


Abstract
We used 2D protein gel electrophoresis and DNA microarray technologies to systematically analyze genes under glucose repression in Bacillus subtilis. In particular, we focused on genes expressed after the shift from glycolytic to gluconeogenic at the middle logarithmic phase of growth in a nutrient sporulation medium, which remained repressed by the addition of glucose. We also examined whether or not glucose repression of these genes was mediated by CcpA, the catabolite control protein of this bacterium. The wild-type and ccpA1 cells were grown with and without glucose, and their proteomes and transcriptomes were compared. 2D gel electrophoresis allowed us to identify 11 proteins, the synthesis of which was under glucose repression. Of these proteins, the synthesis of four (IolA, I, S and PckA) was under CcpA-independent control. Microarray analysis enabled us to detect 66 glucose-repressive genes, 22 of which (glmS, acoA, C, yisS, speD, gapB, pckA, yvdR, yxeF, iolA, B, C, D, E, F, G, H, I, J, R, S and yxbF ) were at least partially under CcpA-independent control. Furthermore, we found that CcpA and IolR, a repressor of the iol divergon, were involved in the glucose repression of the synthesis of inositol dehydrogenase encoded by iolG included in the above list. The CcpA-independent glucose repression of the iol genes appeared to be explained by inducer exclusion.
INTRODUCTION
Glucose repression, the phenomenon of catabolite repression, is a regulatory mechanism for global gene expression by which Bacillus cells, as well as those of other bacteria, coordinate the metabolism of carbon and energy sources for maximum efficiency and regulate other metabolic pathways as well (1–3). The well-characterized mechanisms underlying glucose repression are those of catabolite repression and inducer exclusion. Recently, the mechanism underlying catabolite repression in Bacillus subtilis was extensively investigated. These studies revealed that Bacillus, as well as other low-GC Gram-positive bacteria, possess a negative regulatory mechanism for catabolite repression, which is very different from the positive regulatory mechanism of enteric bacteria, involving cAMP and its receptor protein (4,5). In the low-GC Gram-positive bacteria, the negative regulation of transcription of catabolite-repressive genes occurs through the binding of the catabolite control protein (CcpA) (6), which interacts with allosteric effectors such as P-Ser-HPr (7), to cis-acting catabolite-responsive elements (cres) (8,9). On the other hand, inducer exclusion is independent of catabolite repression mediated by CcpA, which is explained by a specific mechanism to each operon in which its transcriptional repressor is involved.
For physiological and genetic studies on B.subtilis, the cells are often cultivated in various nutrient sporulation media. When grown in such media, the growth of B.subtilis changes from glycolytic to gluconeogenic at the middle logarithmic phase (1). After this shift, many genes that are initially repressed by rapidly metabolizable glycolytic carbon sources, such as glycerol, become induced when these compounds are used up. This induction is due to the presence in the nutrient broth of inducers of catabolic genes or operons, such as the iolG gene encoding inositol dehydrogenase, which is glucose-repressive but can be induced by myo-inositol present in the nutrient broth (1,10–12). Thus, for our studies on the glucose-repressive genes in B.subtilis, we focused on genes that are induced after consumption of glycolytic carbon sources, since the expression of these genes will remain repressed by the addition of glucose.
In 1997, the genome sequence of B.subtilis was completely determined (13). The available nucleotide sequence of all of the genes made it possible to analyze in a systematic way the proteome and transcriptome of B.subtilis cells cultivated under certain physiological conditions. The method most often used for B.subtilis proteome analysis is 2D protein gel electrophoresis (14) in conjunction with N-terminal sequencing of the proteins in the spots on the gels. The proteome analyses provided valuable information about global regulation of the central pathways of carbon catabolism governed by CcpA (15), and important information about extracellular proteins and components of the major secretion pathway, such as SecA and Ffh (16). Furthermore, the DNA chip technology, involving high density arrays of ORF-specific DNA fragments, has been rapidly developed for transcriptome analysis in various microorganisms whose genome sequences have been determined (for instance, Saccharomyces cerevisiae; 17). Very recently, a transcriptional profile of early to middle sporulation of B.subtilis under the control of Spo0A and σE was reported (18), which was obtained from macroarrays on nylon membranes. In addition, global gene expression profiles of this bacterium under anaerobic conditions were obtained from microarrays on microscope glass slides (19). These global gene expression profiles obtained from the large-scale experiments will prompt further investigation of the genes, the expression patterns of which were newly unveiled.
The major aim of the work described here was to detect and study glucose-repressive genes of B.subtilis that are expressed after the shift from glycolytic to gluconeogenic during growth in the nutrient medium. As an approach for this analysis we used a combination of the powerful 2D gel electrophoresis and DNA microarray technologies. In particular, we examined whether or not the glucose repression of the detected genes was mediated by the CcpA protein of this bacterium. This work is heuristic, which may be used to identify the B.subtilis genes under glucose repression, and provides valuable information for further investigation of the transcriptional regulation of several genes from among those that were found to be under glucose repression.
MATERIALS AND METHODS
Bacteria and their construction
Bacillus subtilis strains 168 trpC2 (20), 1A250 (trpC2 alsR1 ilvBΔ1), 1A147 [alsA1 (=ccpA1) trpC2 alsR1 ilvBΔ1] (21), FU706 (iolR::neo trpC2 alsR1 ilvBΔ1) and FU707 (ccpA1 iolR::neo trpC2 alsR1 ilvBΔ1) were used in this work. Strains FU706 and FU707 were constructed as follows. Plasmid pIOLR1 carrying the iolR gene (11) was cleaved at an EcoRV site in iolR and then ligated to the neo cassette, resulting in plasmid pIOLR1::neo. Plasmid pIOLR1::neo was linearized with EcoRI and then mixed with competent cells of strain 1A250 or 1A147. Neomycin-resistant transformants, resulting from a double crossover recombination, were selected.
Cell growth conditions and isolation of total protein
Cells of strains 1A250 and 1A147 were inoculated into 50 ml of Difco sporulation medium (DSM) (20) with and without 10 mM glucose in 200 ml Erlenmeyer flasks and incubated in a water-bath reciprocal shaker until growth to an A600 of 1.0 at 37°C had occurred. Cells in 15 ml portions of the cultures were pelleted and resuspended in 1 ml of 10% sucrose and 50 mM Tris–Cl, pH 8. After pelleting the cells again, 20 µl of the lysis solution containing 10 mg/ml lysozyme, 10% sucrose, 50 mM Tris–Cl, pH 8, and 1 mM phenylmethylsulfonyl fluoride (PMSF) was added to the pellets, followed by thorough mixing and incubation of the cell suspensions at 37°C for 10 min. To completely lyse the cells treated with lysozyme, 90 µl of the lysis buffer [9 M urea, 0.31 M 2-mercaptoethanol, 2.2% (v/v) Triton X-100, 90 µM PMSF and 0.9% (v/v) Pharmalyte 3-10; Amersham Pharmacia Biotech, Buckinghamshire, UK] was added, followed by thorough mixing. After centrifugation, the supernatants were kept at –80°C until used for 2D protein gel electrophoresis.
Analytical 2D protein gel electrophoresis
The first dimension on 2D gel electrophoresis consisted of isoelectrical focusing on an Immobiline DryStrip pH4-7L (11 cm) (Amersham Pharmacia Biotech). For this aim, a solution of 80 µg of total protein was mixed with an equal volume of the sample buffer [1% (v/v) Pharmalyte 3-10, 8 M urea, 0.288 M 2-mercaptoethanol, 0.5% (v/v) Triton X-100 and 7 µM bromophenol blue] and applied to this strip, which had been treated with the rehydration buffer [8 M urea, 0.5% (v/v) Triton X-100, 1.5% dithiothreitol (DTT), 0.5% (v/v) Pharmalyte 3-10 and 0.005% Orange G]. The gels were focused for 4 h at 300 V followed by 16 h at 3500 V and 15°C, using a Multiphor II apparatus (Amersham Pharmacia Biotech). The strips were placed in equilibration buffer A [50 mM Tris–Cl, pH 6.8, 6 M urea, 30% (v/v) glycerol, 1% sodium dodecyl sulfate (SDS) and 0.25% DTT] for 15 min, and next in buffer B [50 mM Tris–Cl, pH 6.8, 6 M urea, 30% (v/v) glycerol, 1% SDS, 4.5% iodoacetic acid and 3.5 µM bromophenol blue] for 15 min. Subsequently, the isoelectric focusing gels were placed onto SDS–polyacrylamide gel sheets (ExcelGel XL SDS 12-14; Amersham Pharmacia Biotech) and the proteins were then resolved in the second dimension with a constant current of 20 mA at 15°C until the bromophenol blue marker entered the gel sheets, followed by a second period of 40 mA for 2 h. The gels were fixed in a 45% ethanol/10% acetic acid solution and then silver-stained as described previously (22).
Preparative 2D protein gel electrophoresis and N-terminal amino acid sequencing
For preparative 2D gel electrophoresis, 1.2 mg of total protein were separated by 2D gel electrophoresis as described above, except that the total protein preparation was scaled up and that an Immobiline DryStrip pH4-7L (18 cm) was used for the first dimension. The proteins in the spots on the gels were transferred to PVDF membranes (Bio-Rad Laboratories, Hercules, CA) by the method of Matsudaira (23), and then stained with 0.1% Coomassie Brilliant Blue R250, 30% (v/v) methanol and 10% (v/v) acetic acid. The proteins were excised from the membranes and sequenced with an Applied Biosystems Protein sequencer (Model 492 Procise; Perkin-Elmer/Applied Biosystems, Foster City, CA).
Preparation of DNA microarrays
PCR primer pairs for the amplification of each of the 4100 ORFs in the B.subtilis 168 trpC2 genome (13) were obtained from Eurogentec (Seraing, Belgium). The primers were designed to amplify each ORF starting from the 5′ portion near a start codon and ending at the 3′ portion near a stop codon. Each primer contained a 9-base non-variable ‘tag’ sequence at its 5′ end, which can be used for normalization of the quantities of PCR products spotted onto microscope glass slides, followed by 16–26 bases of gene-specific sequences, the length of which is dependent on GC content. PCR amplification of each gene was performed with recombinant Taq DNA polymerase (Takara Shuzo, Kyoto, Japan) using B.subtilis 168 trpC2 DNA as the template. All the PCR products were analyzed by electrophoresis on agarose gels to determine their size and purity. Approximately 96% of the genes in B.subtilis were successfully amplified.
DNA microarrays were prepared as described previously (17). The PCR products (0.2 µg/µl for 3898 genes and 0.1–0.2 µg/µl for 43 genes), together with those of the hTFR gene (positive control), the human β-actin gene and calf thymus DNA (negative controls), were spotted onto polylysine-coated glass slides with the robotic printing device of a GMS 417 Arrayer (Affymetrix/Genetic MicroSystems, Woburn, MA) (spot diameter, 150 µm).
RNA isolation for DNA microarray analysis
Total RNA was isolated from cells essentially as described previously (24). Cells of strains 1A250 and 1A147 were inoculated into 250 ml of DSM with and without 10 mM glucose in 1 l Erlenmeyer flasks, and then grown to an A600 of 1.0 at 37°C. Cells of 240 ml portions of the cultures were pelleted, and the pellets were washed with 10 mM Tris–Cl (pH 7.5). The cells were suspended in 4 ml LETS buffer (100 mM LiCl, 10 mM EDTA, 10 mM Tris–Cl pH 7.5, and 1% SDS) and 1 ml aliquots of each suspension were distributed into four 15 ml Corning tubes with 1.0 ml of a phenol/chloroform/iso-amyl alcohol (25:24:1) mixture saturated with TE buffer (50 mM Tris–Cl pH 8, and 1 mM EDTA) and 500 µl of glass beads (0.5 mm diameter; Biospec Products, Bartlesville, OK). The mixtures were vortexed at maximal intensity for 4 min. The aqueous layers were collected after brief centrifugation and then reextracted with an equal volume of the phenol/chloroform/iso-amyl alcohol mixture. Total RNA was pelleted by stepwise mixing with a one-tenth volume of 1 M LiCl and 2.5 vol of ethanol, and subsequent centrifugation. The RNA was dissolved in distilled water and then recovered by ethanol precipitation (70%) in the presence of 0.3 M sodium acetate. The resulting total RNA preparations were treated with RNase-free DNase I (Roche Diagnostics GmbH/Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer’s protocol. The amounts of RNA were then quantified by A260 measurement, and the preparations stored at –80°C. Using this method, ~3 mg of total RNA were obtained from the above cultures (250 ml).
Synthesis of fluorescently-labeled cDNA synthesis
Specific primed cDNA synthesis of total RNA was performed to prepare fluorescently-labeled probes for hybridization to microarrays. RNA (50 µg) and a mixture of 4100 primers complementary to mRNAs (0.5 pmol each), which had been used for the preparation of microarrays, were mixed together with human transferrin receptor (hTFR) mRNA and its complementary hTFR primer (0.5 pmol). hTFR mRNA synthesized in vitro was kindly supplied by Takara Shuzo and was used as a positive control for microarray analysis. Tris–acetate (pH 8.4) (50 mM final concentration), potassium acetate (75 mM), magnesium acetate (8 mM), DTT (10 mM), dATP, dCTP and dGTP (0.5 mM each), dTTP (0.2 mM), Cy3 (or Cy5)-dUTP (Amersham Pharmacia Biotech) (0.1 mM), RNaseOUT (Life Technology, Inc., Rockville, MD) (40 U) and Thermoscript, a heat-resistant reverse transcriptase (Life Technology, Inc.) (30 U), were added to this mixture to a final volume of 40 µl. Reverse transcription for fluorescent-labeling of cDNA was carried out at 60°C for 1 h, and then continued for another hour following the addition of 30 U of Thermoscript. To stop reverse transcription and to degrade total RNA, 5 µl each of 0.5 M Na-EDTA (pH 8) and 2 N NaOH were added to the reaction mixture, which was then incubated for 30 min at 65°C. After neutralization by the addition of 25 µl of 1 M Tris–Cl (pH 7.5), the reaction mixture was applied to a Centri-Sep spin column (Perkin-Elmer/Applied Biosystems) according to the supplier’s instructions. The fluorescently-labeled cDNA was pelleted by ethanol precipitation (70%) in the presence of 0.3 M sodium acetate and subsequent centrifugation, washed with ethanol, air-dried and then dissolved in 8 µl of distilled water. The fluorescently-labeled cDNA was immediately used for hybridization to microarrays.
Hybridization and data analysis
Prior to hybridization, microarrays were pre-hybridized for 2 h at 65°C with 4× standard saline citrate (SSC), 100 µg/ml sonicated salmon sperm DNA (Stratagene, La Jolla, CA), 50× Denhardt’s solution and 0.2% SDS, washed in 3× SSC and 0.1% SDS and then dried. The microarrays were hybridized in a final volume of 20 µl of a hybridization solution containing 5.5 µl each of Cy3- and Cy5-labeled cDNAs prepared as described above (3× SSC, 100 µg/ml sonicated salmon sperm DNA, 10× Denhardt’s solution and 0.1% SDS). Prior to hybridization, the hybridization solutions were heated at 98°C for 2 min and then put onto microarrays. A cover slip was applied and the arrays were placed in hybridization chambers (CMT; Corning Inc., Corning, NY) and hybridization was allowed to take place for 40 h at 70°C. After hybridization, the microarrays were washed in 0.5× SSC and 0.01% SDS at 45°C for 5 min subsequently washed in 0.06× SSC at 25°C for 5 min, and finally dried. The intensities of fluorescence of the microarrays were determined with a confocal laser scanner (GMS 418 Array Scanner, Affymetrix/Genetic MicroSystems). The signal intensity for each spot was determined with ImaGene software (version 3; BioDiscovery, Inc., Los Angeles, CA). The mean fluorescence intensity for each spot was calculated, the background being taken as the average of the intensities of eight spots of calf thymus DNA and four spots of the PCR product of the human β-actin gene as negative controls.
RESULTS
Gene expression and glucose repression during cell growth in DSM
When B.subtilis cells are grown in nutrient sporulation media containing 0.8% nutrient broth (Difco Laboratories, Detroit, MI) such as DSM (20) and nutrient sporulation medium supplemented with K-phosphate buffer (NSMP) (25), the synthesis of various catabolic enzymes is induced after the shift from glycolytic and gluconeogenic at the mid-logarithmic growth phase (A600 = 0.8) (1,10,12).
In order to examine the glucose repression of the synthesis of various proteins induced during cell growth in DSM, cells of strain 1A250 (wild-type) were cultivated in DSM with and without 10 mM glucose, and then harvested at A600 = 1.0 after glucose repression had been relieved only in cells grown without glucose. Lysates prepared from the cells grown with and without glucose were subjected to 2D gel electrophoresis for analysis of their proteomes (Fig. 1A). Comparison of the spot patterns of the cells grown with and without glucose indicated that the synthesis of nearly 80 proteins was glucose repressive.
Figure 1.
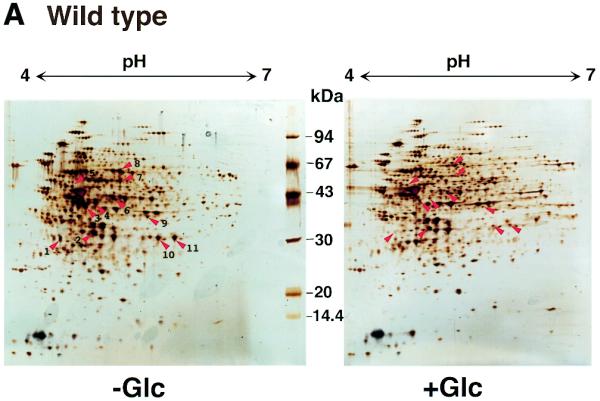
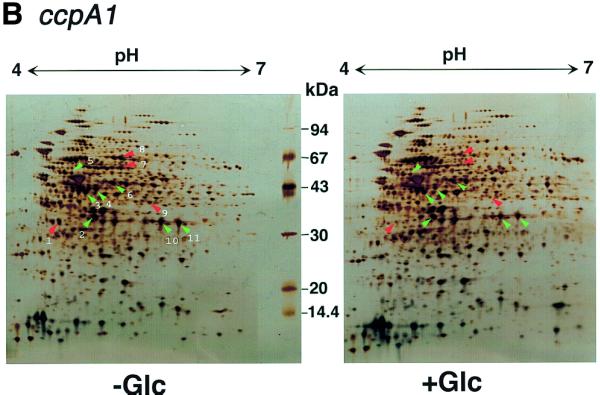
Glucose-repressive gene products on 2D gels induced in B.subtilis cells during growth in DSM. Total proteins from strain 1A250 (wild-type) (A) and strain 1A147 (ccpA1) (B) grown in DSM to an A600 of 1.0 with and without 10 mM glucose (Glc) were subjected to 2D gel electrophoresis as described in the text. Identification of the proteins in 11 glucose-sensitive spots, which are indicated by red arrowheads and numbers, is shown in Table 1. The 2D gel patterns (B) for strain 1A147 cells indicated that out of the 11 spots, four were still glucose-sensitive (indicated by red arrowheads), whereas the others (green arrowheads) had become glucose-insensitive due to the ccpA1 mutation.
To study whether the observed repression was mediated by the CcpA protein, that is, under catabolite repression, strain 1A147 (ccpA1) was also grown in DSM with and without glucose to A600 = 1.0, and then cell lysates were likewise subjected to 2D gel electrophoresis. The majority of the glucose-sensitive spots observed with strain 1A250 (Fig. 1A) turned out to be glucose-insensitive in the ccpA1 strain (Fig. 1B). However, several protein spots in the patterns remained glucose-sensitive even in the ccpA1 background, which indicated that the glucose repression was under CcpA-independent control.
Eleven proteins, whose synthesis had been found to be under glucose repression, were extracted from the corresponding spots on the preparative 2D gel for strain 1A250 (grown without glucose) and sequenced. Of these, the synthesis of four proteins was under CcpA-independent glucose repression. Table 1 shows the glucose-repressive gene products deduced from the sequencing of these proteins, their description and the CcpA dependency of their glucose repression. As shown in Table 1, the glucose repression of the synthesis of RocA, D, F, SucC, D, Pdp and FolD was CcpA-dependent, whereas that of IolA, I, S and PckA was CcpA-independent. Further identification of glucose-repressive gene products was found to be difficult, because other candidate spots, containing sufficient amounts of protein (30 pmol/spot) for the determination of their amino acid sequences, could not be separated pure enough from surrounding spots on the preparative 2D gels.
Table 1. Identification of the B.subtilis glucose-repressive gene products on 2D gelsa.
| Spot no. |
Sequence |
Protein |
Protein descriptionb |
CcpA dependencyc |
| 1 |
MKLXFNEATTLE |
IolI |
Inositol utilization (unspecified) |
– |
| 2 |
MDKTISVIGMPM |
RocF |
Arginase |
+ |
| 3 |
MNIHEYQGKEVL |
SucC |
Succinyl CoA synthetase β chain |
+ |
| 4 |
TALSKSKEIIDQ |
RocD |
Ornithine aminotransferase |
+ |
| 5 |
MRMVDIIIKKQN |
Pdp |
Pyrimidine nucleoside phosphorylase |
+ |
| 6 |
LVKEAGKPXKEA |
RocA |
Pyrroline-5-carboxylate dehydrogenase |
+ |
| 7 |
AEIRKLKNYING |
IolA |
Oxidative decarboxylase in inositol
catabolism |
– |
| 8 |
MNSVDLTADLXA |
PckA |
Phosphoenolpyruvate carboxykinase |
– |
| 9 |
MKKAKLGKSDLQ |
IolS |
Inositol catabolism (unspecified) |
– |
| 10 |
SVFINKDTRVIV |
SucD |
Succinyl CoA synthetase α chain |
+ |
| 11 | TATIIDGKEXAR | FolD | Methylenetetrahydrofolate dehydrogenase | + |
aTotal protein from strain 1A250 (wild-type) cells grown to A600 = 1.0 in DSM without glucose was subjected to preparative 2D gel electrophoresis, as described in the text. The proteins in the spots corresponding to those shown in the left panel of Figure 1A were extracted and sequenced. N-terminal sequencing of the proteins in the spots allowed us to identify the B.subtilis genes encoding these products by means of the SubtiList worldwide web server (http://www.pasteur.fr/Bio/SubtiList) with the FASTA program (48).
bThe protein descriptions were mainly obtained from the SubtiList World Wide Web server.
cThe CcpA dependency of the glucose repression of the synthesis of each protein was obtained from the results (Fig. 1B).
Microarray analysis of glucose repression
In order to more systematically detect glucose-repressive genes, which were derepressed in cells grown in DSM without glucose, we prepared B.subtilis microarrays containing nearly 96% of the B.subtilis genes. Several probe preparation methods were examined, such as direct fluorescent labeling of total RNA (26) and cDNA-labeling by reverse transcription using random hexomers priming (27), but these methods gave unsatisfactory results. Finally, we used cDNA labeling, which involves a heat-resistant reverse transcriptase, Thermoscript (Life Technology, Inc.), and a mixture of 4100 primers complementary to mRNAs. This method appeared to be the most efficient and gave the lowest background in spot intensities without removal of the rRNA from the total RNA preparation.
As an indication of the reliability of the microarray analysis performed as described above, we compared the Cy3 and Cy5 spot intensities on a microarray hybridized with a mixture of Cy3- and Cy5-labeled cDNAs prepared from the same total RNA. The results showed a good correlation between the Cy3 and Cy5 spot intensities (Fig. 2A); the correlation coefficient (r) of spot intensities was 0.937. When microarray analysis with Cy3- and Cy5-labeled probes prepared from total RNAs of strain 1A250 (wild-type) cells, grown with and without glucose, was performed, the correlation coefficient was 0.538 (Fig. 2B), indicating that the transcriptome in the cells grown without glucose was highly affected by the presence of glucose in the medium. Further microarray analysis involving total RNAs of strain 1A147 (ccpA1) (Fig. 2C) revealed a correlation coefficient of 0.826, which is higher than that observed with strain 1A250 RNA and reflects the situation where a considerable number of glucose-repressive genes become glucose-insensitive in this strain.
Figure 2.
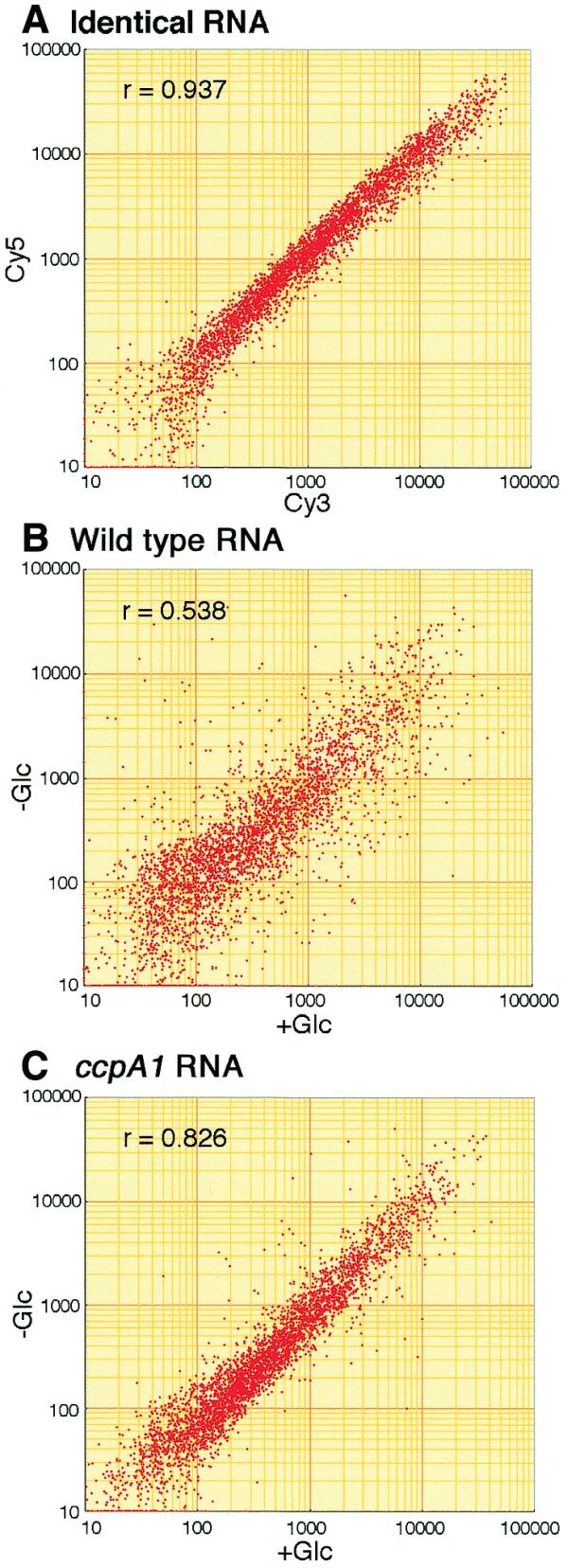
Logarithmic-scale plots of normalized spot intensities (arbitrary units). Spot intensities were normalized after subtracting their backgrounds, i.e. the average spot intensities of 12 mammal gene spots. After substituting the intensities of less than 10 with that of 10, which was less than the standard deviations of their backgrounds, the intensities were plotted logarithmically. The correlation coefficient is indicated by r. (A) RNA of strain 1A250 (wild-type) grown with glucose was used for both Cy3 and Cy5 cDNA-labeling. After hybridization of microarrays with the Cy3 and Cy5 probes, the respective spot intensities for 4100 genes, calculated as above, were plotted. The backgrounds for Cy3 and Cy5 spot intensities were 244.6 ± 42.7 and 278.2 ± 74.4, respectively. (B) RNA of strain 1A250 (wild-type) grown with and without glucose (Glc) was used for Cy3- and Cy5-labeling, respectively. The backgrounds for Cy3 and Cy5 spot intensities were 189.3 ± 30.4 and 249.5 ± 46.7, respectively. (C) RNA of strain 147 (ccpA1) grown with and without glucose was used for Cy3- and Cy5-labeling, respectively. The backgrounds for Cy3 and Cy5 spot intensities were 186.3 ± 21.5 and 206.7 ± 31.4, respectively.
Figure 3 shows the glucose-sensitive spots in red on microarray pseudocolor images obtained for strains 1A250 (wild-type) and 1A147 (ccpA1), where the spot intensities with and without glucose are depicted in green and red, respectively, and are overlaid. The number of red spots in the image for the wild-type cells (Fig. 3A) was much greater than in that for the ccpA1 strain (Fig. 3B). Examination of the red spots on the two overlaid images revealed that tens of red spots were common to the two images, indicating that the corresponding genes were under CcpA-independent glucose repression. Among the common red spots, those for the iol genes are indicated in the images (Fig. 3). These microarray results indicate that negative regulation of catabolite repression mediated by the CcpA protein plays a very important role in the overall control of an efficient carbon flow during cell growth.
Figure 3.
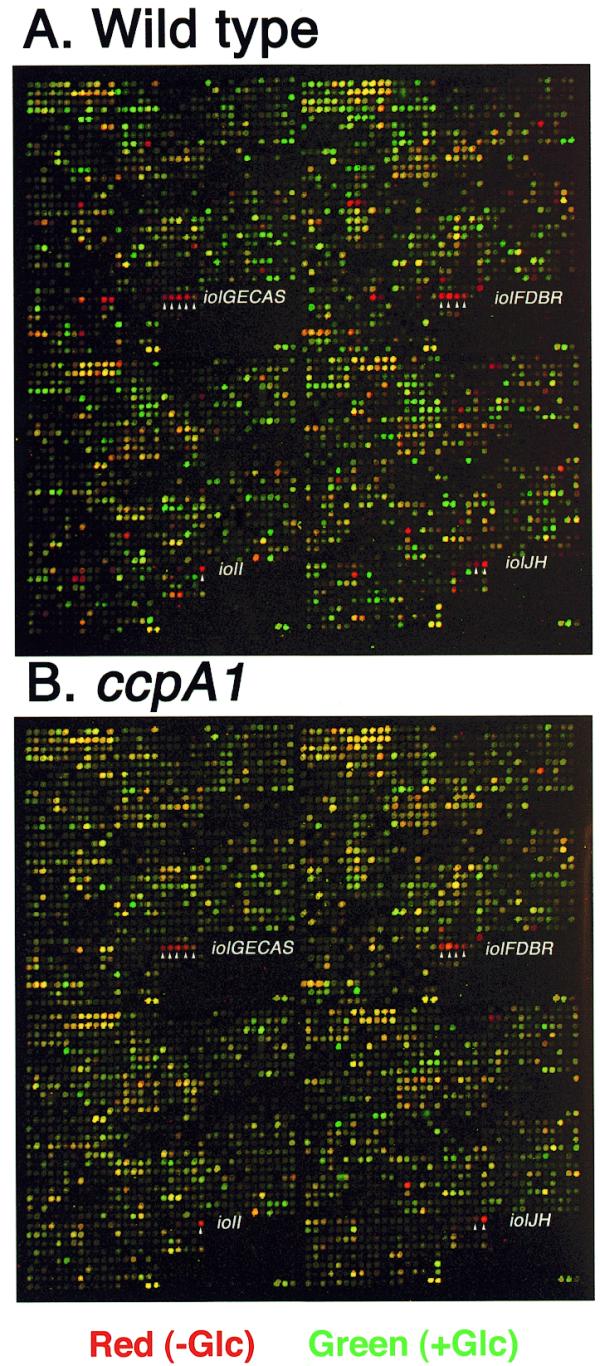
Pseudocolor imaging of glucose-repressive genes. Cy5[–glucose (Glc)] and Cy3 (+ glucose) spot intensities are denoted in red and green, respectively. The red and green images were overlaid, producing pseudocolors for all the spots. Red and green spots correspond to glucose-repressive and -inducible genes, respectively. The 12 iol genes are indicated by white arrowheads. (A) RNA of strain 1A250 (wild-type) grown with and without glucose was used for Cy3- and Cy5-labeling of their cDNAs, respectively (the same analysis as in Fig. 2B). (B) RNA of strain 1A147 (ccpA1) grown with and without glucose was used for Cy3- and Cy5-labeling of their cDNAs, respectively (the same analysis as in Fig. 2C).
Detection of glucose-repressive genes
Judging from the expression levels of lacZ gene fusions in plasmid pMUTIN-integrants during growth in DSM (20), roughly 25% of the B.subtilis genes were well expressed. This corresponds to approximately 1000 genes. Therefore, we detected the glucose-repressive genes from among these 1000 genes, which exhibited high spot intensities with RNA from strain 250 (wild-type) cells grown without glucose (Fig. 2B). Among these spots, none exhibited more than a 2.2-fold difference in ratios of the Cy5 to Cy3 spot intensity ratio (Fig. 2A). Glucose repression of enzyme synthesis is usually >5-fold (1,2,21), as such, we detected 66 genes exhibiting >5-fold repression among the 1000 genes (Table 2) through comparison of the spot intensities in the highest quality representative Cy3 and Cy5 microarray profiles for strain 1A250 cells (wild-type) grown with and without glucose (Figs 2B and 3A). These genes exhibited repression ratios comparable to those shown in Table 2 in two independent microarray analyses of glucose repression. Moreover, of the 11 proteins whose synthesis was found to be under glucose repression through the 2D gel electrophoresis analysis (Fig. 1B and Table 1), nine corresponding genes (iolA, I, S, rocA, D, F, sucC, D and pckA) are included in this set of 66 genes (Table 2).
Table 2. The glucose-repressive genes detected through microarray analysis.
| Genea |
Functiona |
Ratio (wt)b |
Ratio (ccpA1)b |
| glmS | l-glutamine-d-fructose-6-phosphate amidotransferase | 7.2 | 3.5 |
| ybcM | similar to d-glucosamine-d-fructose-6-phosphate aminotransferase | 5.4 | <2 |
| glpQ | glycerophosphoryl diester phosphodiesterase | 5.5 | <2 |
| ycnKc | similar to transcriptional regulator (DeoR family) | 6.4 | <2 |
| yerA | similar to adenine deaminase | >100 | <2 |
| yesL | similar to unknown proteins from B.subtilis | 14.0 | <2 |
| yesM | similar to two-component sensor histidine kinase | 8.2 | <2 |
| yfmQ | unknown function | 12.5 | <2 |
| acoAc,d | acetoin dehydrogenase E1 component (TPP-dependent α subunit) | >100 | 12.9 |
| acoCc | acetoin dehydrogenase E2 component (dihydrolipoamide acetyltransferase) | 58.0 | 5.3 |
| glpF | glycerol uptake facilitator | 5.3 | <2 |
| yisS | similar to myo-inositol 2-dehydrogenase | 20.6 | 4.7 |
| ykuL | unknown function | 6.7 | <2 |
| ctaC | cytochrome caa3 oxidase (subunit II) | 31.3 | <2 |
| ctaDd | cytochrome caa3 oxidase (subunit I) | 37.3 | <2 |
| ctaE | cytochrome caa3 oxidase (subunit I) | 22.6 | <2 |
| ctaG | function unknown | 7.8 | <2 |
| sucC | succinyl-CoA synthetase (β subunit) | 5.2 | <2 |
| sucD | succinyl-CoA synthetase (α subunit) | 5.1 | <2 |
| tdh | threonine 3-dehydrogenase | 10.6 | <2 |
| yocH | similar to cell wall-binding protein | 6.3 | <2 |
| qcrB | menaquinol:cytochrome c oxidoreductase (cytochrome b subunit) | 5.7 | <2 |
| qcrA | menaquinol:cytochrome c oxidoreductase (iron-sulfur subunit) | 16.0 | <2 |
| resE | two-component sensor histidine kinase involved in aerobic and anaerobic respiration | 6.7 | <2 |
| sacC | levanase | 14.4 | <2 |
| cstAd | carbon starvation-induced protein | 6.2 | <2 |
| araA | l-arabinose isomerase | 7.5 | <2 |
| speD (ytcF) | unknown function | 13.2 | 2.0 |
| gapB | glyceraldehyde-3-phosphate dehydrogenase | >100 | 25.2 |
| pckA | phosphoenolpyruvate carboxykinase | 26.8 | 3.7 |
| ytkA | unknown function | 6.1 | <2 |
| dhbC | isochorismate synthase | 5.5 | <2 |
| yvfL | similar to maltodextrin transport system permease | 5.6 | <2 |
| yvfK | similar to maltose/maltodextrin-binding protein | 8.3 | <2 |
| yvdR | similar to molecular chaperone | 26.4 | 7.1 |
| gerBC | germination response to the combination of glucose, fructose, l-asparagine and KCl | 23.0 | <2 |
| ywtD | similar to murein hydrolase | 5.8 | <2 |
| rbsRd | transcriptional repressor of the ribose operon | >100 | <2 |
| rbsK | ribokinase | >100 | <2 |
| rbsD | ribose ABC transporter (membrane protein) | >100 | <2 |
| rbsAd | ribose ABC transporter (ATP-binding protein) | >100 | <2 |
| rbsB | ribose ABC transporter (ribose-binding protein) | >100 | <2 |
| ywsB | similar to unknown proteins from B.subtilis | 32.2 | <2 |
| ywsA | unknown function | 23.2 | <2 |
| ywqM | similar to transcriptional regulator (LysR family) | 16.0 | <2 |
| rocA | pyrroline-5 carboxylate dehydrogenase | 5.6 | <2 |
| rocG (yweB) | glutamate dehydrogenase (major) | 48.6 | <2 |
| licA | PTS lichenan-specific enzyme IIA component | 5.2 | <2 |
| msmXc,d | similar to multiple sugar-binding transport ATP-binding protein | 9.2 | <2 |
| yxeF | unknown function | >100 | 22.3 |
| iolJc | aldolase in myo-inositol catabolism | 28.4 | 4.5 |
| iolIc | unknown function in myo-inositol catabolism | >100 | 9.2 |
| iolHc | unknown function in myo-inositol catabolism | >100 | 22.3 |
| iolGc | myo-inositol 2-dehydrogenase | >100 | 9.2 |
| iolFc | myo-inositol permease | >100 | 6.5 |
| iolEc | 2-inosose dehydratase | >100 | 8.3 |
| iolDc | 2,3-diketo-4-deoxy-epi-inositol hydrolase | >100 | 14.4 |
| iolCc | kinase in myo-inositol catabolism | >100 | 7.0 |
| iolBc,d | unknown function in myo-inositol catabolism | 91.5 | 7.5 |
| iolAc | oxidative decarboxylase in myo-inositol catabolism | 87.8 | 6.0 |
| iolR | transcriptional repressor of the inositol operon | 6.7 | 4.2 |
| iolS | unknown function in myo-inositol catabolism | 5.0 | 2.0 |
| yxbF | unknown function | 15.3 | 3.6 |
| rocF | arginase | 5.5 | <2 |
| rocE | amino acid permease | 7.5 | <2 |
| rocD | ornithine aminotransferase | 8.3 | <2 |
aThe gene names and functions are basically as in the literature (13) and SubtiList (http://genolist.pasteur.fr/SubtiList). The functions of some iol genes were deduced from unpublished results (K.Yoshida and Y.Fujita).
bThe repression ratios [wild-type (wt) and ccpA1] were calculated by dividing the spot intensities obtained for cells grown without glucose by those with glucose after subtraction of their backgrounds and subsequent normalization.
cGlucose repression of the expression of these genes were confirmed on first level analysis of pMUTIN integrants, the results of which are obtainable from JAFAN (http://bacillus.genome.ad.jp).
dPutative catabolite responsible elements (cres) are associated with these genes (9).
A literature survey revealed that 29 of the 66 genes selected here had been reported to be glucose (or catabolite)-repressive with regard to the genes themselves or the operons containing them as constituents and/or synthesis of their products [acoA, C (28), glpF (29), ctaC, D, E, G (30), sucC, D (1), sacC (31,32), araA (33,34), speD (35), gapB (36), pckA (37), rocG (38), iolA, B, C, D, E, F, G, H, I, J (11,39), rocD, E, F (40) and licA (41)]. In addition, glucose repression of the expression of ycnK, acoA, C, iolA, B, C, D, E, F, G, H, I, J and msmX was confirmed in the analysis of lacZ expression in the MUTIN integrants, the results of which are obtainable from JAFAN (http://bacillus.genome.ad.jp). Thus, 31 of the 66 potential glucose-repressive genes found through microarray analysis were somehow proven to be under glucose repression. Therefore, it is likely that among the other 35 genes a considerable number are also glucose-repressive under the growth conditions used.
CcpA dependency of glucose-repressive genes
Among the 66 glucose-repressive genes that were found through microarray analysis (Table 2), seven (acoA, ctaD, cstA, rbsA, R, msmX and iolB) were associated with a putative cre site, which might be the recognition target for CcpA (9). We therefore examined whether or not the glucose repression of these genes was CcpA dependent. This was done through comparison of the spot intensities in representative Cy3 and Cy5 microarray profiles with RNA of strain 1A147 (ccpA1) cells grown with and without glucose (Figs 2C and 3B). As shown in Table 2, 22 of the 66 genes were still glucose-repressed, even in the ccpA1 background, these were glmS, acoA, C, yisS, speD, gapB, pckA, yvdR, yxeF, iolA, B, C, D, E, F, G, H, I, J, R, S and yxbF. This CcpA independency was confirmed by two independent microarray analyses involving RNA of strain 1A147 grown with and without glucose. Furthermore, the results of 2D gel electrophoresis analysis (Fig. 1B and Table 1) also indicated that the synthesis of the products of four genes (iol, A, I, S and pckA) was under CcpA-independent glucose repression, which is consistent with the results of microarray analysis.
Investigation of the glucose repression of the synthesis of inositol dehydrogenase
Table 2 shows that the glucose repression of the genes included in the iolABCDEFGHIJ and iolRS oprons is partially CcpA independent. The two operons constitute the iol divergon involved in myo-inositol catabolism (11,42), and IolR is a transcriptional repressor of this divergon (42). Glucose repression of the synthesis of inositol dehydrogenase encoded by iolG was reported previously (11,39), and here we investigated the CcpA dependency of this repression.
Table 3 shows that the glucose repression ratios of inositol dehydrogenase synthesis were over 350 and 4.9 in the wild-type and ccpA1 backgrounds, respectively, which is well consistent with the data obtained on microarray analysis (Table 2). The remaining glucose repression in the ccpA1 background can presumably be explained by inducer exclusion because the glucose repression ratio in the wild-type background (over 350-fold) was reduced to 6.1-fold in the iolR::neo background, and almost no glucose repression was observed in the doubly mutated iolR::neo and ccpA1 background. The results suggested that the CcpA-independent repression of the iol genes might be exerted by inducer exclusion.
Table 3. Glucose repression of the synthesis of inositol dehydrogenase encoded by iolG.
| Strain |
Relevant genotype |
Glucose repression ratioa (fold, +Iol/+Iol
and Glc) |
| 1A250 |
Wild-type |
>350 |
| 1A147 |
ccpA1 |
4.9 |
| FU706 |
iolR::neo |
6.1 |
| FU707 | ccpA1 iolR::neo | 1.1 |
aCells of B.subtilis strains were grown to an A600 of 0.8 in S6 medium (25) containing 0.5% casamino acids with 10 mM inositol (Iol), or 10 mM each of inositol and glucose (Glc). The cells (4.5 A600 units) were harvested and then treated with lysozyme and sonicated briefly to prepare crude extracts (39). The inositol dehydrogenase activity in the crude extracts was measured as described previously (10). The repression ratios, folds, were obtained by dividing the enzyme activities in the crude extracts prepared from cells grown with glucose by those in the case of growth with inositol and glucose.
DISCUSSION
We have prepared DNA microarrays and developed the microarray techniques for analysis of the B.subtilis transcriptomes. As described above, the cDNA-labeling method used was found to be the most efficient and to give the lowest background with regard to microarray spot intensities. We also raised the hybridization temperature to 70°C, which caused significant reduction of cross-hybridization between paralogous genes (data not shown). Before this microarray analysis for glucose repression, we examined whether or not the results obtained by the microarray techniques used coincide with those of northern analysis (20) for 11 genes exhibiting different expression patterns during growth in DSM. The microarray results coincided well with those of northern analysis (data not shown), indicating that our microarray analysis is reliable. Very recently, Fawcett et al. (18) and Ye et al. (19) reported the macroarray and microarray analyses for transcription profiles of early to middle sporulation, and in aerobic and anaerobic growth conditions, respectively. There is a striking difference between the cDNA-labeling methods; these previous studies used random hexomers to prime the reverse transcription, whereas we used a mixture of specific primers. However, it is very difficult to judge at present which priming method is better for B.subtilis array analysis, because the cDNA-labeling conditions were quite different from each other.
To systematically study B.subtilis glucose-repressive genes, we combined the techniques of 2D protein gel electrophoresis and DNA microarrays to analyze the proteome and transcriptome in cells grown in DSM. We identified 11 glucose-repressive gene products through 2D gel electrophoresis (Table 1), whereas we detected 66 glucose-repressive genes through microarray analysis (Table 2). The CcpA dependency of the repression of these genes was also investigated using the ccpA mutant, indicating that 22 of the 66 genes were partially under CcpA-independent control. Among the 11 proteins whose synthesis was glucose-repressive, nine of the corresponding genes (the exceptions being pdp and folD) were included among the 66 genes found to be glucose-repressive through microarray analysis. The CcpA independency of glucose repression of four gene products identified through 2D gel electrophoresis analysis was also confirmed by microarray analysis. Thus, the patterns of glucose-repressive genes found by proteome and transcriptome analyses were largely similar, although a discrepancy was observed in the glucose repressibility of two genes (pdp and folD). At present, we cannot explain this observation.
We detected the 66 glucose-repressive genes by means of microarray analysis of the transcriptomes in cells grown in DSM with and without glucose (Table 2). These genes form only part of the total number of glucose-repressive genes of B.subtilis, mainly because many glucose-repressive genes are not expressed under the growth conditions used due to the lack of proper inducers, or the presence of corepressors in the medium during the chosen growth phase. For example, the gnt operon is not induced in DSM, which is probably due to the lack of its inducer, gluconate, in the medium (Y.Miwa and Y.Fujita, unpublished results). Furthermore, we were probably unable to detect all the glucose-repressive genes that are expressed under the growth conditions used. This was inferred from the observation that only acoA, C, araA, glpF and glpQ were detected from the genes included in the acoABCL (43), araABDLMNPQ-abfA (34), glpFK (44) and glpTQ (45) polycistronic operons. Moreover, ctaF, which is included in the ctaCDEF operon (30), was missing from the list (Table 2). The following reasons suggest why we are likely to have missed a considerable number of glucose-repressive genes expressed under the growth conditions. (i) We could not amplify 202 genes from the B.subtilis genome properly, so the microarrays would have contained insufficient amounts of their PCR products. (ii) We detected the glucose-repressive genes that exhibited a >5-fold difference in the repression ratio among the 1000 genes giving high spot intensities. Although in this detection we may have missed a substantial number of glucose-repressive genes, we attempted to detect only the genes that are very likely to be glucose-repressive. Furthermore, we considered that only glucose-repressive gene transcripts varied by such a huge factor, so it is likely that the results we provided will hold even though the medium was different from batch to batch of nutrient broth (Difco). (iii) We cannot exclude the possibility that cDNAs of certain genes were not efficiently fluorescently-labeled due to insufficient priming. Moreover, hybridization of labeled cDNAs with the microarray may not in all cases be specific to their PCR products due to cross hybridization to paralogous genes, so the actual glucose repression ratios could not be obtained for certain genes. Even considering these potential problems, our microarray analyses appeared to be reliable for transcriptome analysis, because we could detect the 66 genes that are very likely to be under glucose repression.
The genes listed in Table 2 include those involved in catabolism of various carbohydrates and amino acids such as glycerol (glpF, Q), acetoin (acoA, C), levan (sacC), arabinose (araA), ribose (rbsR, K, D, A, B), lichenan (licA), arginine (rocA, G, F, E, D), threonine (tdh) and inositol (iolA, B, C, D, E, F, G, H, I, J, R, S). yerA, yisS, yvfL, yvfK, ywtD and msmX are also likely to be involved in catabolism of carbohydrates and others according to their similarity search (Table 2). The sucC and sucD genes encode the α and β subunits of succinyl-CoA synthetase, respectively, involved in the citric acid cycle, whereas gapB (36) and pckA are involved in gluconeogenesis. In addition, ctaC, D, E, G, qcrA, B and resE are involved in aerobic growth or its regulation. Therefore, it is not difficult to explain the reasons why expression of these genes was found to be under glucose repression: because they are likely to be needed less as long as glucose is present in the medium. Actually, many of them were reported to be under glucose repression, as described above. However, it is rather difficult to explain the reasons why the other known and unknown genes were found to be under glucose repression.
Glucose repression is the phenomenon of catabolite repression. Glucose repression is mainly exerted by catabolite repression and inducer exclusion. In the low-GC Gram-positive bacteria, catabolite repression is mediated by the CcpA protein. Among the 66 glucose-repressive genes detected through the DNA microarray analysis, 22 were at least partially under CcpA-independent repression (Table 2), which appeared to be explained by CcpA-independent mechanisms such as inducer exclusion. Actually, CcpA and IolR, a repressor for the iol divergon, were involved in glucose repression of the synthesis of inositol dehydrogenase encoded by iolG (Table 3). Thus, both catabolite repression and inducer exclusion appeared to be involved in the glucose repression of the iol genes. Glucose repression of the other 12 genes was also partially CcpA independent (Table 2). However, it is difficult to judge whether or not CcpA is also involved in their glucose repression. The ccpA1 mutation largely affected the overall carbon flow, which was normally induced by the addition of a glycolytic carbon source, glucose (Figs 2 and 3), which might cause the reduction in the glucose repression ratio observed in the wild-type background.
We now point out the interesting features of the expression of several glucose-repressive genes found through microarray analysis. It was reported that glucose repression of sacC is CcpA-dependent (32), whereas that of ctaC, D, E, F (30) and gapB (36) is partially and only indirectly CcpA-dependent, respectively. According to our results of the microarray analysis (Table 2), the CcpA dependency of glucose repression of sacC and gapB was consistent with the published observations. But the glucose repression of ctaC, D, E and G was completely CcpA dependent. Moreover, the syntheses of arginase and ornithine aminotransferase, encoded by rocF and rocD, respectively, were reported to be under only limited glucose repression in arginine-induced cultures with glucose or citrate as the carbon source (46). Nevertheless, we found that not only the syntheses of these enzymes, but also the transcription of their genes was under CcpA-dependent glucose repression (Tables 2 and 3). Also, the derepression of arginase synthesis in cells grown in a nutrient medium was reported previously (40). The discrepancy in these results might be explained by the differences in the minimal and nutrient media; we used DSM containing nutrient broth. Furthermore, speD expression from its own promoter, which was very high in cells growing on glucose (35), might be repressed by an unknown repressor present in the DSM because we did not observe high expression of this gene induced by glucose (Table 2) in the medium. Another complex matter is the transcriptional regulation of the qcrA and B genes. Our results showed that qcrA and B were under CcpA-dependent glucose repression (Table 2), whereas they were previously reported to be under abrB-negative regulation (47). At present, we cannot explain the regulation of these genes properly. Our most interesting finding was that acoA, C, gapB and pckA, which are supposed to play important roles in gluconeogenesis, were under CcpA-independent glucose repression (Table 2). It will be interesting to investigate the mechanism underlying this glucose repression.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank M. Ueda, J. Mineno and K. Asada for their indispensable assistance in the preparation of the B.subtilis microarrays. We are also grateful to Y. Nakaura, H. Kushimoto, C. Mitsuboshi, Y. Okabe and S. Yamasako for their help in the experiments. This work was supported by a Grant-in-Aid for Scientific Research on Priority Areas (C) from the Ministry of Education, Science and Sports and Culture, and the Special Coordination Funds of the Science and Technology Agency of the Japanese Government.
References
- 1.Freese E. and Fujita,Y. (1976) Control of enzyme synthesis during growth and sporulation. In Schlessinger,D. (ed.), Microbiolgy-1976. American Society for Microbiology, Washington, DC, pp. 164–184.
- 2.Fisher S.H. and Sonenshein,A.L. (1991) Control of carbon and nitrogen metabolism in Bacillus subtilis. Annu. Rev. Microbiol., 45, 107–135. [DOI] [PubMed] [Google Scholar]
- 3.Chambliss G.H. (1993) Carbon source-mediated catabolite repression. In Sonenshein,A.L., Hoch,J.A. and Losick R. (eds), Bacillus subtilis and other Gram-positive Bacteria. American Society for Microbiology, Washington, DC, pp. 213–219.
- 4.Saier M.H.,Jr, Chauvaux,S., Cook,G.M., Deutscher,J., Paulsen,I.T., Reizer,J. and Ye,J. (1996) Catabolite repression and inducer control in Gram-positive bacteria. Microbiology, 142, 217–230. [DOI] [PubMed] [Google Scholar]
- 5.Stülke J. and Hillen,W. (1999) Carbon catabolite repression in bacteria. Curr. Opin. Microbiol., 2, 195–201. [DOI] [PubMed] [Google Scholar]
- 6.Henkin T.M., Grundy,F.J., Nicholson,W.L. and Chambliss,G.H. (1991) Catabolite repression of α-amylase gene expression in Bacillus subtilis involves a trans-acting gene product homologous to the Escherichia coli lacI and galR repressors. Mol. Microbiol., 5, 575–584. [DOI] [PubMed] [Google Scholar]
- 7.Fujita Y., Miwa,Y., Galinier,A. and Deutscher,J. (1995) Specific recognition of the Bacillus subtilis gnt cis-acting catabolite-responsive element by a protein complex formed between CcpA and seryl-phosphorylated HPr. Mol. Microbiol., 17, 953–960. [DOI] [PubMed] [Google Scholar]
- 8.Weickert M.J. and Chambliss,G.H. (1990) Site-directed mutagenesis of a catabolite repression operator sequence in Bacillus subtilis. Proc. Natl Acad. Sci. USA, 87, 6238–6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miwa Y., Nakata,A., Ogiwara,A., Yamamoto,M. and Fujita,Y. (2000) Evaluation and characterization of catabolite-responsive elements (cre) of Bacillus subtilis. Nucleic Acids Res., 28, 1206–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramaley R., Fujita,Y. and Freese,E. (1979) Purification and properties of Bacillus subtilis inositol dehydrogenase. J. Biol. Chem., 254, 7684–7690. [PubMed] [Google Scholar]
- 11.Yoshida K., Aoyama,D., Ishio,I., Shibayama,T. and Fujita,Y. (1997) Organization and transcription of the myo-inositol operon, iol, of Bacillus subtilis. J. Bacteriol., 179, 4591–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujita Y., Yoshida,K., Miwa,Y., Yanai,N., Nagakawa,E. and Kasahara,Y. (1998) Identification and expression of the Bacillus subtilis fructose-1,6-bisphosphatase gene (fbp). J. Bacteriol., 180, 4309–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunst F., Ogasawara,N., Moszer,I., Albertini,A.M., Alloni,G., Azevedo,V., Bertero,M.G., Bessieres,P., Bolotin,A., Borchert,S. et al. (1997) The complete genome sequence of the Gram-positive bacterium Bacillus subtilis. Nature, 390, 249–256. [DOI] [PubMed] [Google Scholar]
- 14.O’Farrell P.H. (1975) High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem., 250, 4007–4021. [PMC free article] [PubMed] [Google Scholar]
- 15.Tobisch S., Zühlke,D., Bernhardt,J., Stülke,J. and Hecker,M. (1999) Role of CcpA in regulation of the central pathways of carbon catabolism in Bacillus subtilis. J. Bacteriol., 181, 6996–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirose I., Sano,K., Shioda,I., Kumano,M., Nakamura,K. and Yamane,K. (2000) Proteome analysis of Bacillus subtilis extracellular proteins: a two-dimensional protein electrophoretic study. Microbiology, 146, 65–75. [DOI] [PubMed] [Google Scholar]
- 17.DeRisi J.L., Iyer,V.R. and Brown,P.O. (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science, 278, 680–686. [DOI] [PubMed] [Google Scholar]
- 18.Fawcett P., Eichenberger,P., Losick,R. and Youngman,P. (2000) The transcriptional profile of early to middle sporulation in Bacillus subtilis. Proc. Natl Acad. Sci. USA, 97, 8063–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ye R.W., Tao,W., Bedzyk,L., Young,T., Chen,M. and Li,L. (2000) Global gene expression profiles of Bacillus subtilis grown under anaerobic conditions., J. Bacteriol. 182, 4458–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida K., Ishio,I., Nagakawa,E., Yamamoto,Y., Yamamoto,M. and Fujita,Y. (2000) Systematic study of gene expression and transcription organization in the gntZ-ywaA region of the Bacillus subtilis genome. Microbiology, 146, 573–579. [DOI] [PubMed] [Google Scholar]
- 21.Miwa Y., Saikawa,M. and Fujita,Y. (1994) Possible function and some properties of the CcpA protein of Bacillus subtilis. Microbiology, 140, 2567–2575. [DOI] [PubMed] [Google Scholar]
- 22.Morrissey J.H. (1981) Silver stain for proteins in polyacrylamide gels: a modified procedure with enhanced uniform sensitivity. Anal. Biochem., 117, 307–310. [DOI] [PubMed] [Google Scholar]
- 23.Matsudaira P. (1987) Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membrane. J. Biol. Chem., 262, 10035–10038. [PubMed] [Google Scholar]
- 24.Igo M.M. and Losick,R. (1986) Regulation of a promoter that is utilized by minor forms of RNA polymerase holoenzyme in Bacillus subtilis. J. Mol. Biol., 191, 615–624. [DOI] [PubMed] [Google Scholar]
- 25.Fujita Y. and Freese,E. (1981) Isolation and properties of a Bacillus subtilis mutant unable to produce fructose-bisphosphatase. J. Bacteriol., 145, 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Saizieu A., Certa,U., Warrington,J., Gray,C., Keck,W. and Mous,J. (1998) Bacterial transcript imaging by hybridization of total RNA to oligonucleotide arrays. Nat. Biotechnol., 16, 45–48. [DOI] [PubMed] [Google Scholar]
- 27.Richmond C.S., Glasner,J.D., Mau,R., Jin,H. and Blattner,F.R. (1999) Genome-wide expression profiling in Escherichia coli K-12. Nucleic Acids Res., 27, 3821–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lopez J.M. and Thoms,B. (1976) Beziehungen zwischen katabolischer Repression und Sporulation bei Bacillus subtilis. Arch. Microbiol., 109, 181–186. [DOI] [PubMed] [Google Scholar]
- 29.Lindgren V. and Rutberg,L. (1974) Glycerol metabolism in Bacillus subtilis: gene-enzyme relationships. J. Bacteriol., 119, 431–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu X. and Taber,H.W. (1998) Catabolite regulation of the Bacillus subtilis ctaBCDEF gene cluster. J. Bacteriol., 180, 6154–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin I., Débarbouillé,M., Klier,A. and Rapoport,G. (1989) Induction and metabolite regulation of levanase synthesis in Bacillus subtilis. J. Bacteriol. 171, 1885–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin-Verstraete I., Deutscher,J. and Galinier,A. (1999) Phosphorylation of HPr and Crh by HprK, early steps in the catabolite repression signalling pathway for the Bacillus subtilis levanase operon. J. Bacteriol., 181, 2966–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepesant J. and Dedonder,R. (1967) Biochimie. Metabolisme de L-arabinose chez Bacillus subtilis Marburg Ind168. C. R. Acad. Sci. Paris, 268, 2683–2686. [PubMed] [Google Scholar]
- 34.Sá-Nogueira I., Nogueira,T.V., Soares,S. and de Lencastre,H. (1997) The Bacillus subtilis L-arabinose (ara) operon: nucleotide sequence, genetic organization and expression. Microbiology, 143, 957–969. [DOI] [PubMed] [Google Scholar]
- 35.Sekowska A., Coppée,J., Le Caer,J., Martin-Verstraete,I. and Danchin,A. (2000) S-adenosylmethionine decarboxylase of Bacillus subtilis is closely related to archaebacterial counterparts. Mol. Microbiol., 36, 1135–1147. [DOI] [PubMed] [Google Scholar]
- 36.Fillinger S., Boschi-Muller,S., Azza,S., Dervyn E., Branlant,G. and Aymerich,S. (2000) Two glyceraldehyde-3-phosphate dehydrogenases with opposite physiological roles in a nonphotosynthetic bacterium. J. Biol. Chem., 275, 14031–14037. [DOI] [PubMed] [Google Scholar]
- 37.Diesterhaft M.D. and Freese,E. (1973) Role of pyruvate carboxylase, phosphoenolpyruvate carboxykinase and malic enzyme during growth and sporulation of Bacillus subtilis. J. Biol. Chem., 248, 6062–6070. [PubMed] [Google Scholar]
- 38.Belitsky B.R. and Sonenshein,A.L. (1998) Role and regulation of Bacillus subtilis glutamate dehydrogenase genes. J. Bacteriol., 180, 6298–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nihashi J. and Fujita,Y. (1984) Catabolite repression of inositol dehydrogenase and gluconate kinase syntheses in Bacillus subtilis. Biochim. Biophys. Acta, 798, 88–95. [DOI] [PubMed] [Google Scholar]
- 40.Deutscher M.P. and Kornberg,A. (1968) Biochemical studies of bacterial sporulation and germination. VIII. Patterns of enzyme development during growth and sporulation of Bacillus subtilis. J. Biol. Chem., 243, 4653–4660. [PubMed] [Google Scholar]
- 41.Tobisch S., Glaser,P., Kruger,S. and Hecker,M. (1997) Identification and characterization of a new β-glucoside utilization system in Bacillus subtilis. J. Bacteriol ., 179, 496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshida K., Shibayama,T., Aoyama,D. and Fujita,Y. (1999) Interaction of a repressor and its binding sites for regulation of the Bacillus subtilis iol divergon. J. Mol. Biol., 285, 917–929. [DOI] [PubMed] [Google Scholar]
- 43.Huang M., Oppermann-Sanio,F.B. and Steinbüchel,A. (1999) Biochemical and molecular characterization of the Bacillus subtilis acetoin catabolic pathway. J. Bacteriol., 181, 3837–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beijer L., Nilsson,R., Holmberg,C. and Rutberg,L. (1993) The glpP and glpF genes of the glycerol regulon in Bacillus subtilis. J. Gen. Microbiol., 139, 349–359. [DOI] [PubMed] [Google Scholar]
- 45.Nilsson R., Beijer,L. and Rutberg,B. (1994) The glpT and glpQ genes of the glycerol regulon in Bacillus subtilis. Microbiology, 140, 723–730. [DOI] [PubMed] [Google Scholar]
- 46.Baumberg S. and Harwood,C.R. (1979) Carbon and nitrogen repression of arginine catabolic enzymes in Bacillus subtilis. J. Bacteriol., 137, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu J., Hederstedt,L. and Piggot,P.J. (1995) The cytochrome bc complex (menaquinone:cytochrome c reductase) in Bacillus subtilis has a nontraditional subunit organization. J. Bacteriol., 177, 6751–6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pearson W.R. and Lipman,D.J. (1988) Improved tools for biological sequence comparison. Proc. Natl Acad. Sci. USA, 85, 2444–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]