarbonylase/synthase (ACDS), but, in this case, the enzyme complex has a different subunit organization and is involved in the degradation of acetyl-CoA to form methane and CO2. Additionally, organisms such as the phototrophic anaerobe Rhodospirullum rubrum use monofunctional CODH to grow on CO, a chlorophyll and heme degradation product, when light is not available for photosynthesis. Due to their role in reducing the levels of gaseous pollutants CO and CO2 in our environment, CODH/ACS enzymes have been well studied (reviewed in refs 1–5). One of the best characterized family members, the bifunctional M. thermoacetica enzyme (mtCODH/ACS) is a 310 kDa α2β2 heterotetramer that catalyzes two different reactions using seven metalloclusters (two A-, two B-, two C-, and one D-cluster). While the B- and D-clusters in the CODH (β) subunit are Fe4S4 centers that shuttle electrons, the C-cluster, also in β, generates CO from CO2 (Scheme 1). The CO produced at the C-cluster travels through a channel in the enzyme to the A-cluster of the ACS (α) subunit, where it is combined with a methyl group and coenzyme A (CoA) to form acetyl-CoA (Scheme 1). The methyl group is transferred to the A-cluster from the methyl-Co(III) state of a corrinoid iron–sulfur protein (CFeSP). Thus, the synthesis of acetyl-CoA at the A-cluster involves a methylation reaction, a carbonylation reaction, and the synthesis of a high-energy thioester bond. Acetyl-CoA synthesis also involves substrates of three very different sizes: CO (2 atoms, 28 Da), methylated CFeSP (11911 atoms, 84124 Da), and coenzyme A (84 atoms, 768 Da). Here we use X-ray crystallography and structural comparisons to consider how CO produced at the C-cluster travels to the A-cluster and how the enzyme is able to perform a molecular juggling act such that the A-cluster can react with substrates of such different sizes.
Scheme 1. Reactions of CODH/ACS.
Several lines of evidence indicate that CO is sequestered from solvent as it travels through the enzyme between the C-cluster in CODH where it is produced and the A-cluster in ACS where it is utilized. Studies with labeled CO2 and unlabeled CO show that CO derived from CO2 is preferentially incorporated into acetyl-CoA, suggesting that the main route for CO to access the A-cluster is by way of the C-cluster (6). Also, the rate of acetyl-CoA synthesis is significantly faster with CO2 than CO as a substrate, and hemoglobin, a CO “sponge”, has little effect on the rate, consistent with CO2-derived CO from the C-cluster being the preferred substrate for ACS (7). Similarly, CO does not compete with CO2 as a substrate for an ACS-catalyzed exchange reaction between labeled CO2 and the carbonyl group of acetyl-CoA, and hemoglobin does not affect the rate of this reaction (6). With this strong evidence for a channel between active sites, it came as a surprise when the first crystal structure showed that the C- and A-clusters are almost 70 Å apart (8). How then is the enzyme designed such that CO is sequestered in the enzyme for its 70 Å long journey from one active site to another? The first insights into this query came from cavity calculations on the structure of mtCODH/ACS using the program CAVENV (9). Interior surface calculations revealed a 138 Å long cavity that runs almost the entire length of the enzyme, connecting all A-clusters and C-clusters (8). The radius of this cavity is at least 1.4 Å, except for a contraction at M148 in CODH (Mt numbering is used throughout unless otherwise noted). Mutagenesis experiments of putative channel residues A110C, A222L, and A265M showed decreased acetyl-CoA activity (10), and a preliminary experiment with Xe gas also showed that this cavity could serve as a gas conduit, but the details of the Xe sites were not described (11). Here we use Xe gas to further explore the nature of this amazing 138 Å long channel. We consider how this channel is created given the protein fold and how differences in cavities are generated between a monofunctional CODH (where CO need not be directed to a second active site) and the β-subunits of a bifunctional CODH/ACS where a pathway is required.
The first enzyme channel was discovered in tryptophan synthase by the Davies group (12) and later in carbamoyl phosphate synthase, glutamine phosphoribosylpyrophosphate amidotransferase, asparagine synthetase, imidazole glycerol phosphate synthase, and glucosamine 6-phosphate synthase, among others (13). In many cases, channels exist to allow reactive intermediates to travel from one active site to another without encountering solvent that would lead to their decomposition. In the case of CODH/ACS, however, CO is not a reactive molecule. Instead, one might consider whether its channel protects the environment from this intermediate rather than the other way around. Also, making CO from CO2 and then discarding it, as would occur without a channel, is energetically wasteful since each molecule of lost CO discards two low-potential electrons originally derived from glucose or H2 oxidation. Another function of a channel is to direct molecules to a particular site on an enzyme. For mtCODH/ACS, a channel could direct CO to a metal coordination site on the A-cluster where it would be catalytically competent rather than potentially inhibitory. Finally, the activities of the C- and A-clusters become synchronized when substrates for both reactions are available (14), and a channel connecting the clusters could assist in this coordination. The structural analysis presented here is the most detailed description of Xe sites in a protein channel, allowing us to consider the nature of protein channels in general and the role of this channel in particular.
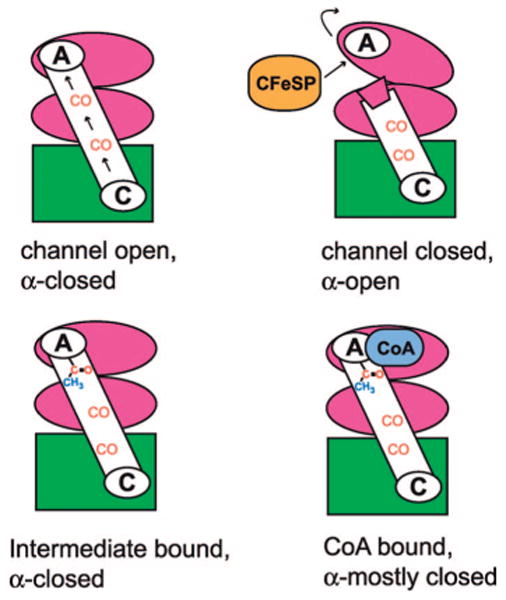
One puzzle is how CO can be so completely sequestered by a channel in mtCODH/ACS when conformational changes must occur to allow this enzyme to react with the large substrate methylated CFeSP and the medium-sized substrate CoA. It seems that at least four different conformations of this enzyme must exist for the A-cluster to access all substrates and perform the required chemistry (Scheme 2). There must be a “channel-open” conformation where CO generated at the C-cluster can travel uninterrupted to the A-cluster. Since biochemical data suggest that CO generated on the enzyme does not leave (6, 7), the A-cluster in the α-subunit must be sealed off from solvent when the CO channel is open (“channel-open; α-closed”). This conformation is presumably the state captured in the structure described here, as well as in our previous structure (8) and the Zn/Ni form of the Fontecilla-Camps structure (5). However, for the CFeSP to transfer a methyl group to the A-cluster, the A-cluster must be solvent accessible. When the A-cluster is accessible for methyl transfer, the channel must be closed or CO would diffuse out of the enzyme, something known not to occur. In this state, the enzyme would be “channel-closed; α-open”. The Ni/Ni form of the Fontecilla-Camps structure is presumed to be in this “channel-closed; α-open” state (5). When CO reacts with a methyl moiety to form an acetyl-metal intermediate, the A-cluster must again be sequestered from solvent (“intermediate-bound; α-closed”). If water were allowed to access the active site at this stage, acetate rather than acetyl-CoA would be formed, leading to an uncoupling of CODH/ACS activity from energy production, as it is the thioester bond of acetyl-CoA from which the organism derives energy. Finally, CoA, a medium-sized substrate, must access the A-cluster in such a way as to prevent solvent entry. This conformation of mtCODH/ACS is expected to be “CoA-bound; α-mostly closed”. Neither an intermediate-bound nor a CoA-bound conformation has been observed crystallographically. Below, we explore the evidence for these different enzyme conformations.
Scheme 2. Cartoon of Required CODH/ACS Conformations.
Materials and Methods
Protein Purification and Crystal Growth
M. thermoacetica (formerly Clostridium thermoaceticum strain ATCC 39073) was purified under strictly anaerobic conditions as described (15, 16). Crystals were grown as described (8), with the addition of 0.25 μL of a solution of crushed microcrystals to aid in nucleation. For data collection, crystals were flash-cooled in liquid nitrogen using 20% glycerol as a cryoprotectant.
Pressurizing mtCODH/ACS Crystals with Xenon
Crystals of CODH/ACS were harvested anaerobically and transported to the Stanford Synchrotron Radiation Laboratory (SSRL) in glass capillaries filled with a stabilizing solution that contained an increased PEG concentration. A Xe pressure cell (smb.slac.stanford.edu/facilities/hardware/pressurecell/ltpc.html), available at SSRL, was used to pressurize the crystals with Xe before cryocooling, according to standard protocols (17). Briefly, a looped crystal was lowered quickly into the pressurizing chamber, and the chamber was locked. The crystal was kept moist by placing ∼500 μL of holding solution at the bottom of the pressurizing chamber. The chamber was purged three times with 50 psi Xe gas, followed by an increase in Xe pressure to 200 psi for 60 s. After quickly depressurizing the chamber (∼20 s), the looped crystal was plunged into liquid nitrogen. Since the Xe pressurization procedure was performed outside of the anaerobic chamber, 2 mM DTT was used to protect the metallocenters from oxygen damage. We know from previous work that crystals exposed to oxygen for more than 26 h show complete destruction of the C- and D-clusters, while the A-cluster's binuclear cluster remains intact (data not shown). Following data collection on the Xe-CODH/ACS crystals, we examined the electron density for the metalloclusters and did not observe any damage to any clusters including the most oxygen sensitive C- and D-centers (Figure S1 in Supporting Information).
Data Collection and Processing
Data on Xe-pressurized CODH/ACS crystals were measured at the SSRL beamline 9-1 (Table 1). The resulting P1 unit cell had the following dimensions: a = 99.54 Å, b = 136.60 Å, c = 141.75 Å, α = 101.29°, β = 109.22°, and γ = 103.91°. The mosaicity of the Xe-pressurized crystal increased to 0.7–0.9 deg from the usual 0.3–0.4 deg. Xe-pressurized crystals diffracted to better than 2.5 Å, allowing for a highly detailed view of the Xe binding sites. Data processing was carried out using the HKL package (18). The published CODH/ACS structure (PDB 1MJG) stripped of water molecules was used for the initial rigid body refinement, followed by slow cool Cartesian dynamics simulated annealing refinement in CNS (19). This first round of refinement alone resulted in R factors that were under 30%, suggesting that the structure with Xe gas was quite similar to the previous native structure. Final rounds of refinement were performed with REFMAC (20) with NCS restraints for each rigid part (two segments in CODH, three in ACS). The final model includes residues 2–674 for CODH chains A–D, residues 2–729 for ACS chains M, N, and P, and residues 2–728 for ACS chain O. Ramachandran analysis of the final model shows that 98.9% of residues lie in the most favored or additionally allowed regions, with 0.4% in disallowed regions. Final data collection and refinement statistics are given in Table 1.
Table 1. Data Collection and Refinement Statistics.
| xenon CODH/ACSa | |
|---|---|
| Data Collection | |
| space group | P1 |
| cell dimensions | |
| a, b, c (Å) | 99.54, 136.60, 141.75 |
| α, β, γ (deg) | 101.29, 109.22, 103.91 |
| wavelength (Å) | 0.9184 |
| resolution (Å) | 49.0–2.51 (2.60–2.51) |
| reflections (total/unique) | 457691/217468 |
| Rsym (%) | 8.3 (42.2) |
| mean I/σI | 10.3 (2.0) |
| completeness | 96.1 (92.0) |
| redundancy | 2.1 (2.0) |
| Refinement | |
| resolution (Å) | 49.0–2.51 |
| reflections (working/test set) | 193081/10717 |
| Rwork/Rfree (%) | 17.8/25.0 |
| no. of atoms | |
| protein | 43321 |
| heteroatoms (Fe, S, Ni, Cu) | 124 |
| waters | 1153 |
| xenon | 42 |
| avg B-factor, all atoms (Å2) | 31.3 |
| protein | 31.5 |
| heteroatoms (Fe, S, Ni, Cu) | 33.8 |
| waters | 23.0 |
| xenon | 28.4 |
| rmsb deviations | |
| bond lengths (Å) | 0.026 |
| bond angles (deg) | 2.28 |
Highest resolution shell is shown in parentheses.
rms, root mean square.
Identifying Xe Binding Sites and Refinement
Following initial rounds of refinement, unaccounted for electron density in the 2|Fo| − |Fc| maps was present in the hydrophobic cavities. |FXe| − |Fnative| difference Fourier maps (Figure 1a) and |Fo| − |Fc| difference Fourier maps showed consistent high σ electron density peaks. Into these electron density peaks, Xe atoms were modeled in XtalView (21). It is important to note that no water molecules are present in these hydrophobic locations in native, high-resolution structures. Electron density only appears in the hydrophobic channels of this enzyme structure when crystals are pressurized with Xe. The model was adjusted, and water molecules were added back to the model. At 2.51 Å resolution, B-factors and occupancies cannot be considered separately; however, Xe sites may not all be fully occupied. Therefore, the B-factors for Xe atoms were set to the average value for the structure (∼30 Å2), and occupancies were refined in CNS (19). When the occupancy of a Xe atom refined to a value greater than 1, then the occupancy for that site was set at 1 and the B-factor was allowed to refine. CNS values for occupancy and the B-factor were then transferred to REFMAC (20) for a final round of refinement.
Figure 1.
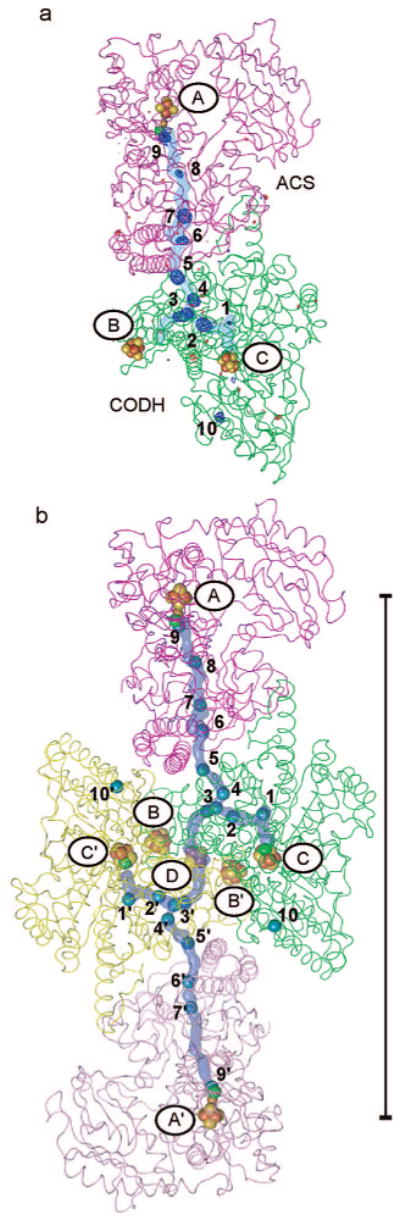
Location of xenon sites within the structure of the CODH/ACS complex. CODH β-subunits are shown in green and yellow, and ACS α-subunits are in pink and magenta. The predicted channel pathway is highlighted in light blue. (a) |FXe| – |Fnative| map contoured to +4σ (blue) and −4σ (red). Positive (blue) density indicates the locations of Xe atoms. (b) Xe sites are shown as blue spheres. The length of the scale bar from the A-cluster to the opposite A-cluster is ∼138 Å.
All figures were created with PyMOL (44).
Results
Prediction versus Experimental Data for Two 70 Å Long Channels
Biochemical data are consistent with the presence of a channel that connects the C-cluster (the site of CO generation) with the A-cluster (the site of CO consumption) (6, 7). The location of this channel has been predicted in this CODH/ACS crystal form by the cavity calculation program CAVENV (9). Here we describe the use of Xe gas to further explore the nature of this predicted cavity. Xe is known to bind to hydrophobic sites on proteins with high enough occupancy to serve as a heavy atom derivative (22). It has also been applied with good success to identify hydrophobic substrate channels (23), and with a van der Waals radius of 2.16 Å, Xe is a reasonable size mimic for CO, which has an effective van der Waals radius on the order of ∼2 Å. Using a SSRL Xe cryocell, we have solved a 2.5 Å resolution structure of Xe-pressurized CODH/ACS and identified 19 Xe sites per α2β2 CODH/ACS complex in locations that are an excellent match with the previously reported cavity calculations (Figure 1b). A total of 38 Xe sites have been identified and refined between the two α2β2 CODH/ACS complexes present in the asymmetric unit. Each α2β2 CODH/ACS complex shows a similar set of sites (Table S1 in Supporting Information). There are four sites distant from the CO channel (sites 10 and 10′ in Figure 1b) at equivalent positions in each of the four CODH subunits. There are eight equivalent sites (1–7 and 9), or total of 32, in each of four CO tunnels, with two sets of these sites (Figure 2b) close enough (2.75 Å) that they are unlikely to be simultaneously occupied. In addition, two minor sites exist that are only present in two of four CO channels (site 8 in Figures 1b and 2f). The Xe-lined channel connects each C-cluster to an A-cluster and spans a distance of ∼70 Å. Xe1 is closest to the Ni of the C-cluster at one end of the 70 Å long channel, and Xe9 is closest to the binuclear metal site of the A-cluster at the other end (Figure 1b). Although the cavity calculations suggest a channel between the two C-clusters of the CODH subunits, no Xe atoms are found there. Other than sites 10 and 10′, all Xe atoms are present in the cavities connecting C- and A-clusters, the route that CO would take under turnover conditions. In addition, no Xe atoms are located in any putative entranceways that substrate CO2 might take from the outside of the protein into the C-cluster. Presumably, the route into and out of the C-cluster is dynamic, allowing Xe to enter but also allowing it to leave. A dynamic channel would not have stable binding sites for gaseous substrates, thus preventing its detection by the Xe method. Since the Xe sites are similar between the two α2β2 CODH/ACS complexes in the asymmetric unit, in the following sections, only one α2β2 CODH/ACS will be discussed.
Figure 2.
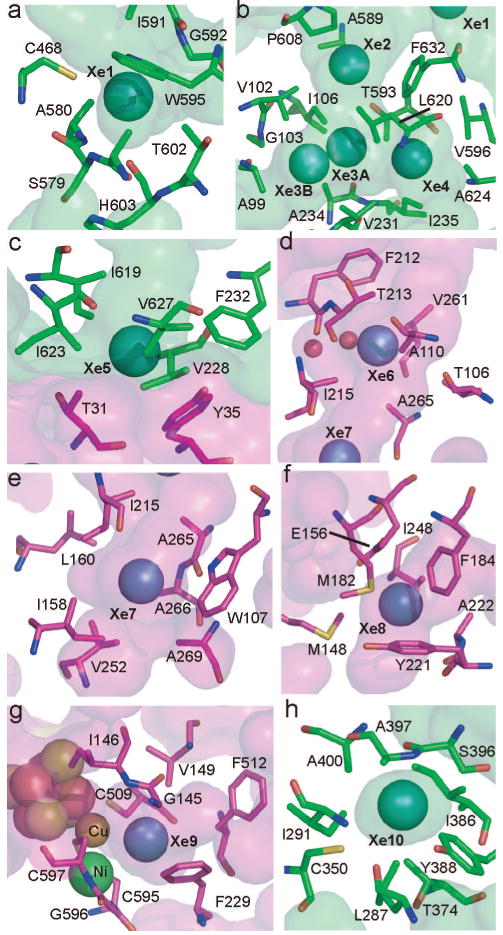
Detailed views of Xe binding sites. The CODH β-subunit is shown in green and the ACS α-subunit in magenta. Residues within 5 Å of xenons are shown as sticks. Interior protein surfaces are lightly shaded. Panels: (a) Xe site 1; (b) Xe sites 2–4; (c) Xe site 5; (d) Xe site 6; (e) Xe site 7; (f) Xe site 8; (g) Xe site 9; (h) Xe site 10.
Nature of the CO Channel
Xe atoms bind in chiefly hydrophobic sites (Figure 2, Table S1 in Supporting Information). Within 5 Å of the Xe atoms are 70 contacting amino acids, of which 50 (71%) are nonpolar, 18 (26%) are polar, and only 2 (3%) are charged. Although there are polar and charged amino acids near the Xe atoms, the polar or charged ends of the side chains usually point away from the channel, while the hydrophobic parts of the side chains are utilized in creating a channel lining that is mostly hydrophobic. The exceptions to this rule are residues C468 (site 1), T593 (site 2), and C350 (site 10), which have polar groups oriented toward the channel (Figure 2). Additionally, there are two water molecules located in the channel near site 6 that were not observed in the native high-resolution structure. The issue of sequence conservation of channel residues is more complex. Analysis of conservation is complicated by the fact that no other CODH/ACS proteins have been experimentally shown to channel CO. Also, a BLAST (24) search reveals only nine pairs of CODH/ACS sequences that contain the channel-forming domain of the α-subunit and are close enough in the genome to be colocalized in a gene cluster. With this limited number of sequences, the channel-lining residues do not exhibit any more significant conservation than the overall level of conservation in the protein. However, the hydrophobic character of residues lining the channel is somewhat conserved (81% conserved for ACS tunnel residues, 67% conserved for CODH residues). The lack of a clear pattern of conservation is not particularly surprising, since a channel might be quite tolerant of mutations that conserve the fold and general hydrophobic character. More detailed analysis of the sequence-based requirements for tunnel formation will require experimental characterization and ideally a crystal structure of another bifunctional CODH/ACS.
Binding of Xe near the A-Cluster
The A-cluster (site of acetyl-CoA synthesis) is a complex metallocofactor, containing a Fe4S4 center bridged through a cysteine residue to a binuclear metal site. One of the positions of the binuclear site, called the proximal metal because it is closest to the Fe4S4 cluster, is subject to substitution and can contain Cu, Ni, and Zn, resulting in binuclear site compositions of Cu–Ni, Ni–Ni, and Zn–Ni (1, 3, 11, 25). The Cu–Ni is the dominant form of the as-isolated enzyme from native M. thermoacetica, and the Ni–Ni form appears to be the catalytically relevant form (11, 25-27) (reviewed in refs 1–3 and 5). Since substrate CO has been proposed to bind to the proximal metal (8, 11), one potential role of the channel is to direct CO to that site. Here we find a high occupancy Xe site (site 9 in Figures 1 and 2) ∼3.9 Å from the proximal metal and ∼3.7 Å from the distal metal. While the Xe packs against the binuclear metal site on one side, protein residues F229, F512, G145, V149, C509, C595, G596, and C597 surround the Xe and contribute to what is a largely hydrophobic binding site (Figure 2g). It is likely that this Xe site represents the entry point to the A-cluster for CO, just prior to ligation of CO to the metal center. The almost identical distance for the Xe to both metals of the binuclear site suggests that the channel alone is not responsible for metal binding selectivity, at least between these two possible sites. The channel may protect against CO binding to the Fe4S4 cluster or to the undersides on the binuclear cluster metal ions, preventing interactions that would not be catalytically relevant. The geometry of the A-cluster is modified by the Xe treatment. Since no oxygen-related damage to the oxygen-sensitive D- and C-clusters is observed (Figure S1 in Supporting Information), the effect must be due to the presence of Xe rather than accidental exposure to oxygen during Xe treatment. A metal–ligand of unknown identity reported previously (8) is displaced by a Xe atom, changing slightly the geometry of the central metal to become more planar and altering the metal–metal distance from ∼2.7 to ∼3.0 Å. These rearrangements demonstrate the plasticity of this metallocenter, a plasticity that is likely to be essential for catalysis.
Protein Architecture That Creates a Channel
Both β-CODH and α-ACS subunits are composed of multiple domains. The CODH subunit has an α-helical domain at the N-terminus (CODH domain 1, residues 1-257), an α/β Rossmann-like domain in the middle (CODH domain 2, residues 262–458), and an α/β Rossmann-like domain at the C-terminus (CODH domain 3, residues 463–674) (Figure 3a). The helical domain contributes ligands to the D-cluster and B-cluster, which are typical Fe4S4 clusters. The two Rossmann-like domains coordinate the C-cluster with loops following the third and fifth β-strands of the middle domain and the first three β-strands of the C-terminal domain. The ACS subunit is composed of three domains (Figure 3a). The first domain consists of a helical region (residues 1–154) at the N-terminus of a Rossmann-like (six-stranded α/β) fold (155–316). Domain 1 resembles the helical region and the C-terminal Rossmann domain of the CODH β-subunit and could have resulted from gene duplication (158 of 316 residues align with an rmsd of 1.83 Å). The second and third domains of the ACS α-subunit are α + β folds, neither of which align well with known structural motifs in the DALI structural database. In both CODH and ACS subunits, the hydrophobic channel passes both through the middle of domains and in between domains. In particular, the pathway starts at the Ni site of the C-cluster in the CODH subunit and then proceeds through the C-terminal Rossmann domain, between the bottom face of its six-stranded β-sheet and its α-helices. After passing through the center of the Rossmann domain, the channel runs alongside this domain, still protected from solvent by the helical domain of the β-subunit. The middle Rossmann domain of the CODH subunit makes no contacts with the channel. The CODH β-subunit part of the pathway contains four Xe sites (sites 1–4), and a single ordered Xe is present at the interface between CODH and ACS subunits (site 5).
Figure 3.
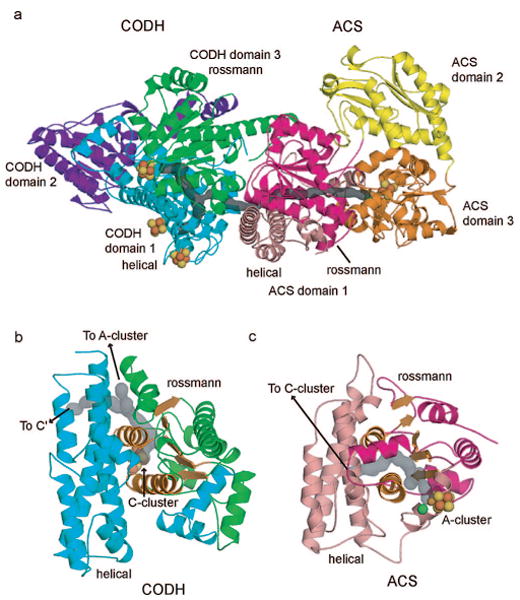
CODH/ACS domain architecture. Metal clusters are shown as spheres; the calculated channel is highlighted in gray. (a) Domain structure of one αβ CODH/ACS complex. CODH domain 1 (helical) is shown in cyan, CODH domain 2 in purple, and CODH domain 3 (Rossmann-like) in green. The ACS domain 1 helical region is shown in pink, the domain 1 Rossmann-like region is in magenta, ACS domain 2 is colored yellow, and domain 3 is colored orange. (b) CODH helical domain 1 (cyan) and Rossmann domain 3 (green) shown in same orientation as ACS domain 1 helical/Rossmann region in (c). The Rossmann β-sheet and two α-helices are shown in orange to highlight the similarities to ACS domain 1. (c) ACS domain 1 helical region shown in pink and Rossmann region shown in magenta. The Rossmann β-sheet and two α-helices are highlighted in orange.
Xe sites 6–9 are found in the channel that runs through the ACS subunit. As the channel enters the ACS subunit, it continues essentially parallel to the β-strands of the Rossmann-like domain from the carboxy side to the N-terminal side (Figure 3a). The channel is sandwiched between the bottom face of this β-sheet and the helical domain, with α-helices from the Rossmann domain on either side. The channel exits the Rossmann domain at the N-terminal side and continues to the A-cluster in the third domain of the ACS subunit. Residues from the third domain and the helical subdomain form the end of the channel; the second domain of the ACS subunit makes no contacts with the channel. While the helical/Rossmann regions of the ACS and CODH subunits are closely related structurally and in both cases are used to form the majority of the channel, the pathway taken by the channel through these very similar folds is completely different (Figure 3b,c).
Channels in Monofunctional versus Bifunctional CODHs
In the bifunctional CODH/ACS, CO produced at the C-cluster must travel to the A-cluster where it is incorporated into acetyl-CoA. In a monofunctional CODH (28, 29) or the monomeric hybrid cluster protein (HCP) from Desulfovibrio vulgaris (30), only one type of active site exists. Gas substrates and products must enter and exit the active site of the monomer, but the location of the exit should not matter. The function of HCP has not been clearly defined, but there is evidence that it may be involved in oxidative stress (31) or nitrogen metabolism (32–35). Structurally, HCP is highly homologous to CODH, with a similar helical domain followed by two Rossmann-like domains (overall 275 α carbons align with an rmsd of 1.8 Å). The Rossmann domains coordinate metalloclusters with nine of twelve cluster ligands conserved, suggesting an evolutionary relationship between HCP and CODH. With this high structural homology between the CODH domain of the bifunctional enzyme, the monofunctional enzyme, and HCP, the protein fold must not be the factor that dictates the directionality of the gaseous product away from the active site metallocluster. Hydrophobic cavities have been identified through cavity calculations in monofunctional CODH from Carboxydothermus hydrogenoformans (28) and R. rubrum (29), and channels have been found through Xe derivatization of HCP (PDB 1E9V) (30, 36). By comparing cavity calculations and Xe experiments, we can identify how the bifunctional enzyme is designed to channel CO from the C-cluster exclusively to the A-cluster.
Not unexpectedly, HCP and monofunctional CODHs show additional cavities compared to the bifunctional enzyme, although the main cavity is duplicated (Figure 4). Three Xe sites in HCP (1E9V) are found in a Rossmann-like domain on the same face of the β-sheet as described for mtCODH/ACS (Figure 4). The other ten Xe sites in HCP identify three additional channels. If these hydrophobic cavities were present in CODH/ACS, CO would not be channeled to the A-cluster as effectively. Structural comparisons show negative design at work: the secondary structure allows for the cavities, but large aromatic residues in mtCODH/ACS block their openings. Blocking a potential channel to the left in Figure 4 is W95 and blocking the one to the right are F632 and F634. A third large channel opens to the right in the HCP structure, above the plane of the CODH channel. Blocking this potential channel are residues W594, W595, and F576 of the mtCODH subunit. Residues F576, W595, and F634 of mtCODH would also block the extra channels that are present in the two available structures (28, 29) of monofunctional CODH enzymes. This analysis does not provide clues as to the entryway for CODH substrate CO2, as several potential cavities appear to be effectively blocked. What is most intriguing is that the protein fold does not dictate channel number or direction; the latter is designed through changes in amino acid side chains. It is interesting that the design principles that nature uses here are similar to ones that a researcher might have tried, blocking potential channel exits with large aromatic residues.
Figure 4.
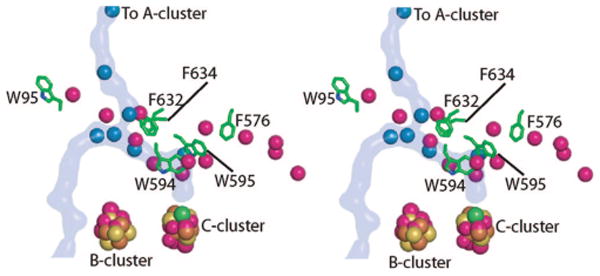
Stereoview comparing Xe sites in M. thermoacetica CODH and HCP. Xenons in the CODH structure are shown as blue spheres, and the CODH/ACS channel is highlighted in blue. The HCP xenons are colored magenta. Aromatic residues in CODH that block channels observed in HCP are shown as green sticks. Metal clusters from HCP (atoms of cluster shown as magenta spheres) align with those from mtCODH (colored by atom with iron in brown, sulfur in yellow, and nickel in green).
Discussion
One hypothesis concerning the origins of life is that nickel–iron–sulfur slurries were able to utilize a largely carbon dioxide environment to create an energy source in the form of acetyl thioesters by reactions similar to those of the modern Wood–Ljungdahl pathway (37, 38), which has been proposed to be the most primitive biochemical pathway of CO2 reduction (39). These concepts are based in part on the activity of CODH/ACS enzymes, which use nickel–iron–sulfur clusters to produce acetyl-CoA from carbon dioxide and coenzyme A. This theory underscores the catalytic role of the metallocenters in CODH/ACS chemistry. Here, we consider the role of the protein. Our studies, taken together with previous work discussed above, suggest that the protein is in fact very important in acetyl-CoA synthesis. Perhaps one reason for the large size of the protein (310 kDa) is that a scaffold is required to encapsulate an interior channel that is hydrophobic and long enough to connect two active sites, allowing CO generated at the C-cluster in the CODH subunit to be used as a substrate for the A-cluster in the ACS subunit.
Here, we describe the use of Xe gas to confirm that a 138 Å long channel identified through cavity searching software can indeed house gas molecules. With its similar size to CO, Xe serves as a good substrate mimic. Ten sites have been described that can stably bind Xe atoms, and nine of these lie within the predicted channel. The ten sites are chiefly hydrophobic, as is the overall channel itself. While some polar and two charged residues are present along the channel, in most cases the polar or charged part of the side chain points away from the channel, and the hydrophobic parts are involved in channel formation. There are a few exceptions, including two waters near Xe site 6 that are absent in the native structure. In this case, it seems that Xe binding causes a rearrangement that creates two water binding sites (Figure 2d). There are no water molecules present in the native structure that are displaced by Xe, presumably because Xe does not bind in hydrophilic sites that water molecules favor. CO would also prefer a more hydrophobic environment than water, and it would be unfavorable from a functional perspective for CO to have to compete with water molecules for channel passage.
As discussed in the Results section above, the analysis of channel residue conservation is complicated by the limited biochemical data on CODH/ACS enzymes from other species; however, it seems unlikely from studies done thus far that residue identity will be particularly important. That is, there is more than one way to line a hydrophobic channel. We see that the use of hydrophobic residues is not even necessary as long as most of the polar parts of residues are pointing away from the channel. The cavities that allow for channel formation are created by the proper juxtaposition of common protein motifs. The CODH/ACS channel runs between helical and Rossmann-like domains as well as through two Rossmann domains. Both helical and Rossmann domains are ubiquitous in nature, showing, as observed before (13), that formation of a channel does not require an unusual protein structure. Interestingly, the helical region and Rossmann region of ACS domain 1, used to form the ACS part of the channel, may have resulted from gene duplication from the CODH helical domain and its C-terminal Rossmann domain, which forms the CODH part of the channel (158 of 316 residues align with an rmsd of 1.83 Å). In analyzing how Xe traveled through the helical and Rossmann motifs of both CODH and ACS subunits, we expected to find channel duplication related to the gene duplication. However, despite the high structural homology, the pathway taken by Xe is completely different in each subunit. Thus, while nature chose the same architecture for both halves of the channel, this architecture was not used in the same manner. As there is more than one way to line a channel, there is apparently more than one way to create a channel using the same ordinary protein structural units.
It is also worth considering how nature uses the same protein structure for monofunctional and bifunctional CODHs, which need to enlist different routes for CO into or out of the C-cluster. In the latter case, the bifunctional enzyme should direct CO to the A-cluster where it is assembled into acetyl-CoA, and in the former, no second active site exists, making substrate/product routes less important. By comparing our CODH/ACS Xe data with cavity calculations of monofunctional CODHs and Xe data from the monofunctional CODH cousin HCP, we can describe the differences in channel compositions between these monofunctional and bifunctional proteins. The solution to this “design” problem appears simple. Monofunctional CODH/HCP's have additional cavities that are absent in the CODH subunit of a CODH/ACS; however, the difference in cavity composition is not due to an elaborate difference in protein structure. Simple placements of aromatic residues block the pathway for Xe, and presumably CO, in the bifunctional enzyme, serving to direct it to the A-cluster. It is important to note that the positions of Xe atoms in this mtCODH/ACS structure do not indicate how CO2, the substrate for the C-cluster, accesses its CODH active site. Xe does access the C-cluster since it is bound within 10 Å of this cluster, but it is unclear how it gets there. We believe that the entrance routes to the C-cluster from the outside of the protein are dynamic and not designed for the stable binding of gaseous molecules and are thus not “visible” through the use of Xe. An alternative hypothesis that has been suggested by Volbeda and Fontecilla-Camps (5) is that Xe or CO2 could enter the enzyme via the A-cluster and travel through the internal channel to the C-cluster (Figure 1b). This hypothesis seems unlikely given that mutants of channel residues between the A- and C-clusters do not appear to affect CODH activity (10), and traveling of CO2 against the flow of CO would require an elaborate gating mechanism (5). Another proposal is that CO2 enters the C-cluster through the channels connecting the C-clusters or through a hydrophilic channel near the CODH dimer interface (40). Entrance through the channel connecting the C-clusters would again require that CO2 travel against the expected path of product CO, even though this distance would be shorter (Figure 1b). The entry of CO2 must not allow escape of CO from the tunnel, so separate “entrance” and “exit” pathways would make sense. At this stage, however, all hypotheses for CO2 entrance should be considered since definitive experimental evidence is difficult to obtain.
Returning to one of the more fundamental questions: why is it necessary for CODH/ACS to have its C- and A-clusters connected via a channel that stretches almost the full length of the 310 kDa protein molecule? Channels often exist to allow reactive intermediates to travel from one active site to another without encountering solvent that would lead to their decomposition; however, CO is very stable in aqueous solution. Instead, a channel might protect the cellular environment from the toxic effects of CO. However, a number of organisms can grow on CO, including M. thermoacetica (41). For example, C. hydrogenoformans, which uses CO as a carbon and energy source, has a doubling time of 2 h in the presence of CO (42). Since these organisms can thrive on growth media containing CO gas, a role for a channel in their CODH/ACS to protect the cellular environment would be unnecessary. We propose that a major role for the channel in CODH/ACS is to ensure that each molecule of CO generated from CO2 is properly directed into cellular metabolism. Being approximately −200 mV more reducing than NAD(P)H, CO is fairly expensive to make from CO2, as it requires low-potential electrons originally derived from substrate (H2, glucose, etc.) oxidation. Thus, the uncoupling of CO production from acetyl-CoA synthesis due to a leaky channel could be life threatening. A similar situation exists for methanogens, since growth on acetate is even more energetically marginal than growth on H2/CO2. Given that the methanogenic ACDS lacks one of the domains that is part of the M. thermoacetica CODH/ACS channel, it will be very interesting to determine if these aceticlastic methanogens have also evolved a CO channel.
Another potential function of a channel is to direct molecules to a particular site on an enzyme. Since many metals are capable of binding CO, a channel in CODH/ACS could direct CO to a metal coordination site on the A-cluster where it would be catalytically competent rather than potentially inhibitory. We find a Xe positioned within van der Waals distance of the proximal and distal metals in the binuclear site of the A-cluster. It is interesting that the channel does not appear to guide CO toward one site of the cluster exclusively; however, it would prevent CO binding to the back sides of both metals and to the Fe4S4 cluster.
An additional role of enzyme channels is to synchronize activities at two active sites. Our Xe data show gas molecules lined up along the full extent of the C- to A-cluster channel, invoking an image of a factory assembly line and suggesting one mechanism by which the activities of the C- and A-clusters become synchronized when substrates for both reactions are available (14). The rates of CO2 reduction by the C-cluster depend on the context of the CODH protein. Monofunctional CODH catalyzes CO2 reduction at a rate of ∼11 s−1 (43), while CODH in complex with ACS catalyzes CO2 reduction at a rate of ∼0.6 s−1 in the absence of all ACS substrates and with a rate of ∼3.3–5.8 s−1 in the presence of ACS substrates (14). Thus, complexation with ACS slows the CO2 reaction rate; however, the rate of CO2 reduction is heavily modulated by the activity of the A-cluster, even at saturating CO2 concentrations (7). In the bifunctional enzyme, if the channel is “full”, catalysis at the C-cluster should temporarily halt until one CO has been removed from the channel by action of the A-cluster. Then, resumption of CODH activity would fill the empty CO binding site and allow continuation of acetyl-CoA synthesis. If the A-cluster is not functioning properly, due for example to lack of substrates, the wait could be even longer. For the monofunctional CODH, CO would simply diffuse into solution, and “buildup” should never occur. It is unknown whether any CO “buildup” would even occur on the bifunctional enzyme under normal physiological conditions, but it has been suggested that the channel “crowding” is responsible for CODH/ACS inhibition observed under high CO concentrations (10).
One danger of keeping a “full” CO channel or in making more CO than is needed at the moment is that CO could escape as the ACS α-subunit opens to react with the methylated CFeSP for methyl transfer, thereby wasting energy. The formidable challenge of reacting with three substrates of very different dimensions requires that the enzyme perform a molecular juggling act in which the ACS α-subunit must open and close to allow the largest substrate to access the A-cluster. The fact that the A-cluster is coordinated directly by only one domain of ACS (the C-terminal domain) allows for a domain rearrangement to occur without damaging the A-cluster (8). Our cavity calculations shown in Figure 5 compare and contrast the “channel open” and “channel closed” states of the ACS subunit. As discussed previously (11), helix 7 of domain 1 moves to seal the channel upon subunit opening. Thus, the channel in CODH/ACS comes complete with a plug.
Figure 5.
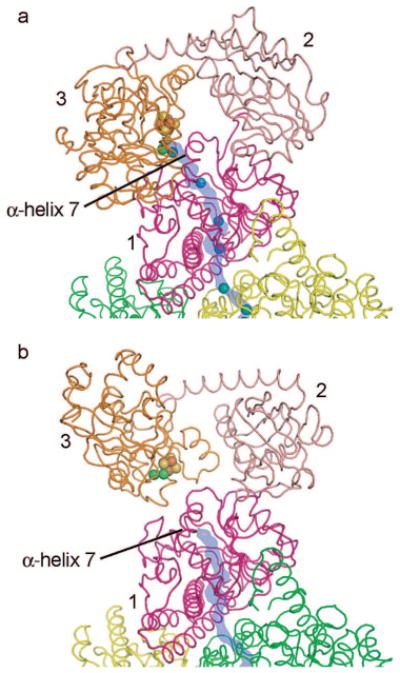
Comparison of “open” and “closed” α-subunit structures. ACS α-subunit domains are labeled and colored magenta (domain 1), pink (domain 2), and orange (domain 3); CODH β-subunits are colored yellow and green. The A-cluster is shown as spheres, Xe sites are shown as blue spheres, and the predicted channel pathway is highlighted in blue. (a) Xe-bound structure with closed α-subunit and open channel. (b) Structure with open β-subunit and closed channel (PDB 1OAO).
While “channel open” and “channel closed” states are two necessary forms of the CODH/ACS enzyme, there are at least two others (Scheme 2). After methylation and carbonylation, an acetyl intermediate is formed, coordinated to a metal of the A-cluster. At this stage, opening of the ACS subunit could be disastrous since the access of water to the A-cluster would result in formation of acetate rather than the desired acetyl-CoA. How then is the ACS subunit prevented from opening? We have proposed (8) that the intermediate itself might restrict the motion of the domains of the ACS subunit by jutting across the interface between domains 1 and 3. It is interesting to think about when and how often the ACS opens. Is it opening and closing all the time under physiological conditions or does it only open in the presence of methylated CFeSP? When methylated CFeSP interacts with ACS, CFeSP may help to slide ACS domain 1 from 3, creating access to the A-cluster for methyl transfer. When the A-cluster is unliganded or when the small substrate CO is bound, this sliding motion should be unrestricted and easily prompted by the presence of CFeSP. In contrast, if a branched acetyl intermediate is present, extending across the domain–domain interface of ACS, sliding may be restricted such that subunit opening would not occur until turnover is complete. Thus, ACS itself may have a turnover-dependent locking device. To date, no one has captured a structure of CODH/ACS with CoA (Scheme 2, “CoA-bound; α-mostly closed”), although proposals have been made as to where it binds, including the suggestion that it binds to the cavity between the three domains of ACS (8). Thus, in CODH/ACS, we have a protein with a channel for a small substrate, a channel plug, a flexible ACS subunit that can open to interact with a large substrate, and an interdomain cavity to putatively bind a medium-sized substrate.
While Ni–S–Fe slurries could have been involved in the origins of life by producing acetate from CO2, current day CODH/ACS chemistry requires a protein scaffold. The enzymatic machine that is CODH/ACS is fueled by electrons from glucose oxidation, transforms CO2 into CO, and funnels CO down an assembly line as ACS opens and closes to catalyze a condensation of CO with a methyl moiety and finally “pump” out acetyl-CoA.
Supplementary Material
Acknowledgments
We thank Matthew H. Sazinsky at Pomona College and Aina Cohen at the Stanford Synchrotron Radiation Laboratory (SSRL) for the collection of the Xe data. SSRL, a national user facility, is operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences.
Footnotes
This work is supported by NIH Grants GM69857 (C.L.D.) and GM39451 (S.W.R.).
The atomic coordinates and structure factors (accession number 2Z8Y) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics (http://www.pdb.org).
Abbreviations: ACS, acetyl-coenzyme A synthase; ACDS, acetyl-coenzyme A decarbonylase/synthase; CoA, coenzyme A; CFeSP, corrinoid iron–sulfur protein; CODH, carbon monoxide dehydrogenase; HCP, hybrid cluster protein; NAD(P)H, nicotinamide adenine dinucleotide (phosphate), reduced form; NCS, noncrystallographic symmetry.
Supporting Information Available: Figure S1 showing electron density for metalloclusters C and D and Table S1 describing the B-factors, occupancies, and neighboring residues of the Xe sites. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Ragsdale SW. Life with carbon monoxide. Crit Rev Biochem Mol Biol. 2004;39:165–195. doi: 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]
- 2.Hegg EL. Unraveling the structure and mechanism of acetyl-coenzyme A synthase. Acc Chem Res. 2004;37:775–783. doi: 10.1021/ar040002e. [DOI] [PubMed] [Google Scholar]
- 3.Drennan CL, Doukov TI, Ragsdale SW. The metalloclusters of carbon monoxide dehydrogenase/acetyl-CoA synthase: a story in pictures. J Biol Inorg Chem. 2004;9:511–515. doi: 10.1007/s00775-004-0563-y. [DOI] [PubMed] [Google Scholar]
- 4.Lindahl PA. Acetyl-coenzyme A synthase: the case for a Ni. (p)(0)-based mechanism of catalysis. J Biol Inorg Chem. 2004;9:516–524. doi: 10.1007/s00775-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 5.Volbeda A, Fontecilla-Camps JC. Crystallographic evidence for a CO/CO(2) tunnel gating mechanism in the bifunctional carbon monoxide dehydrogenase/acetyl coenzyme A synthase from Moorella thermoacetica. J Biol Inorg Chem. 2004;9:525–532. doi: 10.1007/s00775-004-0565-9. [DOI] [PubMed] [Google Scholar]
- 6.Seravalli J, Ragsdale SW. Channeling of carbon monoxide during anaerobic carbon dioxide fixation. Biochemistry. 2000;39:1274–1277. doi: 10.1021/bi991812e. [DOI] [PubMed] [Google Scholar]
- 7.Maynard EL, Lindahl PA. Evidence of a molecular tunnel connecting the active sites for CO2 reduction and acetyl-CoA synthesis in acetyl-CoA synthase from Clostridium thermoaceticum. J Am Chem Soc. 1999;121:9221–9222. [Google Scholar]
- 8.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 9.Collaborative Computational Project Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 10.Tan X, Loke HK, Fitch S, Lindahl PA. The tunnel of acetyl-coenzyme a synthase/carbon monoxide dehydrogenase regulates delivery of CO to the active site. J Am Chem Soc. 2005;127:5833–5839. doi: 10.1021/ja043701v. [DOI] [PubMed] [Google Scholar]
- 11.Darnault C, Volbeda A, Kim EJ, Legrand P, Vernede X, Lindahl PA, Fontecilla-Camps JC. Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat Struct Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 12.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 13.Raushel FM, Thoden JB, Holden HM. Enzymes with molecular tunnels. Acc Chem Res. 2003;36:539–548. doi: 10.1021/ar020047k. [DOI] [PubMed] [Google Scholar]
- 14.Maynard EL, Lindahl PA. Catalytic coupling of the active sites in acetyl-CoA synthase, a bifunctional CO-channeling enzyme. Biochemistry. 2001;40:13262–13267. doi: 10.1021/bi015604+. [DOI] [PubMed] [Google Scholar]
- 15.Andreesen JR, Ljungdahl LG. Formate dehydrogenase of Clostridium thermoaceticum: incorporation of selenium-75, and the effects of selenite, molybdate, and tungstate on the enzyme. J Bacteriol. 1973;116:867–873. doi: 10.1128/jb.116.2.867-873.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ragsdale SW, Ljungdahl LG, DerVartanian DV. Isolation of carbon monoxide dehydrogenase from Acetobacterium woodii and comparison of its properties with those of the Clostridium thermoaceticum enzyme. J Bacteriol. 1983;155:1224–1237. doi: 10.1128/jb.155.3.1224-1237.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soltis SM, Stowell MHB, Wiener MC, Phillips GN, Rees DC. Successful flash-cooling of xenon-derivatized myoglobin crystals. J Appl Crystallogr. 1997;30:190–194. [Google Scholar]
- 18.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 19.Brünger AT, Adams PD, Clore GM, Delano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges N, Pannu NS, Read RJ, Rice LM, Simonson T, W GL. Crystallography and NMR system (CNS): A new software system for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.McRee DE. XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 22.Schiltz M, Fourme R, Prangé T. Use of noble gases xenon and krypton as heavy atoms in protein structure determination. Methods Enzymol. 2003;374:83–119. doi: 10.1016/S0076-6879(03)74004-X. [DOI] [PubMed] [Google Scholar]
- 23.Prangé T, Schiltz M, Pernot L, Colloc'h N, Longhi S, Bourguet W, Fourme R. Exploring hydrophobic sites in proteins with xenon or krypton. Proteins. 1998;30:61–73. [PubMed] [Google Scholar]
- 24.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Seravalli J, Xiao Y, Gu W, Cramer SP, Antholine WE, Krymov V, Gerfen GJ, Ragsdale SW. Evidence that NiNi acetyl-CoA synthase is active and that the CuNi enzyme is not. Biochemistry. 2004;43:3944–3955. doi: 10.1021/bi036194n. [DOI] [PubMed] [Google Scholar]
- 26.Svetlitchnyi V, Dobbek H, Meyer-Klaucke W, Meins T, Thiele B, Romer P, Huber R, Meyer O. A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc Natl Acad Sci U S A. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu W, Gencic S, Cramer SP, Grahame DA. The A-cluster in subunit beta of the acetyl-CoA decarbonylase/synthase complex from Methanosarcina thermophila: Ni and Fe K-edge XANES and EXAFS analyses. J Am Chem Soc. 2003;125:15343–15351. doi: 10.1021/ja036602a. [DOI] [PubMed] [Google Scholar]
- 28.Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science. 2001;293:1281–1285. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 29.Drennan CL, Heo J, Sintchak MD, Schreiter E, Ludden PW. Life on carbon monoxide: X-ray structure of Rhodospirillum rubrum Ni-Fe-S carbon monoxide dehydrogenase. Proc Natl Acad Sci U S A. 2001;98:11973–11978. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper SJ, Garner CD, Hagen WR, Lindley PF, Bailey S. Hybrid-cluster protein (HCP) from Desulfovibrio vulgaris (Hildenborough) at 1.6 Å resolution. Biochemistry. 2000;39:15044–15054. doi: 10.1021/bi001483m. [DOI] [PubMed] [Google Scholar]
- 31.Almeida CC, Romao CV, Lindley PF, Teixeira M, Saraiva LM. The role of the hybrid cluster protein in oxidative stress defense. J Biol Chem. 2006;281:32445–32450. doi: 10.1074/jbc.M605888200. [DOI] [PubMed] [Google Scholar]
- 32.Cabello P, Pino C, Olmo-Mira MF, Castillo F, Roldan MD, Moreno-Vivian C. Hydroxylamine assimilation by Rhodobacter capsulatus E1F1. Requirement of the hcp gene (hybrid cluster protein) located in the nitrate assimilation nas gene region for hydroxylamine reduction. J Biol Chem. 2004;279:45485–45494. doi: 10.1074/jbc.M404417200. [DOI] [PubMed] [Google Scholar]
- 33.Wolfe MT, Heo J, Garavelli JS, Ludden PW. Hydroxylamine reductase activity of the hybrid cluster protein from Escherichia coli. J Bacteriol. 2002;184:5898–5902. doi: 10.1128/JB.184.21.5898-5902.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He Q, Huang KH, He Z, Alm EJ, Fields MW, Hazen TC, Arkin AP, Wall JD, Zhou J. Energetic consequences of nitrite stress in Desulfovibrio vulgaris Hildenborough, inferred from global transcriptional analysis. Appl Environ Microbiol. 2006;72:4370–4381. doi: 10.1128/AEM.02609-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flatley J, Barrett J, Pullan ST, Hughes MN, Green J, Poole RK. Transcriptional responses of Escherichia coli to S-nitrosoglutathione under defined chemostat conditions reveal major changes in methionine biosynthesis. J Biol Chem. 2005;280:10065–10072. doi: 10.1074/jbc.M410393200. [DOI] [PubMed] [Google Scholar]
- 36.Macedo S, Mitchell EP, Romao CV, Cooper SJ, Coelho R, Liu MY, Xavier AV, LeGall J, Bailey S, Garner DC, Hagen WR, Teixeira M, Carrondo MA, Lindley P. Hybrid cluster proteins (HCPs) from Desulfovibrio desulfuricans ATCC 27774 and Desulfovibrio vulgaris (Hildenborough): X-ray structures at 1.25 Å resolution using synchrotron radiation. J Biol Inorg Chem. 2002;7:514–525. doi: 10.1007/s00775-001-0326-y. [DOI] [PubMed] [Google Scholar]
- 37.Huber C, Wachtershauser G. Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science. 1997;276:245–247. doi: 10.1126/science.276.5310.245. [DOI] [PubMed] [Google Scholar]
- 38.Russell MJ, Martin W. The rocky roots of the acetyl-CoA pathway. Trends Biochem Sci. 2004;29:358–363. doi: 10.1016/j.tibs.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 39.Fuchs G, Stupperich E. Evolution of autotrophic CO2 fixation. In: Schleifer KH, Stackebrandt E, editors. FEMS Symposium No. 29: Evolution of prokaryotes; London, U.K.: Academic Press; 1985. pp. 235–251. [Google Scholar]
- 40.Tan X, Volbeda A, Fontecilla-Camps JC, Lindahl PA. Function of the tunnel in acetylcoenzyme A synthase/carbon monoxide dehydrogenase. J Biol Inorg Chem. 2006;11:371–378. doi: 10.1007/s00775-006-0086-9. [DOI] [PubMed] [Google Scholar]
- 41.Kerby R, Zeikus JG. Growth of Clostridium thermoaceticum on H2/CO2 or CO as energy source. Curr Microbiol. 1983;8:27–30. [Google Scholar]
- 42.Svetlichny VA, Sokolova TG, Gehardt M, Ringpfeil M, Kostrikina NA, Zavarzin GA. Carboxydothermus hydrogenoformans gen. nov., sp nov., a CO-utilizing thermophilic anaerobic bacterium from hydrothermal environments of Kunashir Island. Syst Appl Micobiol. 1991;14:254–260. [Google Scholar]
- 43.Kumar M, Lu WP, Ragsdale SW. Binding of carbon disulfide to the site of acetyl-CoA synthesis by the nickel-iron-sulfur protein, carbon monoxide dehydrogenase, from Clostridium thermoaceticum. Biochemistry. 1994;33:9769–9777. doi: 10.1021/bi00198a048. [DOI] [PubMed] [Google Scholar]
- 44.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; Palo Alto, CA: 2002. http://www.pymol.org. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.