

Abstract
Cocaine induces plasticity at glutamatergic synapses in the nucleus accumbens (NAc). Withdrawal was suggested to play an important role in the development of this plasticity by studies showing that some changes only appear several weeks after the final cocaine exposure. In this study, the requirement for prolonged withdrawal was evaluated by comparing the changes in glutamatergic transmission induced by two different noncontingent cocaine treatments: a short treatment followed by prolonged withdrawal, and a longer treatment without prolonged withdrawal. Recordings were performed from mouse medium spiny neurons (MSNs) in the NAc at the same time after the first cocaine injection under both treatments. A similar increase in the frequency of glutamate-mediated miniature EPSCs was observed in D1-expressing MSNs after both cocaine treatments, demonstrating that prolonged withdrawal was not required. Furthermore, larger AMPA receptor-to-NMDA receptor ratios, higher spine density, and enlarged spine heads were observed in the absence of withdrawal after a long cocaine treatment. These synaptic adaptations expressed in D1-containing MSNs of the NAc core were not further enhanced by protracted withdrawal. In conclusion, a few repeated cocaine injections are enough to trigger adaptations at glutamatergic synapses in D1-expressing MSNs, which, although they take time to develop, do not require prolonged cocaine withdrawal.
Introduction
The nucleus accumbens (NAc) is implicated in reward-motivated learning when involving both natural and pathological rewards (Hyman et al., 2006; Thomas et al., 2008). It is divided into core and shell subregions (Zahm, 1999; Humphries and Prescott, 2010) and receives major excitatory inputs from the hippocampus, amygdala, and prefrontal cortex (Phillipson and Griffiths, 1985; Sesack et al., 1989; McDonald, 1991). These glutamatergic inputs form synapses onto the spines of medium spiny neurons (MSNs). The NAc also receives dopaminergic innervation from the ventral tegmental area (Bouyer et al., 1984; Freund et al., 1984), which modulates glutamatergic transmission and mediates the rewarding actions of psychostimulants (Fields et al., 2007; Kalivas, 2009; Kalivas et al., 2009).
Two subpopulations of GABAergic MSNs have been distinguished based on the expression of D1 or D2 dopamine receptors (D1Rs, D2Rs) in the dorsal striatum and NAc (Kebabian and Calne, 1979; Sibley and Monsma, 1992). It remains unclear which of these two receptors is responsible for the different actions of cocaine in these regions. On one hand, pharmacological, imaging, and genetic approaches suggest an involvement of D2Rs in cocaine responses in the CNS (Caine et al., 2002; Thanos et al., 2008; Asensio et al., 2010). On the other hand, D1Rs play a role in the response to cocaine (Zhang et al., 2002; Zhang and Xu, 2006; Heiman et al., 2008), D1R knock-out mice fail to self-administer cocaine (Caine et al., 2007), and repeated administration of D1R antagonists block the cocaine-induced increase in spine density in the NAc (Ren et al., 2010).
Repeated cocaine administration causes plasticity at glutamatergic synapses in the NAc that is expressed as changes in glutamate receptor surface expression, density of dendritic spines, and synaptic function (Zhang et al., 1998; Robinson et al., 2001; Li et al., 2004; Boudreau and Wolf, 2005; Martin et al., 2006; Kourrich et al., 2007; Huang et al., 2009; Kourrich and Thomas, 2009; Moussawi et al., 2009). Many of these changes only develop several weeks after the final cocaine exposure, suggesting that abstinence is an important mediator of the plasticity (Robinson et al., 2001; Li et al., 2004; Boudreau and Wolf, 2005; Boudreau et al., 2007; Kourrich et al., 2007; Guan et al., 2009). These observations raised the possibility that withdrawal itself might be the trigger for the reported functional and morphological changes in the NAc.
Some recent studies, however, have challenged this notion by demonstrating increased spine density 2 d after the final cocaine injection (Lee et al., 2006; Kim et al., 2009; Ren et al., 2010). The requirement for withdrawal has not been addressed previously with regard to the functional plasticity. In this study, we test the role of withdrawal on glutamatergic plasticity and perform behavioral, morphological, and electrophysiological analysis to the same set of animals to correlate functional with morphological changes and evaluate cell-specific actions of cocaine. The results show that D1R-expressing MSNs in the NAc core are particularly susceptible to cocaine exposure and cocaine withdrawal is not required for the functional and morphological adaptations in the NAc.
Materials and Methods
Animals.
All experiments were performed in accordance with guidelines from the National Institute on Alcohol Abuse and Alcoholism Animal Care and Use Committee. Male and female mice were maintained on a 12 h light/dark cycle and housed in groups of two to four with ad libitum access to food and water. Bacterial artificial chromosome transgenic mice (Swiss Webster background) expressing enhanced green fluorescent protein (EGFP) under the control of the D1a dopamine receptor promoter (Drd1a-EGFP; GENSAT) were used in the study (Gong et al., 2003). Wild-type Swiss Webster mice were also used for one of the morphological experiments.
Drug treatment.
Male and female mice (6–9 weeks of age) were randomly assigned to one of five groups shown in Table 1 and were subject to one of the three following treatments: (1) short treatment (7 consecutive days; groups A and B), (2) long treatment (20 injections: 5 consecutive days followed by 2 injection-free days for 4 weeks; groups C and D), and (3) long uninterrupted treatment (28 consecutive injections; group E). The groups were balanced with respect to gender, age, and weight. Mice received daily intraperitoneal injections of saline or cocaine (30 mg/kg; Sigma-Aldrich) in a novel cage. Injections were performed in small cohorts of animals (two to six) to allow for electrophysiology and morphology study to occur within a narrow range of time (±1 d). Several cohorts were run over a period of 12 months during which time the recorded behavior was very stable.
Table 1.
Experimental design for the short and long treatments of animals with either saline or cocaine
| Group | No. injections | Withdrawal | Study |
|---|---|---|---|
| A | 7 d, i.p. | 2 d | Electrophysiology |
| B | 7 d, i.p. | 21 d | Electrophysiology |
| C | 20 d, i.p. (5 d ON, 2 d OFF × 4 weeks) | 2 d | Electrophysiology and morphology |
| D | 20 d, i.p. (5 d ON, 2 d OFF × 4 weeks) | 30 d | Electrophysiology and morphology |
| E | 28 d, i.p. | 1 d | Electrophysiology |
Locomotor activity.
All cages used for behavioral testing were constructed of clear polycarbonate walls (10 × 6.5 inch) with perforated stainless-steel flooring. Horizontal activity was detected as infrared beam crosses (1 inch spacing, 10 beams per cage) using Opto M3 activity monitors (Columbus Instruments). Mice were allowed to run freely in the cage for 5 min before each injection. They were then injected with cocaine or saline and placed back into the same cage while ambulatory counts were recorded for 20 min under dim illumination (100 lux).
Electrophysiology.
Animals were killed 1, 20, or 30 d (+1 or 2) after the last cocaine injection according to the group described in Table 1. Alternating sagittal brain slices of the NAc were prepared for the electrophysiological recording (250 μm) or neuronal morphology (200 μm), using a vibrating slicer (Leica VT1200) in choline solution containing the following (in mm): 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7 MgCl2, 25 glucose, 0.5 CaCl2, 110 (CH3)3N(Cl)CH2CH2OH, 11.6 C6H7NaO6, 3.1 C3H4O3. For recordings, slices were allowed to recover in oxygenated artificial CSF (ACSF) containing the following (in mm): 127 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 25 glucose for 30 min at 33°C, and then transferred to a recording chamber. Neurons in NAc core were recorded from slices in which the rostral and caudal limbs of the anterior commissure, and the dorsal striatum were present. Shell neurons were recorded from medial NAc slices that did not contain dorsal striatal tissue [as described by Thomas et al. (2001)]. D1(+) MSNs were identified based on the green fluorescence, average soma size of ∼35 μm, and high resting membrane potential (−75 to −85 mV). Whole-cell voltage-clamp recordings were performed using patch electrodes (4–6 MΩ) filled with internal solution containing the following (in mm): 135 CsMeSO4, 4 MgCl2, 10 HEPES, 0.5 EGTA, 0.4 GTP-sodium salt, 4 ATP-Na2, 10 phosphocreatine disodium salt, pH 7.2–7.4 (295–300 mOsm) at 25°C. A Multiclamp 700B amplifier (Molecular Devices) was used and currents were filtered at 2 kHz and digitized at 5 kHz. Membrane potential was held at −80 mV and series resistance (6–25 MΩ) and input resistance were monitored with a +20 mV (100 ms) depolarizing step, throughout the experiment. Miniature EPSCs were recorded for 5 min in the presence of tetrodotoxin (1 μm) and bicuculine (20 μm). Quantal events were detected and analyzed blind to treatment using Mini Analysis software (Synaptosoft) with an amplitude threshold that was three times the noise amplitude. Evoked EPSCs were generated by afferents stimulation (0.05 Hz; intensity, 30 μA) with a glass monopolar microelectrode filled with ACSF and placed 100–150 μm rostral to the recorded neurons in the presence of GABAA blocker gabazine (5 μm) alone. Evoked EPSCs were recorded when holding at +40 mV, and AMPA receptor (AMPAR)/NMDA receptor (NMDAR) ratios were measured by a pharmacological dissection as described previously (Thomas et al., 2001). Briefly, the AMPA receptor-mediated component was measured (3–6 ms around peak) in the presence of d-AP-5 (50 μm) and NMDA receptor-mediated response was determined by subtracting the AMPA component from the total (mean at 20 ms after stimulation).
Diolistic labeling.
Sagittal slices of NAc (200 μm) were fixed in 4% paraformaldehyde/4% sucrose for 30 min and washed with PBS thoroughly. Particle-mediated ballistic delivery of fluorescent dyes was used to label medium spiny neurons. Tungsten beads (1.7 μm in diameter; Bio-Rad) coated with 1–1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI) (Invitrogen) were shot through a membrane filter with a 3 μm pore size (Millipore) using a biolistic Helios gene gun (Bio-Rad) (180 psi helium gas pressure) as described previously (Seabold et al., 2010). DiI-labeled slices were permeabilized with 0.01% Triton X-100 in PBS for 15 min and then incubated in blocking solution (0.01% Triton X-100, 10% normal goat serum in PBS) for 30 min (Lee et al., 2006). Slices were then incubated with primary antibody anti-green fluorescent protein (GFP) (1:1000–1:2000; AB3080P; Millipore Bioscience Research Reagents) for 1 h, rinsed with PBS, and incubated with FITC-conjugated secondary antibody (1:1000; Invitrogen). All antibodies were dissolved in blocking solution, and incubations were performed at room temperature. Slices were then rinsed with PBS and mounted on slides using ProLong Antifade Gold (Invitrogen). The specificity of primary and secondary antibodies was confirmed by the lack of immunostaining observed in GFP-negative littermate mice.
Morphological analysis.
Image acquisition and analysis were performed in a systematic way and blind from the treatment. Image stacks of the distal portion of three dendrites (secondary and tertiary dendrites only) per cell were collected in at least four cells (two from core, two from shell) per mouse. Regions with dense DiI staining in which individual neurons could not be distinguished were avoided. Image stacks (512 × 512; z spacing, 0.7 μm; x–y scaling, 0.14 μm/pixel) were acquired using a confocal microscope (Zeiss LSM 510 META) with a 63× water objective [numerical aperture (NA), 1.2] at 1.0 μm optical section and 2× zoom corresponding to a 71.4 × 71.4 μm image field. DiI was excited using a DPS 561 nm laser line. Dendritic spine morphology was analyzed using ImageViewer (Steiner et al., 2008), a custom software for spine analysis written in Matlab (The MathWorks). Spines were identified manually in the three-dimensional image stacks but spines protruding in the z-axis cannot be distinguished clearly and are not counted. Thus, although the error is constant for all conditions and treatments, spine density values are an underestimation of the real value. Spine head width and spine length were also measured in the three-dimensional stacks by drawing two lines, one longitudinal along the whole spine and the other transversal across the thickest part of the spine head (parallel to the dendrite). Length and head width were then automatically determined from the fluorescent distribution along the longitudinal and transversal lines as the length at the 30% fluorescence of the maximum. For dendrite morphology and branching, whole-cell image stacks (512 × 512; z spacing, 3.5 μm; x–y scaling, 0.879 μm/pixel) were acquired with a 20× air objective (NA, 0.8) at 3.8 μm optical section and 1× zoom corresponding to a 450 × 450 μm image field. Imaris filament tracer module (Bitplane) was used for Scholl analysis and measurements of branch points and total dendrite length.
Statistical analysis.
Results are shown as mean ± SEM. Statistical analysis was performed using Igor Pro software (Wavemetrics). Student's t test was used unless noted. Kolmogorov–Smirnov (K-S) or Mann–Whitney test was performed to compare distributions depending on the normality. ANOVA followed by Tukey's test was used for all multiple comparisons.
Results
Transgenic mice expressing the fluorescent reporter EGFP under the control of D1 dopamine receptor promoter (Drd1a-EGFP) were used in this study to identify MSNs of the direct pathway. A sagittal brain section from these mice shows green fluorescent labeling in the dorsal striatum and the NAc (Fig. 1A). These mice have been characterized (Kramer et al., 2011) and used successfully in the past for this purpose (Kreitzer and Malenka, 2005; Day et al., 2006, 2008; Lee et al., 2006; Wang et al., 2006; Surmeier et al., 2007; Heiman et al., 2008).
Figure 1.
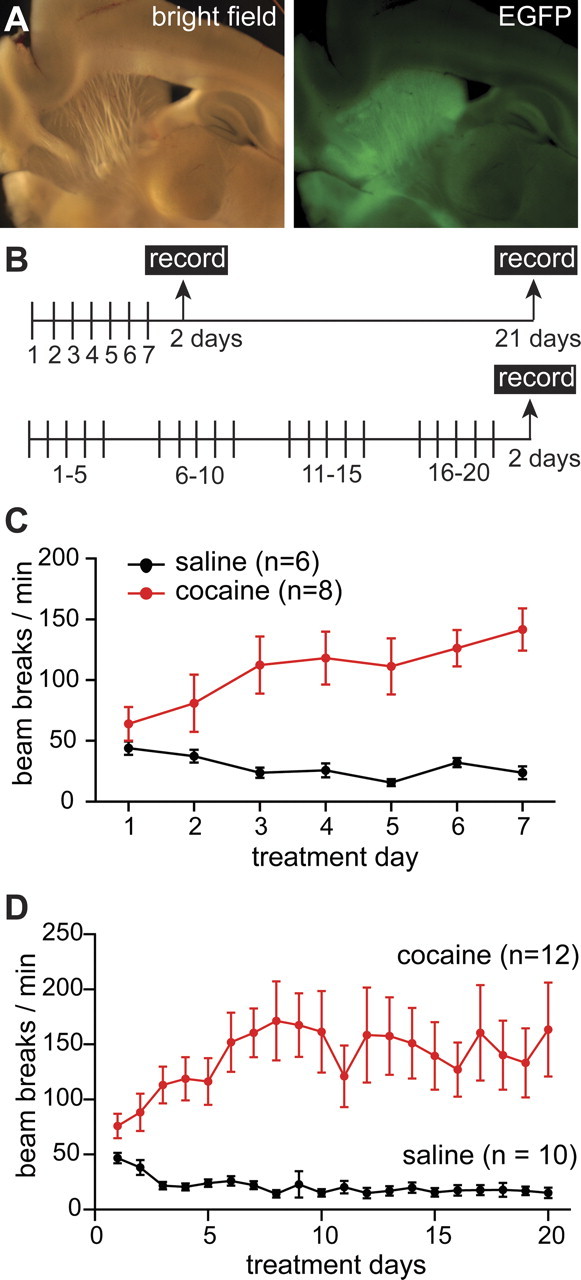
Locomotor response to repeated cocaine injections in Drd1-GFP mice during the short and long treatment. A, A bright-field (top) and a fluorescent image (bottom) of a sagittal brain section from a Drd1a-GFP mouse showing green fluorescence in the dorsal striatum and nucleus accumbens regions. B, Schematic diagram of the main two intraperitoneal injections treatments used: short (7 consecutive injections) and long (20 injections: 5 d ON–2 d OFF). C, Daily locomotor activity (mean ± SEM) during the short treatment measured as beam breaks per minute during 20 min after the saline (black) or cocaine (red) injections for mice from groups A and B, respectively. D, Daily locomotor activity (mean ± SEM) during the long treatment measured as beam breaks per minute during 20 min after the saline (black) or cocaine (red) injection for mice from groups C and D, respectively.
Comparable locomotor responses to cocaine in short and long treatment
Mice were assigned to groups (Table 1) and received saline or cocaine (30 mg/kg) daily injections according to a short (7 consecutive days) or long treatment (20 injections: 5 d ON–2 d OFF for 4 weeks). Under these two treatments, mice received different total doses of cocaine (210 mg/kg in the short and 600 mg/kg in the long treatment). To address the requirement for cocaine withdrawal, mice that received the short treatment were studied at 2 and 21 d after the last cocaine injection and compared with mice that received the long treatment and were studied 2 d after the last cocaine injection (Fig. 1B).
Horizontal locomotor response was measured for 20 min after each injection. On the first day of the short treatment, mice that received a cocaine injection displayed an acute locomotor response to the drug manifested by a mild increase in beam breaks per minute when compared with littermates that received saline injections (day 1 saline: 43 ± 5.6 beam breaks/min, n = 6; cocaine: 64.1 ± 13.7 beam break/min, n = 8) (Fig. 1C). Consecutive daily cocaine injections caused a larger increase in locomotor response, a phenomenon known as locomotor sensitization to cocaine (day 7 cocaine: 141.4 ± 17.3 beam break/min, n = 8; F(3,28) = 25.3, ANOVA, and p < 0.01 for cocaine day 7 vs saline day 1, vs saline day 7, and vs cocaine day 1 by Tukey's test) (Fig. 1C), whereas repeated saline injections led to a decline in the activity as the animals habituated to the cage and the procedure (day 7 saline: 23.7 ± 5.2 beam breaks/min, n = 6, p > 0.05 for saline day 7 vs saline day 1 by ANOVA and Tukey).
Animals exposed to the long drug treatment showed locomotor responses to saline and cocaine that were undistinguishable, during the first 7 d, from those who received the short treatment (Fig. 1D). Mice showed a mild acute response to cocaine in day 1 (saline: 46 ± 4.8 breaks/min, n = 10; cocaine: 75.7 ± 10.9 breaks/min, n = 12) (Fig. 1D) and repeated cocaine injections led to the development of psychomotor sensitization that was expressed as a larger increase in locomotor response to cocaine. The cocaine-induced increase in locomotor activity reached a maximum by day 7 and remained elevated throughout the treatment (cocaine day 7, 160.3 ± 22.1 breaks/min; day 20, 163.4 ± 42.7; n = 12; F(4,48) = 6.4 by ANOVA and p < 0.05 for cocaine day 7 vs saline day 1 and not significant for cocaine day 7 vs cocaine day 20, p > 0.05 by Tukey's test).
Outcome of short treatment in the NAc core depends on the presence or absence of a prolonged withdrawal
Mice that received short treatment were killed after 2 d (group A) or after 21 d (group B) from the last injection (Fig. 2A). Acute brain slices were prepared and whole-cell voltage-clamp recordings were made from D1(+) MSNs in the core, which were identified based on the medium size of the cell bodies, their green fluorescence, and a small holding current when voltage was held at −80 mV (Fig. 2B). The detection of green fluorescence in fresh acute slices was limited by the sensitivity of the electrophysiology microscope [optimized for differential interference contrast (DIC) visualization] and the medium-to-low levels of EGFP expression that are achieved when expression is driven by endogenous protein promoters. As a consequence of these, the number of EGFP-positive neurons in fresh acute slices was underestimated, and in this study, recordings were made only from D1 receptor-expressing neurons [D1(+) MSNs] showing clear green fluorescence in the soma (Fig. 2B).
Figure 2.
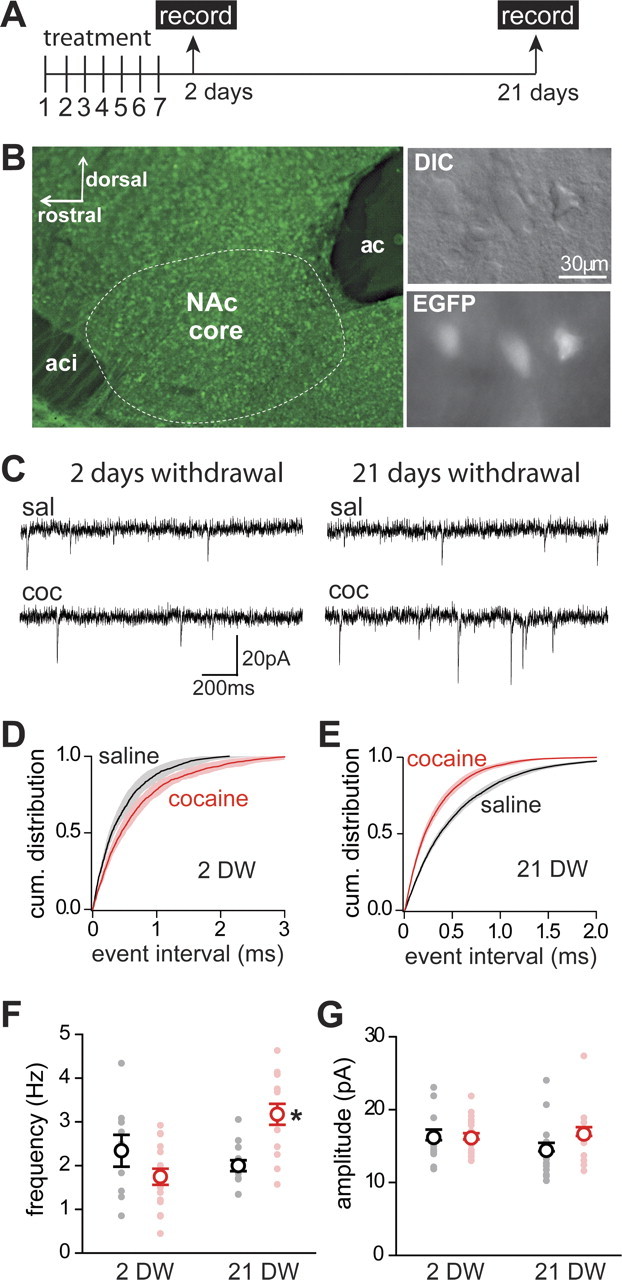
Withdrawal from short cocaine treatment is required for increased frequency of mEPSCs in D1(+) MSNs. A, Time line of short treatment (7 consecutive injections of saline or cocaine) followed by recordings 2 or 21 d after the last injection (arrows). B, Left, Green fluorescence from an acute sagittal slice from Drd1a-GFP mice showing the core subregion of the NAc (dotted line) located between the rostral (aci) and caudal (ac) limbs of the anterior commissure. Right, DIC image (top) of MSNs in a slice identified based on size and the expression of EGFP (bottom). C, Sample recording traces from D1(+) MSNs in saline- and cocaine-treated mice studied at 2 or 21 d after the last injection (groups A and B). D, E, Cumulative distribution of mEPSC interevent time from core D1(+) MSNs in saline-treated (black; n = 11–13 cells, 4 mice) and cocaine-treated mice (red; n = 16 cells, 4 mice; K-S test, p < 0.01) after 2 d withdrawal (D) and 21 d withdrawal (E). The solid lines and shadows represent the mean distribution and ±SEM for each condition, respectively. F, Frequency of mEPSCs of individual cells (filled circle) and mean ± SEM values per condition (open circles) recorded in D1(+) MSNs from NAc core in saline-treated (black) and cocaine-treated (red) mice at 2 d (2 DW) and 21 d (21 DW) of withdrawal. *p < 0.05. G, mEPSC amplitude of individual cells (filled circle) and mean ± SEM values per condition (open circle) recorded in D1(+) MSNs from NAc core in saline-treated (black) and cocaine-treated (red) mice at 2 d (2 DW) and 21 d (21 DW) of withdrawal (n = 11–16 cells, 4 mice).
Miniature EPSCs (mEPSCs) were recorded in the presence of sodium channel and GABAA receptor blockers to isolate glutamate-mediated transmission. Two days after the last cocaine injection, the mean distribution of mEPSC interevent intervals showed no significant difference from the mean distribution in saline-treated animals (Fig. 2D) and the mean frequency of mEPSC was similar to the mean frequency in saline-treated mice (2.1 ± 0.2 Hz for saline and 1.7 ± 0.2 Hz for cocaine; n = 23–16; p > 0.3 by K-S test) (Fig. 2F, left).
However, in agreement with previous reports showing potentiation of glutamatergic transmission after several weeks of withdrawal from a short cocaine treatment (Kourrich et al., 2007), the mean frequency of mEPSCs in D1(+) MSNs was significantly increased in the cocaine group 3 weeks after last cocaine injection (3.1 ± 0.2 Hz, for cocaine, n = 14; p < 0.01 by K-S test) (Fig. 2F, right). Consequently, a shift to shorter interevent intervals was detected in the mean distribution in the cocaine group after 3 weeks of withdrawal (Fig. 2E) (p < 0.05, K-S test). The mean amplitude of mEPSC events was not significantly changed by the cocaine treatment either 2 d or 3 weeks of withdrawal. (Fig. 2G).
Enhanced glutamatergic transmission in core and shell without cocaine withdrawal after long treatment
The increase in mEPSC frequency observed 3 weeks, but not 2 d, after last cocaine injection could have several possible explanations. One possibility is that extended withdrawal after repeated cocaine injections is the signal that triggers the cocaine-induced plasticity. Another hypothesis is that the events leading to the changes in glutamatergic transmission in the NAc take several weeks to develop, and thus they are not expressed right after a short cocaine treatment. In this case, the changes would be observed even if the cocaine treatment is extended for 2–3 weeks and protracted withdrawal is avoided. To discriminate between these two hypotheses, a long drug treatment was used (group C) (Fig. 1B).
Two days after the last injection of the long treatment, acute brain slices were prepared from saline- and cocaine-treated mice. D1(+) MSNs from core and shell were recorded under the same conditions described for mice that received short treatment (Fig. 3A,B). The frequency (freq) of mEPSCs was significantly increased in MSNs from the core and the shell in cocaine-treated mice compared with saline-treated mice (core freq: 2.7 ± 0.3 Hz for saline and 3.7 ± 0.3 Hz for cocaine, n = 16–20; shell freq: 1.8 ± 0.3 Hz for saline and 3.9 ± 0.7 Hz for cocaine, n = 13–16; *p < 0.05) (Fig. 3C). An significant increase in the amplitude of the events was seen in shell D1(+) MSNs after the cocaine treatment, and no change was detected in the amplitude (amp) of the events in the core (shell amp: 13.2 ± 1.0 pA for saline and 18.8 ± 1.4 pA for cocaine, n = 13–16 neurons; p < 0.05; core amp: 16.9 ± 1.0 pA for saline and 18.2 ± 1.0 pA for cocaine, n = 16–20 neurons) (Fig. 3D).
Figure 3.

Increased mEPSC frequency and AMPA/NMDA ratio after long cocaine treatment without protracted withdrawal. A, Schematic diagram of long treatment (20 injections: 5 d ON–2 d OFF) followed by recordings 2 d after the last injection (arrow). B, Sample traces of mEPSCs recordings from D1(+) MSNs in slices from saline- and cocaine-treated mice. C, mEPSC frequency values of individual cells (filled circle) and mean ± SEM per condition (open circle) recorded in D1(+) MSNs from core and shell of saline-treated (black) and cocaine-treated (red) mice (n = 16–20 cells). *p < 0.05. D, mEPSC amplitude of individual cells (filled circle) and mean ± SEM values per condition (open circle) recorded in D1(+) MSNs from core and shell of saline-treated (black) and cocaine-treated (red) mice. *p < 0.05. E, Time line of long uninterrupted treatment (28 consecutive injections) and recordings 1 d after the last injection (arrow). F, mEPSC frequency and amplitude (mean ± SEM) recorded from core D1(+) MSNs of saline- and cocaine-treated mice (n = 8–9 cells, 3 mice each group). *p < 0.05. G, Exemplary traces of evoked EPSCs showing isolated AMPA and NMDA receptor-mediated currents recorded at Vh = +40 mV. H, AMPAR/NMDAR ratio of individual cells (filled circles) and mean ± SEM values per condition (open circle) recorded in core D1(+) MSNs from saline (black)- and cocaine-treated (red) mice at 1 d of withdrawal (n = 5–6 cells, 3–4 mice). *p < 0.05.
A long uninterrupted treatment (28 consecutive injections) (Table 1, group E) was also performed to rule out any possible contribution of the three brief periods of withdrawal (2 d OFF) on the cocaine-induced effect on glutamatergic transmission. Two days after the last injection of the long uninterrupted treatment, recordings were obtained from MSNs in NAc core. This subregion was chosen first because similar changes in mEPSC frequency were seen in core and shell after the long (20 d) treatment, and second because of the larger yield of slices containing NAc core. Frequency of mEPSCs was significantly increased in core D1(+) MSNs from cocaine-treated mice compared with saline animals (2.0 ± 0.2 Hz for saline and 3.6 ± 0.6 Hz for cocaine; n = 8–9; *p < 0.05) (Fig. 3F) and no effect was seen in the amplitude (14.4 ± 1.7 pA for saline and 11.5 ± 1.7 pA for cocaine; n = 8–9; p > 0.05) (Fig. 3F). The magnitude of the mEPSC frequency increase was similar between the two long treatments, indicating that the brief withdrawal periods were not required for the cocaine-induced synaptic adaptation.
Evoked EPSCs were measured in core D1(+) MSNs after the long uninterrupted treatment and AMPA/NMDA ratios were calculated from saline- and cocaine-treated mice. Cocaine-treated mice showed a significant increased in AMPAR/NMDAR ratios, indicating that the synaptic adaptations extend beyond miniature event (1.1 ± 0.1 for saline and 1.5 ± 0.1 for cocaine, n = 5–6; three mice each group; p < 0.05) (Fig. 3G,H). Together, these experiments showed that the potentiation of glutamatergic transmission triggered by cocaine develops and expresses itself without the need for protracted or brief withdrawal after a long repeated treatment. Furthermore, when compared with the results of the short treatment, these results indicate that seven repeated cocaine injections are sufficient for triggering these changes and that it is the time, but not the prolonged withdrawal, that is required for the induction of this cocaine-induced plasticity in the NAc.
Concurrent increase in spine density and glutamatergic transmission in D1(+) MSNs
The next experiments addressed whether the increase in mEPSC frequency detected in D1(+) MSNs from cocaine-treated mice could reflect the presence of more glutamatergic synapses in these neurons after cocaine. Because the density of the dendritic spines can be a morphological readout for the abundance of glutamatergic synapses, the morphology of dendrites and dendritic spines was studied in parallel with the electrophysiology. We focused on mice exposed to the long cocaine treatment (20 injections: 5 d ON–2 d OFF for 4 weeks) and studied spine morphology 2 d after the last cocaine injection.
Diolistic labeling was used and combined with immunostaining to further amplify the EGFP signal at the cell bodies. DiI-labeled MSNs with positive immunostaining were identified as D1(+) MSNs and those lacking EGFP immunostaining were classified as D1 receptor-negative MSNs [D1(−) MSNs] (Fig. 4B). Confocal images were acquired from the distal portion of MSN dendrites in the core and shell of the NAc, and they were analyzed blind to the drug treatments. An increased density of spines was detected in D1(+) MSNs from in cocaine-treated animals (core and shell combined) compared with saline-treated mice (0.91 ± 0.03 μm−1 for saline and 1.10 ± 0.08 μm−1 for cocaine; n = 13–14 neurons, 3–4 mice; p < 0.05) (Fig. 4E,F). No significant change was detected in the spine density of D1(−) MSNs between saline- and cocaine-treated mice (0.91 ± 0.06 μm−1 for saline and 0.96 ± 0.07 μm−1 for cocaine; n = 11–13 neurons, 3–4 mice; p > 0.05). Note that there was no significant difference in the mean density of spine in D1(+) and D1(−) MSNs in saline-injected animals (Fig. 4F).
Figure 4.
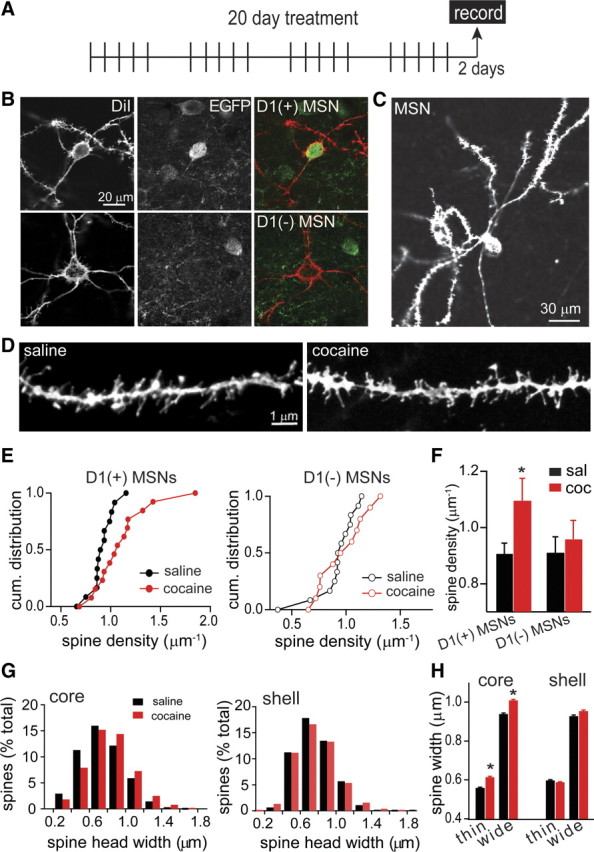
Paralleled increase in spine density in D1(+) MSNs after long cocaine treatment without protracted withdrawal. A, Schematic diagram of long treatment (20 injections: 5 d ON–2 d OFF) followed by morphology analysis 2 d after the last injection (arrow). B, Top panels, Confocal images of a D1(+) MSN that is labeled with DiI (left, red) and displays EGFP immunostaining (middle, green). Bottom panels, Confocal image of a D1(−) MSN showing DiI labeling (left, red) and undetectable EGFP immunostaining (middle). C, DiI-labeled MSN from the shell of the NAc. D, Confocal images of distal dendrites from saline- and cocaine-treated mice 2 d after the last injection (groups C). E, Cumulative distribution of the densities of dendritic spine for each D1(+) MSN (left) and for each D1(−) MSN (right) from both core and shell subregions in saline (black)- and cocaine (red)-treated mice. Significant difference was found between the density distribution of saline and cocaine D1(+) MSNs (n = 13–14 neurons, 3–4 mice; p < 0.05, K-S test) not in D1(−) MSNs (n = 11–13 neurons, 3–4 mice; p > 0.05, K-S test). F, Spine density (mean ± SEM) in D1(+) MSNs and D1(−) MSNs in saline (black)- and cocaine (red)-treated mice (black) core and shell combined. *p < 0.05. G, Histogram distribution for spine head width in MSNs from the NAc core (left) or shell (right). H, Mean and SEM values of the spine head width of thin (smaller half) and wide (larger half) dendritic spines that were defined using a median split of the width distributions. *p < 0.01, ANOVA followed by Tukey.
These experiments showed that functional and structural plasticities of glutamatergic synapses happen simultaneously in D1(+) MSNs shortly after a long cocaine treatment.
Enlarged spine heads in cocaine-treated animals
The morphology of dendritic spines is diverse and it can be linked to the functional diversity of glutamatergic synapses (Alvarez and Sabatini, 2007). Correlations between the size of the spine head and postsynaptic density (Harris and Stevens, 1989) and between the size of the spine head and the amplitude of AMPA currents evoked at each spine (Matsuzaki et al., 2001) have been determined in the past for glutamatergic synapses in the hippocampus.
The structural diversity of spines was studied here by measuring the spine length and width of the spine head. In saline-treated animals, the distribution of spine head width for MSNs [D1(+) and D1(−) MSNs combined] was similar from neurons in the core and shell with a mean width value of 0.75 ± 0.05 μm for core and 0.76 ± 0.06 μm for the shell (n = 1703–784 spines/16–12 neurons). Repetitive cocaine injections caused a rightward shift in the normalized distribution of spine head width in the core MSNs [core width, 0.81 ± 0.06 μm in core; n = 1620 spines/12 neurons D1(+) and D1(−) combined] (Fig. 4G). There were less thin spines and larger percentage of wide spines in core MSNs 2 d after the last cocaine injection but there was no detectable difference in the spine width distribution in shell MSNs after cocaine treatment (shell width, 0.77 ± 0.005 μm; n = 2125 spines/15 neurons). When a median split was used to look at thin spines (the smaller half) and wide spines (the larger half), both thin and wide spines showed larger heads in the core, but not the shell, of cocaine-treated animals (ANOVA, F = 991.8, p < 0.01) (Fig. 4H). No change in the mean spine length was observed after cocaine treatment in core or shell MSNs (core spine length, 1.86 ± 0.02 μm in saline and 1.83 ± 0.02 μm in cocaine; shell spine length, 1.77 ± 0.03 μm in saline and 1.81 ± 0.02 μm in cocaine; n = 784–2125 spines).
Dendrite morphology after long cocaine treatment
A model of each neuron was constructed from confocal image stacks of D1(+) and D1(−) MSNs acquired at low magnification to study dendrite branching and morphology (Fig. 5A,B). The mean number of dendrite branch points per neuron was similar for D1(+) and D1(−) MSNs from saline- and cocaine-treated animals [D1(+) MSNs, 13.8 ± 5.6 and 12.6 ± 6.6 for saline and cocaine, respectively; n = 9–10; D1(−) MSNs, 12.1 ± 5.8 and 11.6 ± 6.2 for saline and cocaine, respectively; n = 14–17] (Fig. 5C). Scholl analysis showed no significant change in the number of intersections or total dendrite lengths in D1(+) MSNs and a small decrease in the total dendrite lengths in D1(−) MSNs after cocaine treatment [D1(+) MSNs length, 1.72 ± 0.52 and 1.31 ± 0.43 mm for saline and cocaine, respectively; D1(−) MSNs length, 1.35 ± 0.48 and 1.05 ± 0.26 mm for saline and cocaine, respectively; p < 0.05] (Fig. 5D,E).
Figure 5.
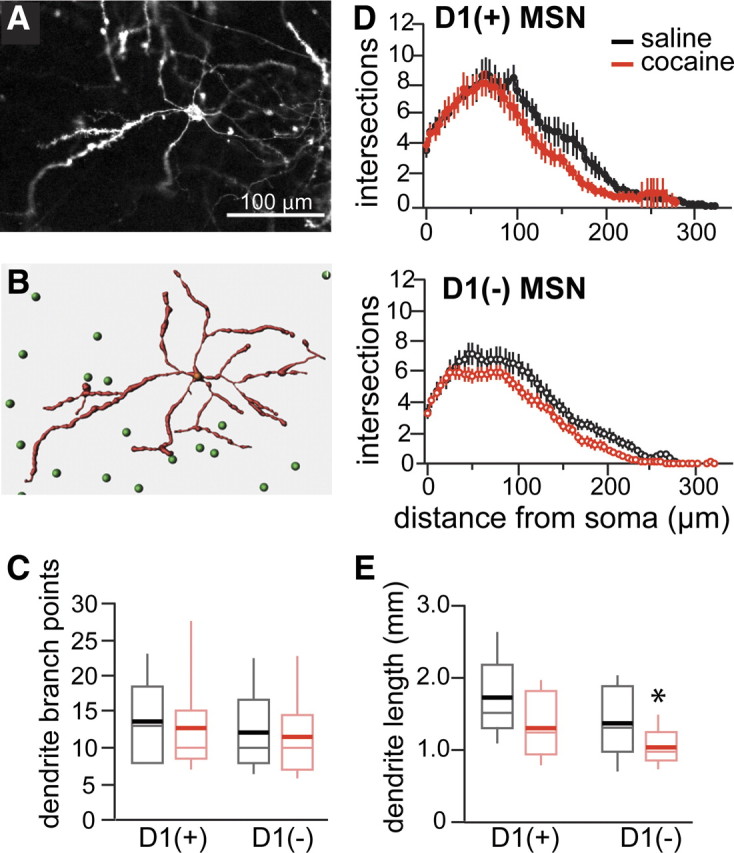
Dendrite morphology after long cocaine treatment without protracted withdrawal. A, Confocal image of a DiI-labeled MSN. B, Model obtained by Imaris software showing a DiI-labeled neuron in red and, in green, the location of the GFP-positive cell soma in the imaged field. C, Dendrite branch points per neuron (mean ± SEM) for D1(+) MSNs and D1(−) MSNs in saline (black)- and cocaine (red)-treated mice. Box plot shows mean (thick line) and median (thin line) for the data of each group (n = 9–10 and 14–17). D, Scholl analysis of dendrite branches for D1(+) (top) and D1(−) (bottom) MSNs in the NAc (core and shell combined) after saline (black) or cocaine (red) treatment. E, Box plots showing mean (thick line) and median (thin line) for the total dendrite length of D1(+) and D1(−) MSNs of saline (black)- or cocaine (red)-treated mice (n = 9–10 and 14–17). *p < 0.01.
Protracted withdrawal after long cocaine treatment does not enhance plasticity
The next experiments investigated whether prolonged withdrawal after the long cocaine treatment can further enhance the functional and morphological plasticity observed in D1(+) MSNs. With this purpose, we measured in parallel mEPSC frequency and spine density 30 d after the last injection of the long treatment (Fig. 6A, group D). The physiology and morphology data were sorted in core and shell D1(+) MSNs and compared with the previous values obtained 2 d after the last injections from the long treatment (Fig. 6B,C). Although the frequency of mEPSCs was significantly increased 2 d after the long cocaine treatment (2 dw) in both core and shell, mEPSC frequency was not different from saline values 30 d (30 dw) after the last injection in both subregions of the NAc [core freq: 2.5 ± 0.2, 3.7 ± 0.3, and 2.7 ± 0.3 Hz for saline, cocaine 2 dw and 30 dw, respectively; n = 41, 20, 17 neurons; F(2,75) = 4.5, ANOVA, and p < 0.05 for cocaine 2 dw vs saline (q = 4.2), Tukey's test; shell freq: 2.2 ± 0.2, 3.9 ± 0.7, and 2.0 ± 0.4 Hz for saline, cocaine 2 dw and 30 dw, respectively; n = 39, 15, 16 neurons; F(2,67) = 6.4, ANOVA, and p < 0.01 for cocaine 2 dw vs saline (q = 4.6) and for cocaine 2 dw vs 30 dw (q = 4.3), Tukey's test] (Fig. 6B).
Figure 6.
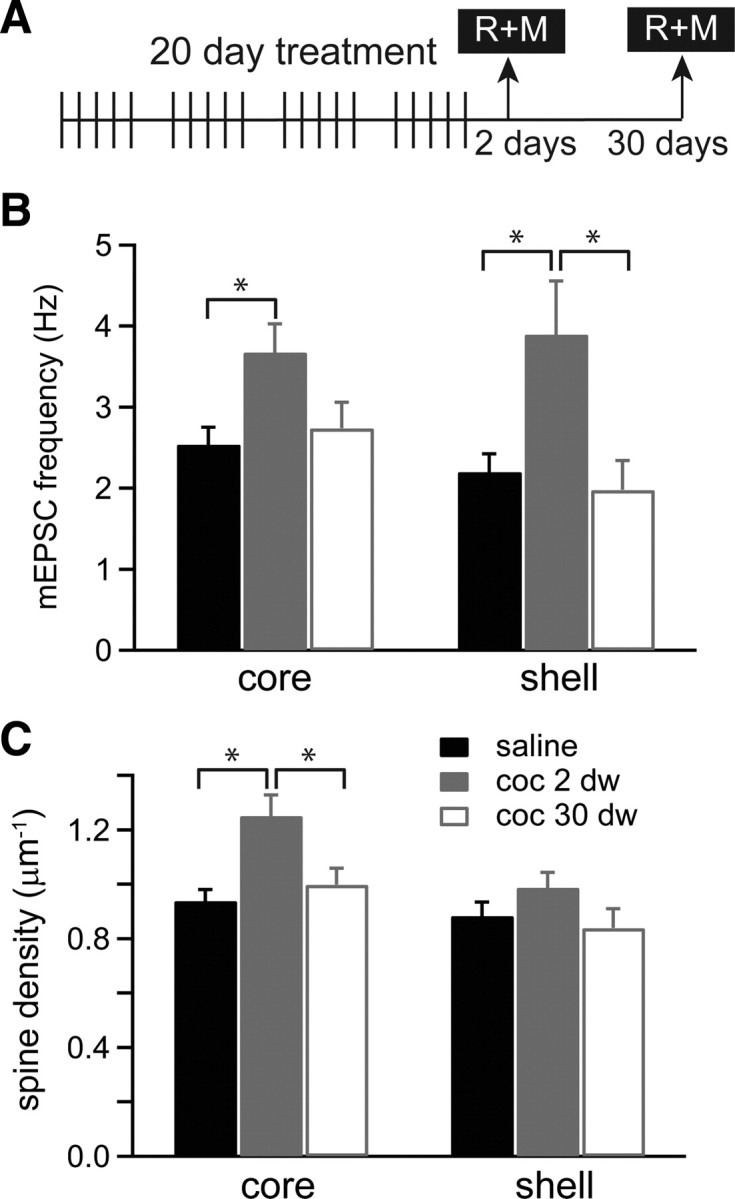
Protracted withdrawal after long cocaine treatment does not enhance plasticity. A, Schematic diagram of long cocaine treatment (20 injections: 5 d ON–2 d OFF) followed by recordings and morphology (R+M) at 2 or 30 d of withdrawal. B, mEPSCs frequency (mean ± SEM) recorded from D1(+) MSNs in core and shell of saline-treated (black bar; 2–30 d combined) and cocaine-treated mice 2 d (gray bar) or 30 d (white bar) after the last cocaine injection (n = 15–41 neurons/4–7 mice). *p < 0.05 by ANOVA and Tukey. C, Spine density (mean ± SEM) in D1(+) MSNs from core and shell in saline-treated mice or cocaine-treated mice 2 d (gray bar) and 30 d (white bar) after the last cocaine injection (n = 6–8 neurons, 3–5 mice). *p < 0.05 by ANOVA and Tukey.
Spine morphology was studied in parallel in D1(+) MSNs after the long treatment, and consistently with the physiological observations, spine density was not different from saline levels 30 d after the end of the long cocaine treatment (core density: 0.94 ± 0.04, 1.23 ± 0.13, and 0.91 ± 0.06 μm−1 for saline, cocaine 2 dw and 30 dw, respectively; n = 6–8 neurons; F(2,56) = 8.4, ANOVA, and q = 5.6 and 3.9, p < 0.01 and 0.05 for cocaine 2 dw vs saline and cocaine 2 dw vs cocaine 30 dw, respectively, by Tukey's test; shell density: 0.87 ± 0.07, 1.0 ± 0.8, and 0.79 ± 0.06 μm−1 for saline, cocaine 2 dw and 30 dw, respectively; n = 6–7 neurons; F(2,55) = 1.5, ANOVA) (Fig. 6C). Interestingly, the increase in spine densities seen in D1(+) MSN 2 d after the long treatment were mainly driven by changes in density of core D1(+) MSN. D1(−) MSNs from neither core or shell showed changes in spine density after the long cocaine treatment (F(2,14) = 2.6 for core and F(2,15) = 0.24 for shell) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Finally, an independent set of experiments was performed in wild-type Swiss Webster mice, which also lacked the increase in the spine density 30 d after the same long cocaine treatment (1.08 ± 0.12 and 0.89 ± 0.07 μm−1 for saline and cocaine, respectively; n = 8–10 neurons, 4–5 mice).
In conclusion, the results of this study show that functional and structural plasticity of glutamatergic synapses happen simultaneously in D1(+) MSNs shortly after a long cocaine treatment and do not require withdrawal.
Discussion
This study investigated the role of prolonged cocaine withdrawal in the development of glutamatergic plasticity in the accumbens by performing behavioral, morphological, and electrophysiological analysis to the same set of animals to correlate functional with morphological changes. These parallel studies are important in light of previous studies that indicated that some parameters of the cocaine treatment would affect the outcome of behavioral and cellular studies, such as home cage versus novel cage injections or the presence of a challenge cocaine injection (Li et al., 2004; Boudreau et al., 2007; Kourrich et al., 2007). The results showed that functional and morphological changes can develop right after a long cocaine treatment without the need for withdrawal. These synaptic changes include increased frequency of AMPA-mediated mEPSC, enhanced AMPA/NMDA ratio of the evoked responses, higher density of dendritic spines, and larger spine heads in D1(+) MSNs of the NAc. Although these changes were expressed independently of withdrawal, other synaptic adaptations, such as long-term potentiation, long-term depression, and AMPAR surface expression, might still require cocaine withdrawal, especially after cocaine self-administration (Grimm et al., 2001; Lu et al., 2003).
A few repeated cocaine injections were sufficient for triggering these synaptic adaptations, but the expression was time dependent and changes were not detected until several weeks have elapsed. This is particularly interesting because it speaks to the possible mechanism(s) underlying the changes in the NAc. Classic forms of plasticity, such as long-term potentiation and depression, are also expressed by changes in AMPA/NMDA ratios and spine morphology, but they develop within minutes to hours after the stimuli. In addition, generation of new synapses could also occur within hours to a few days. Then, why do these cocaine-induced changes in the NAc take so long to develop?
One plausible answer to this question is that cocaine could be altering neuronal connectivity in the ventral striatum region, rather than potentiating existing synapses. It has been suggested that cocaine could trigger a shift in the preponderance of inputs to MSNs in the NAc (Belin et al., 2009; Kalivas, 2009). For example, cocaine could weaken cortical inputs and strengthen those from amygdala, a process that would require synapse formation and elimination. With both these processes happening simultaneously, changes in the number of total inputs, AMPA/NMDA current ratios, and spine morphology could be undetectable until the new balance is achieved. Another possible scenario is that cocaine could be inducing the formation of new connections between MSNs and neurons that were not projecting to the NAc before cocaine exposure. Thus, depending on the distance between the projecting neurons and the core of the NAc, the extension of axons and the formation of synapses can easily account for many days to weeks. Yet a third possible explanation for the fact that potentiation takes time to develop is that the enduring effects of cocaine in strengthening synapses are masked by its acute, depressing action on glutamatergic synapses. The latter is supported by two studies showing that the long-term increase in AMPA/NMDA ratios and GluR1 surface expression are reversed by a cocaine challenge injection (Boudreau et al., 2007; Kourrich et al., 2007). Either way, these conclusions support existing views about possible mechanisms of cocaine action, and at the same time, they provide a frame for the generation of novel hypotheses concerning the impact of cocaine on the mesolimbic system.
By comparing the short and long cocaine treatments, we found that the extent of the treatment was another influential factor. Two days after the last injection, opposite changes in excitatory transmission were observed after the short or long treatments: a mild decrease or no change in mEPSC frequency after 7 daily cocaine injections versus increased mEPSC frequency and AMPA/NMDA ratio after 20–28 injections. This highlights the importance of performing lengthened cocaine treatments when the goal is to address chronic cocaine actions as short exposures can lead to different outcomes. The potentiation of glutamatergic transmission seen after the long treatment was accompanied by larger density of dendritic spines particularly in the core and bigger spine heads throughout the accumbens. Finally, protracted withdrawal after the long cocaine treatment failed to enhance the functional and morphological plasticity.
Three previous studies also showed increased spine density 1–2 d after the last cocaine injection and provided evidence for the involvement of D1(+) MSNs. The most recent and convincing study by Ren et al. (2010) showed increased spine density after 28 consecutive injections in mice. D1R knock-out mice failed to display these morphological changes, and additionally, exposure to D1R antagonist, but not D2R blockers, prevented the increase in spine density (Ren et al., 2010). However, although this study pointed to a requirement for D1R activation, it was unable to determine whether the increase in spine density was restricted to D1(+) MSNs. The other two studies used BAC transgenic mice to determine cell specificity, but they did so by comparing the effect of cocaine on two different lines of mice: Drd1-EGFP and Drd2-EGFP mice (Kim et al., 2009; Lee et al., 2006). In general, this is not a good experimental design, but it is particularly worrisome in light of the finding that Drd2-EGFP mice overexpress D2R and display a paradoxical acute response to cocaine and failed to show locomotor sensitization (Kramer et al., 2011). The current study used Drd1-EGFP transgenic mice that we have previously characterized behaviorally and neurochemically and were shown to be undistinguishable from wild-type mice in their response to cocaine. More importantly, the current study addresses the cell-specific actions of cocaine within the same animals. Although it is worth noting that a percentage of D1(+) MSNs might also express other dopamine receptors (Shetreat et al., 1996; Hasbi et al., 2010) (but see Wong et al., 1999), the results here showed that MSNs expressing D1R in the NAc core are particularly susceptible to effects of cocaine on spine density.
Other studies have shown increased dendrite branching in NAc after chronic cocaine (Robinson et al., 2001; Ren et al., 2010), but in this study we could not detect changes in branching. Some possible explanations for this disparity are the difference in animal species and/or mouse strains used or the fact that analysis of a larger population of cells might be required to determine statistical difference. Also, this study was unable to detect increased spine density 30 d after the last injection from the long treatment. This was consistent with the concomitant return to baseline of the mEPSC frequency seen in this study (Fig. 6) and also consistent with results previously reported using these same mice (Drd1-EGFP mice) that showed a decreased spine density by 30 d compared with the spines density at 2 d withdrawal (Lee et al., 2006). Nonetheless, the main goal of this set of experiments was to test whether withdrawal after the long cocaine treatment could further enhance the plasticity, and the results clearly indicated this was not the case.
In conclusion, the results from this study show that repeated cocaine administration leads to cell-specific changes at excitatory synapses in the NAc that affect both the function and the morphology of the synapse onto D1-expressing MSNs. These changes need time to develop and they appear even when drug exposure was carried on and protracted withdrawal was avoided.
Footnotes
This work was funded by the intramural programs of National Institute on Alcohol Abuse and Alcoholism and National Institute of Neurological Disorders and Stroke at the National Institutes of Health. We are grateful to Dr. Fumi Ono for sharing his confocal microscope. We thank John T. Williams, Christina Gremel, Christopher Ford, and the members of the Alvarez Laboratory for the helpful comments and discussions of this manuscript.
References
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Asensio S, Romero MJ, Romero FJ, Wong C, Alia-Klein N, Tomasi D, Wang GJ, Telang F, Volkow ND, Goldstein RZ. Striatal dopamine D2 receptor availability predicts the thalamic and medial prefrontal responses to reward in cocaine abusers three years later. Synapse. 2010;64:397–402. doi: 10.1002/syn.20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin D, Jonkman S, Dickinson A, Robbins TW, Everitt BJ. Parallel and interactive learning processes within the basal ganglia: relevance for the understanding of addiction. Behav Brain Res. 2009;199:89–102. doi: 10.1016/j.bbr.2008.09.027. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Wolf ME. Behavioral sensitization to cocaine is associated with increased AMPA receptor surface expression in the nucleus accumbens. J Neurosci. 2005;25:9144–9151. doi: 10.1523/JNEUROSCI.2252-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer JJ, Joh TH, Pickel VM. Ultrastructural localization of tyrosine hydroxylase in rat nucleus accumbens. J Comp Neurol. 1984;227:92–103. doi: 10.1002/cne.902270110. [DOI] [PubMed] [Google Scholar]
- Caine SB, Negus SS, Mello NK, Patel S, Bristow L, Kulagowski J, Vallone D, Saiardi A, Borrelli E. Role of dopamine D2-like receptors in cocaine self-administration: studies with D2 receptor mutant mice and novel D2 receptor antagonists. J Neurosci. 2002;22:2977–2988. doi: 10.1523/JNEUROSCI.22-07-02977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine SB, Thomsen M, Gabriel KI, Berkowitz JS, Gold LH, Koob GF, Tonegawa S, Zhang J, Xu M. Lack of self-administration of cocaine in dopamine D1 receptor knock-out mice. J Neurosci. 2007;27:13140–13150. doi: 10.1523/JNEUROSCI.2284-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat Neurosci. 2006;9:251–259. doi: 10.1038/nn1632. [DOI] [PubMed] [Google Scholar]
- Day M, Wokosin D, Plotkin JL, Tian X, Surmeier DJ. Differential excitability and modulation of striatal medium spiny neuron dendrites. J Neurosci. 2008;28:11603–11614. doi: 10.1523/JNEUROSCI.1840-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Hjelmstad GO, Margolis EB, Nicola SM. Ventral tegmental area neurons in learned appetitive behavior and positive reinforcement. Annu Rev Neurosci. 2007;30:289–316. doi: 10.1146/annurev.neuro.30.051606.094341. [DOI] [PubMed] [Google Scholar]
- Freund TF, Powell JF, Smith AD. Tyrosine hydroxylase-immunoreactive boutons in synaptic contact with identified striatonigral neurons, with particular reference to dendritic spines. Neuroscience. 1984;13:1189–1215. doi: 10.1016/0306-4522(84)90294-x. [DOI] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, Heintz N. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Grimm JW, Hope BT, Wise RA, Shaham Y. Neuroadaptation. Incubation of cocaine craving after withdrawal. Nature. 2001;412:141–142. doi: 10.1038/35084134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X, Zhang R, Xu Y, Li S. Cocaine withdrawal enhances long-term potentiation in rat hippocampus via changing the activity of corticotropin-releasing factor receptor subtype 2. Neuroscience. 2009;161:665–670. doi: 10.1016/j.neuroscience.2009.04.035. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbi A, O'Dowd BF, George SR. Heteromerization of dopamine D2 receptors with dopamine D1 or D5 receptors generates intracellular calcium signaling by different mechanisms. Curr Opin Pharmacol. 2010;10:93–99. doi: 10.1016/j.coph.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N. A translational profiling approach for the molecular characterization of CNS cell types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schlüter OM, Zukin RS, Dong Y. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries MD, Prescott TJ. The ventral basal ganglia, a selection mechanism at the crossroads of space, strategy, and reward. Prog Neurobiol. 2010;90:385–417. doi: 10.1016/j.pneurobio.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Lalumiere RT, Knackstedt L, Shen H. Glutamate transmission in addiction. Neuropharmacology. 2009;56(Suppl 1):169–173. doi: 10.1016/j.neuropharm.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature. 1979;277:93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P. Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens. Proc Natl Acad Sci U S A. 2009;106:2915–2920. doi: 10.1073/pnas.0813179106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Thomas MJ. Similar neurons, opposite adaptations: psychostimulant experience differentially alters firing properties in accumbens core versus shell. J Neurosci. 2009;29:12275–12283. doi: 10.1523/JNEUROSCI.3028-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PF, Christensen CH, Hazelwood L, Dobi A, Bock R, Sibley DR, Mateo Y, Alvarez VA. Dopamine D2 receptor overexpression alters behavior and physiology in Drd2-EGFP mice. J Neurosci. 2011;31:126–132. doi: 10.1523/JNEUROSCI.4287-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kim Y, Kim AM, Helmin K, Nairn AC, Greengard P. Cocaine-induced dendritic spine formation in D1 and D2 dopamine receptor-containing medium spiny neurons in nucleus accumbens. Proc Natl Acad Sci U S A. 2006;103:3399–3404. doi: 10.1073/pnas.0511244103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Acerbo MJ, Robinson TE. The induction of behavioural sensitization is associated with cocaine-induced structural plasticity in the core (but not shell) of the nucleus accumbens. Eur J Neurosci. 2004;20:1647–1654. doi: 10.1111/j.1460-9568.2004.03612.x. [DOI] [PubMed] [Google Scholar]
- Lu L, Grimm JW, Shaham Y, Hope BT. Molecular neuroadaptations in the accumbens and ventral tegmental area during the first 90 days of forced abstinence from cocaine self-administration in rats. J Neurochem. 2003;85:1604–1613. doi: 10.1046/j.1471-4159.2003.01824.x. [DOI] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ. Topographical organization of amygdaloid projections to the caudatoputamen, nucleus accumbens, and related striatal-like areas of the rat brain. Neuroscience. 1991;44:15–33. doi: 10.1016/0306-4522(91)90248-m. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson OT, Griffiths AC. The topographic order of inputs to nucleus accumbens in the rat. Neuroscience. 1985;16:275–296. doi: 10.1016/0306-4522(85)90002-8. [DOI] [PubMed] [Google Scholar]
- Ren Z, Sun WL, Jiao H, Zhang D, Kong H, Wang X, Xu M. Dopamine D1 and N-methyl-d-aspartate receptors and extracellular signal-regulated kinase mediate neuronal morphological changes induced by repeated cocaine administration. Neuroscience. 2010;168:48–60. doi: 10.1016/j.neuroscience.2010.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Gorny G, Mitton E, Kolb B. Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse. 2001;39:257–266. doi: 10.1002/1098-2396(20010301)39:3<257::AID-SYN1007>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Seabold GK, Daunais JB, Rau A, Grant KA, Alvarez VA. DiOLISTIC labeling of neurons from rodent and non-human primate brain slices. J Vis Exp. 2010;pii:2081. doi: 10.3791/2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- Shetreat ME, Lin L, Wong AC, Rayport S. Visualization of D1 dopamine receptors on living nucleus accumbens neurons and their colocalization with D2 receptors. J Neurochem. 1996;66:1475–1482. doi: 10.1046/j.1471-4159.1996.66041475.x. [DOI] [PubMed] [Google Scholar]
- Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- Steiner P, Higley MJ, Xu W, Czervionke BL, Malenka RC, Sabatini BL. Destabilization of the postsynaptic density by PSD-95 serine 73 phosphorylation inhibits spine growth and synaptic plasticity. Neuron. 2008;60:788–802. doi: 10.1016/j.neuron.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Thanos PK, Michaelides M, Umegaki H, Volkow ND. D2R DNA transfer into the nucleus accumbens attenuates cocaine self-administration in rats. Synapse. 2008;62:481–486. doi: 10.1002/syn.20523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Kai L, Day M, Ronesi J, Yin HH, Ding J, Tkatch T, Lovinger DM, Surmeier DJ. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Wong AC, Shetreat ME, Clarke JO, Rayport S. D1- and D2-like dopamine receptors are co-localized on the presynaptic varicosities of striatal and nucleus accumbens neurons in vitro. Neuroscience. 1999;89:221–233. doi: 10.1016/s0306-4522(98)00284-x. [DOI] [PubMed] [Google Scholar]
- Zahm DS. Functional-anatomical implications of the nucleus accumbens core and shell subterritories. Ann N Y Acad Sci. 1999;877:113–128. doi: 10.1111/j.1749-6632.1999.tb09264.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xu M. Opposite regulation of cocaine-induced intracellular signaling and gene expression by dopamine D1 and D3 receptors. Ann N Y Acad Sci. 2006;1074:1–12. doi: 10.1196/annals.1369.001. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang D, Xu M. Identification of chronic cocaine-induced gene expression through dopamine D1 receptors by using cDNA microarrays. Ann N Y Acad Sci. 2002;965:1–9. doi: 10.1111/j.1749-6632.2002.tb04146.x. [DOI] [PubMed] [Google Scholar]
- Zhang XF, Hu XT, White FJ. Whole-cell plasticity in cocaine withdrawal: reduced sodium currents in nucleus accumbens neurons. J Neurosci. 1998;18:488–498. doi: 10.1523/JNEUROSCI.18-01-00488.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]