

Abstract
The biochemistry of acetogenesis is reviewed. The microbes that catalyze the reactions that are central to acetogenesis are described and the focus is on the enzymology of the process. These microbes play a key role in the global carbon cycle, producing over 10 trillion kilograms of acetic acid annually. Acetogens have the ability to anaerobically convert carbon dioxide and CO into acetyl-CoA by the Wood–Ljungdahl pathway, which is linked to energy conservation. They also can convert the six carbons of glucose stoichiometrically into 3 mol of acetate using this pathway. Acetogens and other anaerobic microbes (e.g., sulfate reducers and methanogens) use the Wood–Ljungdahl pathway for cell carbon synthesis. Important enzymes in this pathway that are covered in this review are pyruvate ferredoxin oxidoreductase, CO dehydrogenase/acetyl-CoA synthase, a corrinoid iron-sulfur protein, a methyltransferase, and the enzymes involved in the conversion of carbon dioxide to methyl-tetrahydrofolate.
Keywords: acetogenic bacteria, carbon dioxide fixation, carbon monoxide, cobalamin
Introduction
Some anaerobic microbes, including acetogenic bacteria and methanogenic archaea, convert CO2 to cellular carbon by the Wood–Ljungdahl pathway (Fig. 1).1,2 The global importance of acetogens is covered fully in Harold Drake’s chapter.3 Acetogenic bacteria use this pathway as their major means of generating energy for growth. Moorella thermoacetica, isolated in 1942,4 is the model acetogen and is the organism on which most studies of the Wood–Ljungdahl have been performed; its genome was recently sequenced. Methanogens growing on H2/CO2 use the pathway for generating cell carbon; however, those that can grow on acetate, essentially run the pathway in reverse and generate energy by oxidizing acetate to 2 mol of CO2.5 Acetoclastic methanogens also convert acetate into acetyl-CoA for cell carbon synthesis through the combined actions of acetate kinase6,7 and phosphotransacetylase.8
FIGURE 1.
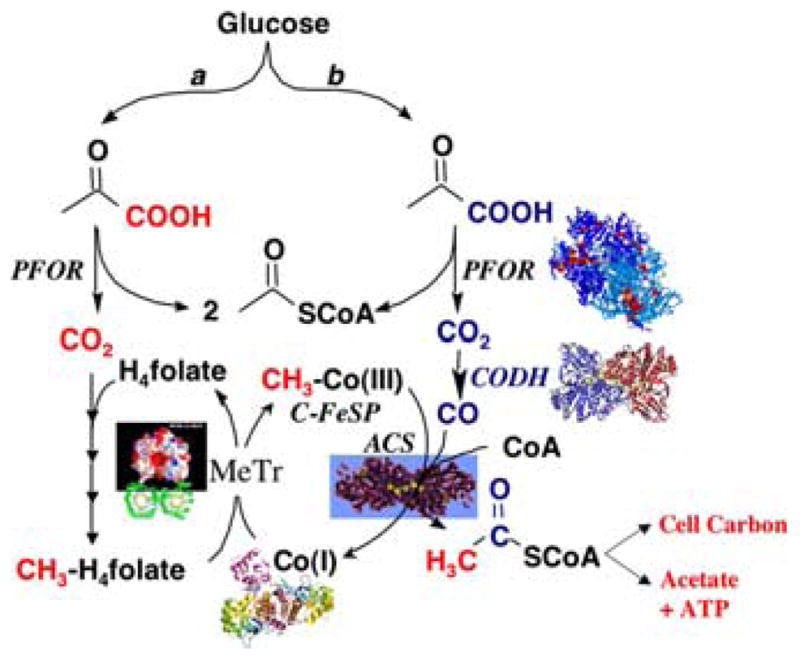
The Wood–Ljungdahl pathway with the Eastern and Western branches depicted in red and blue, respectively. (In color in Annals on line.)
The Wood–Ljungdahl pathway contains an Eastern (in red) and a Western (in blue) branch (Fig. 1), as originally described.9 The Eastern branch is essentially the folate-dependent one-carbon metabolic pathway that is present from bacteria to humans and recapitulated with methanopterin in methanogens. The Western branch is unique to acetogens, methanogens, and sulfate reducers, and exhibits novel mechanistic features. Acetogenic bacteria (e.g., acetogens or homoacetogens) synthesize acetic acid as their sole or primary metabolic end-product. Globally, acetogens produce over 1013 kg (100 billion U.S. tons) of acetic acid annually,10 which dwarfs the total output of the world’s chemical industry.
Figure 1 depicts growth of acetogens on glucose. However, these organisms can use a variety of substrates, including the biodegradation products of most natural polymers, such as cellulose, lignin (sugars, alcohols, aromatic compounds), and inorganic gases (CO, H2, CO2). When acetogens grow on H2/CO2, carbon enters the Wood–Ljungdahl pathway at the CO2 reduction step, with H2 serving as the electron donor. Acetogens are important in the biology of the soil, of extreme environments, and of organisms that house them in their digestive tract, such as humans, termites, and ruminants.11–13
Pyruvate Ferredoxin Oxidoreductase
Pyruvate ferredoxin oxidoreductase (PFOR) was reviewed relatively recently.14 Catalyzing the oxidative decarboxylation of pyruvate to form acetyl-CoA and CO2, PFOR links heterotrophic metabolism to the Wood–Ljungdahl pathway (Fig. 1). PFOR is also key to cell carbon synthesis since, besides its catabolic function, PFOR catalyzes pyruvate formation by reductive carboxylation of acetyl-CoA.15,16 Pyruvate can then enter the incomplete tricarboxylic acid cycle17,18 to generate intermediates for cell carbon synthesis. PFOR is found in archaea, bacteria, and even anaerobic protozoa like Giardia.19
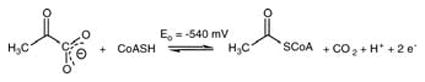 |
(1) |
The structure of the Desulfovibrio africanus PFOR reveals seven domains.20 Thiamine pyrophosphate (TPP) and a proximal [4Fe-4S] cluster (Cluster A) are deeply buried within the protein, while the other two clusters (B and C) lead to the surface (Fig. 2). The TPP binding site consists of two domains that bear strong structural similarity to those in other TPP enzymes21 and contains conserved residues that interact with Mg++, pyrophosphate, and the thiazolium ring. Rapid kinetic studies indicate that the initial steps in oxidative decarboxylation of pyruvate by PFOR are similar to those of other TPP enzymes.22 Deprotonation of TPP yields the active ylide, which forms an adduct with pyruvate. Then, CO2 is released, forming the 2α-hydroxyethylidene-TPP adduct (HE-TPP).23 In acetogens, the CO2 feeds into the Wood–Ljungdahl pathway24,25 (Fig. 1) and, based on isotope-exchange studies,26 may be channeled directly to CO dehydrogenase/acetyl-CoA synthase (CODH/ACS).
FIGURE 2.
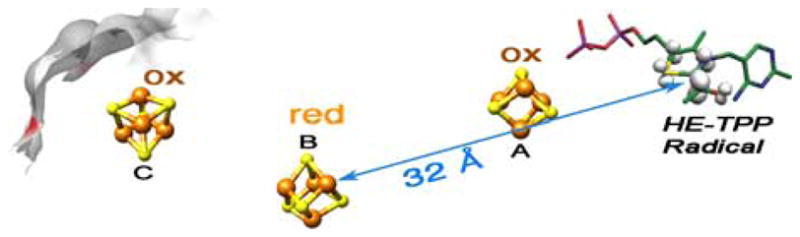
PFOR state including the HE-TPP radical, with the structure based on spectroscopic results, showing highly delocalized spin distribution (From Astashkin et al.29 Used with permission.), and the distance from HE-TPP radical to coupled cluster. Location of the clusters is based on the structure (PDB 1KEK).
After forming the HE-TPP intermediate, the negative charge on C-2 of HE-TPP promotes one-electron reduction of a proximal FeS cluster, forming an HE-TPP radical intermediate (Fig. 2). Based on the crystal structure, this intermediate was proposed to be a novel sigma-type acetyl radical27; however, recent studies show that it is a pi radical with spin density delocalized over the aromatic thiazolium ring, as shown in the figure.28,29
In the next step of the PFOR mechanism, the HE-TPP radical transfers an electron to the Fe-S electron-transfer chain, presumably through the oxidized A-cluster (Fig. 2). The rate of this electron-transfer reaction is CoA dependent.30 In the absence of CoA, the half-life of the HE-TPP radical intermediate is ~2 min; however, in the presence of CoA, the rate of radical decay increases 100,000-fold.22 By studying various CoA analogues, it was shown that the thiol group of CoA alone lowers the transition state barrier for electron transfer by 40.5 kJ/mol. The final step in the PFOR mechanism is electron transfer through Clusters A, B, and C (Fig. 2) to ferredoxin,20 the terminal electron acceptor for PFOR. This electron-transfer reaction occurs extremely rapidly, with a second-order rate constant of 2–7 × 107 M−1 s−1.22,31
CODH/ACS
CODH/ACS was recently reviewed,32 so this part of the Wood–Ljungdahl pathway will be treated rather briefly. As shown in Figure 1, when acetogens are grown heterotrophically, the CO2 and electrons generated by the PFOR reaction are utilized by CODH/ACS and formate dehydrogenase to generate CO and formate, respectively. When they are grown on CO, CODH generates CO2, which is then converted to formate in the Eastern branch of the pathway and CO is incorporated directly as the carbonyl group of acetyl-CoA. Both CO and CO2 are unreactive without a catalyst, but the enzyme-catalyzed reactions are fast, with turnover numbers as high as 40,000 s−1 reported for CO oxidation by the Ni-CODH from Carboxydothermus hydrogenoformans at its physiological growth temperature.33 Even the least active CODHs catalyze CO oxidation at rates of ~50 s−1.34 There are two major classes of CODHs: the aerobic Mo-Cu-Se CODH from carboxydobacteria and the Ni-CODHs. Found in aerobic bacteria that oxidize CO with O2,35,36 the Mo-CODH contains FAD,37 Fe/S centers, Cu, and 2 Mo atoms bound by molybdopterin cytosine dinucleotide, and its structure has been solved.38 This enzyme will not be discussed further in this chapter, since only the Ni-CODH/ACS is involved in the Wood–Ljungdahl pathway. Ni-CODHs are divided into two classes: the monofunctional nickel CODH, which catalyzes the reaction shown in Equation 2, and the bifunctional CODH/ACS, which couples Equation 2 (CO formation) with Equation 3 (acetyl-CoA synthesis). The monofunctional CODH functions physiologically in the direction of CO oxidation, allowing microbes to take up and oxidize CO at the low levels found in the environment, while the CODH in the bifunctional protein converts CO2 into acetyl-CoA.
| (2) |
| (3) |
The crystal structures of the monofunctional NI-CODH and the CODH component of the bifunctional enzymes are very similar.39–42 These mushroom-shaped, homodimeric enzymes contain five metal clusters per dimer: two C-clusters, two B-clusters, and a bridging D-cluster. The C-cluster is the catalytic site for CO oxidation and is buried 18† below the surface. This cluster can be described as a [3Fe-4S] cluster bridged to a binuclear NiFe cluster. The Rhodospirillum rubrum CODH structure with a bridging Cys between Ni and a special iron atom called ferrous component II (FCII), while the C. hydrogenoformans protein appears to have a sulfide bridge between Ni and FCII.43,44 Furthermore, there is evidence for a catalytically important persulfide at the C-cluster.45 Issues related to the different structures are discussed in a recent review.46 Cluster B is a typical [4Fe-4S]2+/1+ cluster, while Cluster D is a [4Fe-4S]2+/1+ cluster that bridges the two identical subunits, similar to the [4Fe-4S]2+/1+ cluster in the iron protein of nitrogenase.
The CODH mechanism involves a Ping-Pong reaction: CODH is reduced by CO in the “ping” step and the reduced enzyme transfers electrons to an external redox mediator like ferredoxin in the “pong” step. The reduced electron acceptors then couple to other reduced nicolinamide adenine dinucleotide phosphate (NAD(P)H) or ferredoxin-dependent cellular processes that require energy. Details of the CODH reaction have been reviewed.32 Recent studies of CODH linked to a pyrolytic graphite electrode show complete reversibility of CO oxidation and CO2 reduction; in fact, at low pH values, the rate of CO2 reduction exceeds that of CO oxidation.47
The association of ACS with CODH forms a bi-functional CODH/ACS machine that is encoded by the acsA/acsB genes, respectively, and plays the key role in the Wood–Ljungdahl pathway.32,48,49 The CODH component catalyzes the conversion of CO2 to CO (Eq. 2) to generate CO as a metabolic intermediate.25 The CO2 comes from the growth medium or from decarboxylation of pyruvate.25,30 Then, ACS catalyzes the condensation of CO,50 CoA, and the methyl group of a methylated corrinoid iron–sulfur protein (CFeSP) to generate acetyl-CoA (Eq. 3),25 a precursor of cellular material and a source of energy.
CODH/ACS contains a 140-† channel that delivers CO generated at the C-cluster to the A-cluster.40,51,52 The only metallocenter in ACS is the A-cluster, which consists of a [4Fe-4S] cluster bridged to a Ni site (Nip) that is thiolate bridged to another Ni ion in a thiolato-and carboxamido-type N2S2 coordination environment.40,42,53 Thus, one can describe the A-cluster as a binuclear NiNi center bridged by a cysteine residue (Cys509) to a [4Fe-4S] cluster, an arrangement similar to the Fe-Fe hydrogenases in which a [4Fe-4S] cluster and a binuclear Fe site are bridged by a Cys residue.87,88
Two mechanisms for acetyl-CoA synthesis have been proposed that differ mainly in the electronic structure of the intermediates: one proposes a paramagnetic Ni(I)-CO species as a central intermediate,2 and the other proposes a Ni(0) intermediate.54,55 A generic mechanism that emphasizes the organometallic nature of this reaction sequence is described in Figure 3.
FIGURE 3.
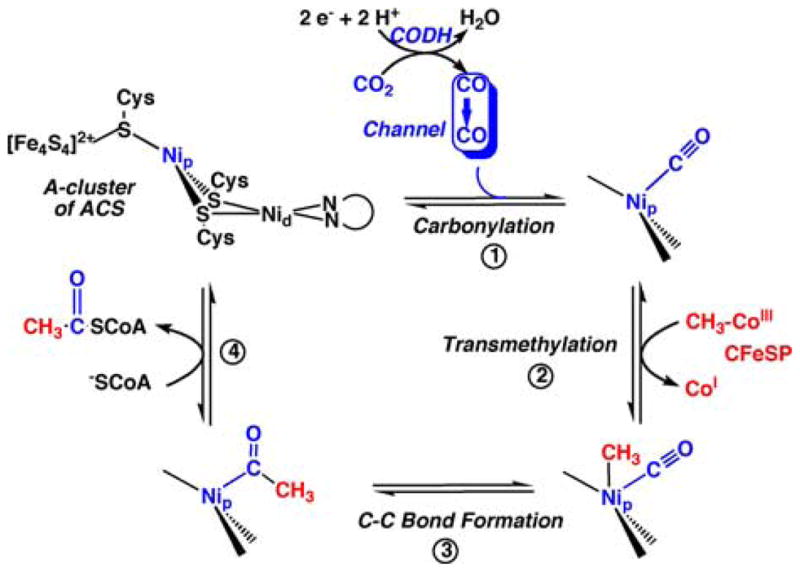
ACS mechanism emphasizing the organometallic nature of the reaction sequence and the channel to deliver CO from the CODH active site to the A-cluster.
Step 1, as shown in the figure, involves the migration of CO, derived from CO2 reduction, through the inter-subunit channel to bind to the Nip site in the A-cluster. The binding of CO to ACS forms an organometallic complex, called the NiFeC species that has been characterized by a number of spectroscopic approaches.2 The electronic structure of the NiFeC species is described as a [4Fe-4S]2+ cluster linked to a Ni1+ center at the Nip site, while the other Ni apparently remains redox-inert in the Ni2+ state.56 Lindahl’s group has argued that the NiFeC species is not a true catalytic intermediate in acetyl-CoA synthesis, that it may represent an inhibited state, and that the Ni(0) state is the catalytically relevant one.54,55 See the recent review for a discussion of these issues.32 Step 2 in the ACS mechanism involves methylation of the A-cluster,57 which involves the conversion of one organometallic species (methyl-Co) to another (methyl-Ni). There is evidence that the methyl group binds to the Nip site of the A-cluster.58–60 Carbon-carbon bond formation, Step 3 in the catalytic cycle, occurs by condensation of the methyl and carbonyl groups to form an acetyl-metal species. In the last step, CoA binds to ACS, triggering thiolysis of the acetyl-metal bond to form the C-S bond of acetyl-CoA, completing the reactions of the Western branch of the Wood–Ljungdahl pathway. Figure 3 indicates, for simplicity, that the ACS reaction sequence is ordered with CO binding before the methyl group. Conversely, Lindahl has argued for a strictly ordered binding mechanism, with methyl binding first, then CO, and finally CoA, as described in a recent review.55 In the author’s opinion, there is insufficient evidence to exclude the 1991 proposal that the carbonylation and methylation steps occur randomly61 (Fig. 4).
FIGURE 4.
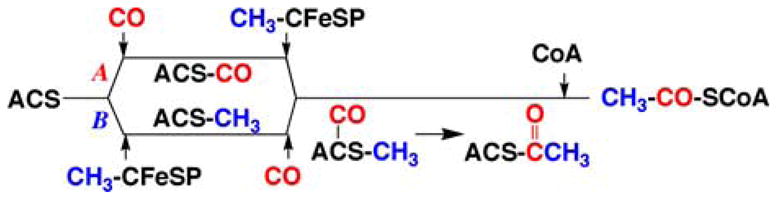
Random mechanism of acetyl-CoA synthesis.
Tetrahydrofolate-Dependent Enzymes
Most of the work on the folate enzymes involved in the Eastern branch of the pathway has been performed in the Ljungdahl laboratory. The methyl group of acetyl-CoA is formed by the six-electron reduction of CO2 in the reactions of the Eastern branch of the acetyl-CoA pathway (Fig. 1).2,62 First, formate dehydrogenase converts CO2 to formate,63 which is condensed with H4folate to form 10-formyl-H4folate.64,65 The latter is then converted by a cyclohydrolase to 5,10-methenyl-H4folate. Next, a dehydrogenase reduces methenyl- to 5,10-methylene-H4folate,66 which is reduced to (6S)-5-CH3-H4folate by a reductase.67,68
Methyltransferase (MeTr, AcsE) and Corrinoid Iron Sulfur Protein (CFeSP, AcsCD)
The methyl group of CH3-H4folate is transferred to the cobalt site in the cobalamin cofactor bound to the CFeSP69,70 to form an organometallic methyl-Co(III) intermediate in the Wood–Ljungdahl pathway (Fig. 1). This reaction is catalyzed by MeTr, encoded by the acsE gene.24 MeTr belongs to the B12-dependent methyltransferase family that includes methionine synthase and related enzymes from methanogens.71 We have cloned, sequenced, and actively overexpressed MeTr72,73 and the CFeSP74 in E coli, making them amenable for site-directed mutagenesis studies. In collaboration with Cathy Drennan (MIT, Cambridge, Massachusetts), we also have determined the structure of MeTr and a site-directed variant in its uncomplexed75 and CH3-H4folate–bound76 states. It was concluded that CH3-H4folate binds tightly (Kd < 10 μM77) to MeTr within a negatively charged crevice of the triose phosphate isomerase (TIM) barrel.78 The structure of the CFeSP was also recently solved.79
A key step in the MeTr mechanism is activation of the methyl group of CH3-H4folate, since the re-action involves displacement at a tertiary amine and because the CH3-N bond is much stronger than the product CH3-Co bond. Of the activation mechanisms that have been considered, protonation at N5 of the pterin seems to be most plausible.71 Generation of a positive charge on N5 would lower the activation barrier for nucleophilic displacement of the methyl group by the Co(I) nucleophile. There is significant experimental support for protonation at N5 of the pterin, including proton uptake studies,77,80 pH dependencies of the steady state, and transient reaction kinetics of MeTr81 and methionine synthase,82 and studies of variants that are compromised in acid–base catalysis.76 A question that has not been resolved is whether the proton transfer takes place upon formation of the binary complex, as indicated by studies with MeTr from M. thermoacetica,49,77 or the ternary complex (with the methyl acceptor), as concluded from studies on E. coli methionine synthase.80 Recent studies indicate that this protonation step relies on an H-bonding network, instead of a single acid–base catalyst and that an Asn residue is a key component of that network.76
As shown in Figure 1, the CFeSP interfaces between CH3-H4folate/MeTr and CODH/ACS. This 88-kDa heterodimeric protein contains a [4Fe-S]2+/1+ cluster and a cobalt cobamide.70,74 The Fe-S cluster plays a role in reductive activation of the cobalt to the active Co(I) state.83,84 Svetlitchnaia et al.79 proposed that the C-terminal domain (Fig. 5) of the large subunit is a mobile element that interacts alternatively with the A-cluster domain of ACS and with MeTr. Three major conformers or complexes are described: (1) a methylation complex, in which the Co(I)-CFeSP binds MeTr and accepts the methyl group of CH3-H4folate; (2) the methylated CFeSP; (3) a complex between ACS and the methylated CFeSP. A fourth conformation, which is not shown here, would be a reductive activation conformer, in which the corrinoid is in the inactive Co(II) state. This molecular juggling proposed for the CFeSP shown in Figure 5 has precedent in the related mechanism involving the various domains of methionine synthase, as shown by the elegant structure–function studies of Matthews and Ludwig.82,85,86
FIGURE 5.
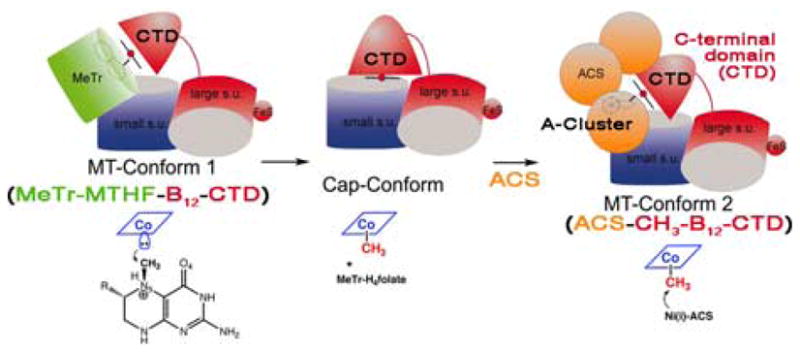
Proposed conformers and interactions of the CFeSP. (Modified from Svetlitchnaia et al.79)
Summary
Studies of the enzymes involved in the Wood–Ljungdahl pathway have elucidated new roles of metal ions in biology (including the formation of bioorganometallic intermediates, discovery of new heterometallic clusters, and nucleophilic metal ions), uncovered novel substrate-derived radical intermediates, and revealed channeling of gaseous substrates. These new outcomes and mechanisms will likely be applicable to other currently less well-studied metal-dependent enzyme systems. Now that detailed structures of PFOR, CODH/ACS, the CFeSP, and MeTr are available to provide a structural framework for these novel and important chemical reactions, mechanistic hypotheses can be posed and tested at a deeper level using a variety of biochemical and biophysical methods.
Acknowledgments
Work on the enzymology of acetogenesis has been funded by the NIH (GM39451). I am grateful for the contributions of Javier Seravalli and Elizabeth Pierce in my laboratory to the recent work on acetogenesis described here.
Footnotes
Conflict of Interest
The author declares no conflicts of interest.
References
- 1.Ragsdale SW. Metalloenzymes in the reduction of one-carbon compounds. In: Bertini I, et al., editors. Biological Inorganic Chemistry: Structure and Reactivity. University Science Books; Mill Valley, CA: 2006. pp. 452–467. [Google Scholar]
- 2.Ragsdale SW. Life with carbon monoxide. CRC Crit Rev Biochem Mol Biol. 2004;39:165–195. doi: 10.1080/10409230490496577. [DOI] [PubMed] [Google Scholar]
- 3.Drake HL. Ann NY Acad Sci Incredible Anaerobes: From Physiology to Genomics to Fuels. 2008. Old acetogens, new light. In Press. [Google Scholar]
- 4.Fontaine FE, et al. A new type of glucose fermentation by Clostridium thermoaceticum. J Bacteriol. 1942;43:701–715. doi: 10.1128/jb.43.6.701-715.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferry JG. Biochemistry of methanogenesis. Crit Rev Biochem Mol Biol. 1992;27:473–502. doi: 10.3109/10409239209082570. [DOI] [PubMed] [Google Scholar]
- 6.Ingram-Smith C, et al. Characterization of the acetate binding pocket in the Methanosarcina thermophila acetate kinase. J Bacteriol. 2005;187:2386–2394. doi: 10.1128/JB.187.7.2386-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorrell A, et al. Structural and kinetic analyses of arginine residues in the active site of the acetate kinase from Methanosarcina thermophila. J Biol Chem. 2005;280:10731–10742. doi: 10.1074/jbc.M412118200. [DOI] [PubMed] [Google Scholar]
- 8.Iyer PP, et al. Crystal structure of phosphotransacetylase from the methanogenic archaeon Methanosarcina thermophila. Structure. 2004;12:559–567. doi: 10.1016/j.str.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 9.Ragsdale SW. The Eastern and Western branches of the Wood/Ljungdahl pathway: how the East and West were won. BioFactors. 1997;9:1–9. doi: 10.1002/biof.5520060102. [DOI] [PubMed] [Google Scholar]
- 10.Drake HL, et al. Acetogenesis, acetogenic bacteria, and the acetyl-CoA pathway: past and current perspectives. In: Drake HL, editor. Acetogenesis. Chapman and Hall; New York: 1994. pp. 3–60. [Google Scholar]
- 11.Lajoie SF, et al. Acetate production from hydrogen and [13C]carbon dioxide by the microflora of human feces. Appl Environ Microbiol. 1988;54:2723–2727. doi: 10.1128/aem.54.11.2723-2727.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Breznak JA, et al. Acetate synthesis from H2 plus CO2 by termite gut microbes. Appl Environ Microbiol. 1986;52:623–630. doi: 10.1128/aem.52.4.623-630.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breznak JA, et al. Microbial H2/CO2 acetogenesis in animal guts: nature and nutritional significance. FEMS Microbiol, Rev. 1990;87:309–314. doi: 10.1111/j.1574-6968.1990.tb04929.x. [DOI] [PubMed] [Google Scholar]
- 14.Ragsdale SW. Pyruvate:ferredoxin oxidoreductase and its radical intermediate. Chem Rev. 2003;103:2333–2346. doi: 10.1021/cr020423e. [DOI] [PubMed] [Google Scholar]
- 15.Furdui C, et al. The role of pyruvate: ferredoxin oxidoreductase in pyruvate synthesis during autotrophic growth by the Wood-Ljungdahl pathway. J Biol Chem. 2000;275:28494–28499. doi: 10.1074/jbc.M003291200. [DOI] [PubMed] [Google Scholar]
- 16.Bock AK, et al. Catalytic properties, molecular composition and sequence alignments of pyruvate: ferredoxin oxidoreductase from the methanogenic archaeon Methanosarcina barkeri (strain Fusaro) Eur J Biochem. 1996;237:35–44. doi: 10.1111/j.1432-1033.1996.0035n.x. [DOI] [PubMed] [Google Scholar]
- 17.Simpson PG, et al. Anabolic pathways in methanogens. In: Ferry JG, editor. Methanogenesis: Ecology Physiology, Biochemistry & Genetics. Chapman & Hall; London: 1993. pp. 445–472. [Google Scholar]
- 18.Yoon KS, et al. Rubredoxin from the green sulfur bacterium Chlorobium tepidum functions as an electron acceptor for pyruvate ferredoxin oxidoreductase. J Biol Chem. 1999;274:29772–29778. doi: 10.1074/jbc.274.42.29772. [DOI] [PubMed] [Google Scholar]
- 19.Horner DS, et al. A single eubacterial origin of eukaryotic pyruvate: ferredoxin oxidoreductase genes: Implications for the evolution of anaerobic eukaryotes. Mol Biol Evol. 1999;16:1280–1291. doi: 10.1093/oxfordjournals.molbev.a026218. [DOI] [PubMed] [Google Scholar]
- 20.Chabriere E, et al. Crystal structures of the key anaerobic enzyme pyruvate:ferredoxin oxidoreductase, free and in complex with pyruvate. Nat Struct Biol. 1999;6:182–190. doi: 10.1038/5870. [DOI] [PubMed] [Google Scholar]
- 21.Muller YA, et al. A thiamin diphosphate binding fold revealed by comparison of the crystal structures of transketolase, pyruvate oxidase and pyruvate decarboxylase. Structure. 1993;1:95–103. doi: 10.1016/0969-2126(93)90025-c. [DOI] [PubMed] [Google Scholar]
- 22.Furdui C, et al. The roles of coenzyme A in the pyruvate:ferredoxin oxidoreductase reaction mechanism: rate enhancement of election transfer from a radical intermediate to an iron-sulfur cluster. Biochemistry. 2002;41:9921–9937. doi: 10.1021/bi0257641. [DOI] [PubMed] [Google Scholar]
- 23.Breslow R. Rapid deuterium exchange in thiazolium salts. J Am Chem Soc. 1957;79:1762–1763. [Google Scholar]
- 24.Drake HL, et al. Purification of five components from Clostridium thermoacticum which catalyze synthesis of acetate from pyruvate and methyltetrahydrofolate. Properties of phosphotransacetylase. J Biol Chem. 1981;256:11137–11144. [PubMed] [Google Scholar]
- 25.Menon S, et al. Evidence that carbon monoxide is an obligatory intermediate in anaerobic acetyl-CoA synthesis. Biochemistry. 1996;35:12119–12125. doi: 10.1021/bi961014d. [DOI] [PubMed] [Google Scholar]
- 26.Schulman M, et al. Total synthesis of acetate from CO2. VII. Evidence with Clostridium thermoaceticum that the carboxyl of acetate is derived from the carboxyl of pyruvate by transcarboxylation and not by fixation of CO2. J Biol Chem. 1973;248:6255–6261. [PubMed] [Google Scholar]
- 27.Chabriere E, et al. Crystal structure of the free radical intermediate of pyruvate:ferredoxin oxidoreductase. Science. 2001;294:2559–2563. doi: 10.1126/science.1066198. [DOI] [PubMed] [Google Scholar]
- 28.Mansoorabadi SO, et al. EPR spectroscopic and computational characterization of the hydroxyethylidene-thiamine pyrophosphate radical intermediate of pyruvate: ferredoxin oxidoreductase. Biochemistry. 2006;45:7122–7131. doi: 10.1021/bi0602516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Astashkin AV, et al. Pulsed electron paramagnetic resonance experiments identify the paramagnetic intermediates in the pyruvate ferredoxin oxidoreductase catalytic cycle. J Am Chem Soc. 2006;128:3888–3889. doi: 10.1021/ja0585275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menon S, et al. Mechanism of the Clostridium thermoaceticum pyruvate:ferredoxin oxidoreductase: evidence for the common catalytic intermediacy of the hydroxyethylthiamine pyropyrosphate radical. Biochemistry. 1997;36:8484–8494. doi: 10.1021/bi970403k. [DOI] [PubMed] [Google Scholar]
- 31.Pieulle L, et al. Structural and kinetic studies of the pyruvate-ferredoxin oxidoreductase/ferredoxin complex from Desulfovibrio africanus. Eur J Biochem. 1999;264:500–508. doi: 10.1046/j.1432-1327.1999.00648.x. [DOI] [PubMed] [Google Scholar]
- 32.Ragsdale SW. Nickel and the carbon cycle. J Inorg Biochem. 2007;101:1657–1666. doi: 10.1016/j.jinorgbio.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svetlitchnyi V, et al. Two membrane-associated NiFeS-carbon monoxide dehydrogenases from the anaerobic carbon-monoxide-utilizing eubacterium Carboxydothermus hydrogenoformans. J Bacteriol. 2001;183:5134–5144. doi: 10.1128/JB.183.17.5134-5144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meyer O, et al. The role of Se, Mo and Fe in the structure and function of carbon monoxide dehydrogenase. Biol Chem. 2000;381:865–876. doi: 10.1515/BC.2000.108. [DOI] [PubMed] [Google Scholar]
- 35.Meyer O, et al. Biochemistry of the aerobic utilization of carbon monoxide. In: Murrell JC, Kelly DP, editors. Microbial Growth on C1 Compounds. Intercept, Ltd; Andover, MA: 1993. pp. 433–459. [Google Scholar]
- 36.Gnida M, et al. A novel binuclear [CuSMo] cluster at the active site of carbon monoxide dehydrogenase: characterization by X-ray absorption spectroscopy. Biochemistry. 2003;42:222–230. doi: 10.1021/bi026514n. [DOI] [PubMed] [Google Scholar]
- 37.Bates DM, et al. Substitution of leucine 28 with histidine in the Escherichia coli transcription factor FNR results in increased stability of the [4Fe-4S](2+) cluster to oxygen. J Biol Chem. 2000;275:6234–6240. doi: 10.1074/jbc.275.9.6234. [DOI] [PubMed] [Google Scholar]
- 38.Dobbek H, et al. Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc Natl Acad Sci USA. 1999;96:8884–8889. doi: 10.1073/pnas.96.16.8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drennan CL, et al. Life on carbon monoxide: X-ray structure of Rhodospirillum rubrum Ni-Fe-S carbon monoxide dehydrogenase. Proc Natl Acad Sci USA. 2001;98:11973–11978. doi: 10.1073/pnas.211429998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doukov TI, et al. A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 41.Dobbek H, et al. Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science. 2001;293:1281–1285. doi: 10.1126/science.1061500. [DOI] [PubMed] [Google Scholar]
- 42.Darnault C, et al. Ni-Zn-[Fe(4)-S(4)] and Ni-Ni-[Fe(4)-S(4)] clusters in closed and open alpha subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat Struct Biol. 2003;10:271–279. doi: 10.1038/nsb912. [DOI] [PubMed] [Google Scholar]
- 43.Dobbek H, et al. Carbon monoxide induced decomposition of the active site [Ni-4Fe-5S] cluster of CO dehydrogenase. J Am Chem Soc. 2004;126:5382–5387. doi: 10.1021/ja037776v. [DOI] [PubMed] [Google Scholar]
- 44.Sun J, et al. Sulfur ligand substitution at the nickel(II) sites of cubane-type and cubanoid NiFe3S4 clusters relevant to the C-clusters of carbon monoxide dehydrogenase. Inorg Chem. 2007;46:2691–2699. doi: 10.1021/ic062362z. [DOI] [PubMed] [Google Scholar]
- 45.Kim EJ, et al. Evidence for a proton transfer network and a required persulfide-bond-forming cysteine residue in ni-containing carbon monoxide dehydrogenases. Biochemistry. 2004;43:5728–5734. doi: 10.1021/bi036062u. [DOI] [PubMed] [Google Scholar]
- 46.Drennan CL, et al. The metalloclusters of carbon monoxide dehydrogenase/acetyl-CoA synthase: a story in pictures. J Biol Inorg Chem. 2004;9:511–515. doi: 10.1007/s00775-004-0563-y. [DOI] [PubMed] [Google Scholar]
- 47.Parkin A, et al. Rapid electrocatalytic CO2/CO interconversions by Carboxydothermus hydrogenoformans CO dehydrogenase I on an electrode. J Am Chem Soc. 2007;129:10328–10329. doi: 10.1021/ja073643o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fontecilla-Camps J-C, et al. Nickel-iron-sulfur active sites: hydrogenase and CO dehydrogenase. In: Sykes AG, Cammack R, editors. Advances in Inorganic Chemistry. Vol. 47. Academic Press, Inc; San Diego: 1999. pp. 283–333. [Google Scholar]
- 49.Seravalli J, et al. Mechanism of transfer of the methyl group from (6S)-methyltetrahydrofolate to the corrinoid/iron-sulfur protein catalyzed by the methyl-transferase from Clostridium thermoaceticum: a key step in the Wood-Ljungdahl pathway of acetyl-CoA synthesis. Biochemistry. 1999;38:5728–5735. doi: 10.1021/bi982473c. [DOI] [PubMed] [Google Scholar]
- 50.Ragsdale SW, et al. EPR evidence for nickel substrate interaction in carbon monoxide dehydrogenase from Clostridium thermoaceticum. Biochem Biophys Res Commun. 1982;108:658–663. doi: 10.1016/0006-291x(82)90880-4. [DOI] [PubMed] [Google Scholar]
- 51.Seravalli J, et al. Channeling of carbon monoxide during anaerobic carbon dioxide fixation. Biochemistry. 2000;39:1274–1277. doi: 10.1021/bi991812e. [DOI] [PubMed] [Google Scholar]
- 52.Maynard EL, et al. Evidence of a molecular tunnel connecting the active sites for CO2 reduction and acetyl-CoA synthesis in acetyl-CoA synthase from Clostridium thermoaceticum. J Am Chem Soc. 1999;121:9221–9222. [Google Scholar]
- 53.Svetlitchnyi V, et al. A functional Ni-Ni-[4Fe-4S] cluster in the monomeric acetyl-CoA synthase from Carboxydothermus hydrogenoformans. Proc Natl Acad Sci USA. 2004;101:446–451. doi: 10.1073/pnas.0304262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bramlett MR, et al. Mossbauer and EPR study of recombinant acetyl-CoA synthase from Moorella thermoacetica. Biochemistry. 2006;45:8674–8685. doi: 10.1021/bi060003+. [DOI] [PubMed] [Google Scholar]
- 55.Lindahl PA. Acetyl-coenzyme A synthase: the case for a Nip0-Based mechanism of catalysis. J Biol Inorg Chem. 2004;9:516–524. doi: 10.1007/s00775-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 56.Brunold TC. Spectroscopic and computational insights into the geometric and electronic properties of the A cluster of acetyl-coenzyme A synthase. J Biol Inorg Chem. 2004;9:533–541. doi: 10.1007/s00775-004-0566-8. [DOI] [PubMed] [Google Scholar]
- 57.Seravalli J, et al. Rapid kinetic studies of acetyl-CoA synthesis: evidence supporting the catalytic intermediacy of a paramagnetic NiFeC species in the autotrophic Wood-Ljungdahl pathway. Biochemistry. 2002;41:1807–1819. doi: 10.1021/bi011687i. [DOI] [PubMed] [Google Scholar]
- 58.Shin W, et al. Heterogeneous nickel environments in carbon monoxide dehydrogenase from Clostridium thermoaceticum. J Am Chem Soc. 1993;115:5522–5526. [Google Scholar]
- 59.Barondeau DP, et al. Methylation of carbon monoxide dehydrogenase from Clostridium thermoaceticum and mechanism of acetyl coenzyme A synthesis. J Am Chem Soc. 1997;119:3959–3970. [Google Scholar]
- 60.Seravalli J, et al. Evidence that Ni-Ni acetyl-CoA synthase is active and that the Cu-Ni enzyme is not. Biochemistry. 2004;43:3944–3955. doi: 10.1021/bi036194n. [DOI] [PubMed] [Google Scholar]
- 61.Lu WP, et al. Reductive activation of the coenzyme A/acetyl-CoA isotopic exchange reaction catalyzed by carbon monoxide dehydrogenase from Clostridium thermoaceticum and its inhibition by nitrous oxide and carbon monoxide. J Biol Chem. 1991;266:3554–3564. [PubMed] [Google Scholar]
- 62.Ragsdale SW. Enzymology of the acetyl-CoA pathway of CO2 fixation. CRC Crit Rev Biochem Mol Biol. 1991;26:261–300. doi: 10.3109/10409239109114070. [DOI] [PubMed] [Google Scholar]
- 63.Ljungdahl LG, et al. Formate dehydrogenase, a selenium-tungsten enzyme from Clostridium thermoaceticum. Methods Enzymol. 1978;53:360–372. doi: 10.1016/s0076-6879(78)53042-5. [DOI] [PubMed] [Google Scholar]
- 64.Lovell CR, et al. Cloning and expression in Escherichia coli of the Clostridium thermoaceticum gene encoding thermostable formyltetrahydrofolate synthetase. Arch Microbiol. 1988;149:280–285. doi: 10.1007/BF00411642. [DOI] [PubMed] [Google Scholar]
- 65.Lovell CR, et al. Primary structure of the thermostable formyltetrahydrofolate synthetase from Clostridium thermoaceticum. Biochemistry. 1990;29:5687–5694. doi: 10.1021/bi00476a007. [DOI] [PubMed] [Google Scholar]
- 66.Moore MR, et al. Purification and characterization of nicotinamide adenine dinucleotide-dependent methylenetetrahydrofolate dehydrogenase from Clostridium formicoaceticum. J Biol Chem. 1974;249:5250–5253. [PubMed] [Google Scholar]
- 67.Clark JE, et al. Purification and properties of 5,10-methylenetetrahydrofolate reductase, an iron-sulfur flavoprotein from Clostridium formicoaceticum. J Biol Chem. 1984;259:10845–10889. [PubMed] [Google Scholar]
- 68.Park EY, et al. 5,10-methylenetetrahydrofolate reductases: iron-sulfur-zinc flavoproteins of two acetogenic clostridia. In: Miller F, editor. Chemistry and Biochemistry of Flavoenzymes. Vol. 1. CRC Press; Boca Raton, FL: 1991. pp. 389–400. [Google Scholar]
- 69.Hu SI, et al. Acetate synthesis from carbon monoxide by Clostridium thermoaceticum. Purification of the corrinoid protein. J Biol Chem. 1984;259:8892–8897. [PubMed] [Google Scholar]
- 70.Ragsdale SW, et al. Mössbauer, EPR, and optical studies of the corrinoid/Fe-S protein involved in the synthesis of acetyl-CoA by Clostridium thermoaceticum. J Biol Chem. 1987;262:14289–14297. [PubMed] [Google Scholar]
- 71.Banerjee R, et al. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Ann Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 72.Roberts DL, et al. Cloning and expression of the gene cluster encoding key proteins involved in acetyl-CoA synthesis in Clostridium thermoaceticum: CO dehydrogenase, the corrinoid/Fe-S protein, and methyltransferase. Proc Natl Acad Sci USA. 1989;86:32–36. doi: 10.1073/pnas.86.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roberts DL, et al. The reductive acetyl-CoA pathway: sequence and heterologous expression of active CH3-H4folate:corrinoid/iron sulfur protein methyltransferase from Clostridium themoaceticum. J Bacteriol. 1994;176:6127–6130. doi: 10.1128/jb.176.19.6127-6130.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu WP, et al. Sequence and expression of the gene encoding the corrinoid/iron-sulfur protein from Clostridium thermoaceticum and reconstitution of the recombinant protein to full activity. J Biol Chem. 1993;268:5605–5614. [PubMed] [Google Scholar]
- 75.Doukov T, et al. Preliminary X-ray diffraction analysis of the methyltetrahydrofolate:corrinoid/iron sulfur protein methyltransferase from Clostridium themoaceticum. Acta Crystallographa D51: Part. 1995;6:1092–1093. doi: 10.1107/S0907444995005403. [DOI] [PubMed] [Google Scholar]
- 76.Doukov TI, et al. Structural and kinetic evidence for an extended hydrogen bonding network in catalysis of methyl group transfer: role of an active site asparagine residue in activation of methyl transfer by methyltransferases. J Biol Chem. 2007;282:6609–6618. doi: 10.1074/jbc.M609828200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seravalli J, et al. Binding of (6R,S)-methyltetrahydrofolate to methyltransferase from Clostridium thermoaceticum: role of protonation of methyltetrahydrofolate in the mechanism of methyl transfer. Biochemistry. 1999;38:5736–5745. doi: 10.1021/bi9824745. [DOI] [PubMed] [Google Scholar]
- 78.Doukov T, et al. Crystal structure of a methyltetrahydrofolate and corrinoid dependent methyltransferase. Structure. 2000;8:817–830. doi: 10.1016/s0969-2126(00)00172-6. [DOI] [PubMed] [Google Scholar]
- 79.Svetlitchnaia T, et al. Structural insights into methyltransfer reactions of a corrinoid iron-sulfur protein involved in acetyl-CoA synthesis. Proc Natl Acad Sci USA. 2006;103:14331–14336. doi: 10.1073/pnas.0601420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith AE, et al. Protonation state of methyltetrahydrofolate in a binary complex with cobalamin-dependent methionine synthase. Biochemistry. 2000;39:13880–13890. doi: 10.1021/bi001431x. [DOI] [PubMed] [Google Scholar]
- 81.Zhao S, et al. Mechanistic studies of the methyltransferase from Clostridium thermoaceticum: origin of the pH dependence of the methyl group transfer from methyltetrahydrofolate to the corrinoid/iron-sulfur protein. Biochemistry. 1995;34:15075–15083. doi: 10.1021/bi00046a013. [DOI] [PubMed] [Google Scholar]
- 82.Matthews RG. Cobalamin-dependent methyltransferases. Acc Chem Res. 2001;34:681–689. doi: 10.1021/ar0000051. [DOI] [PubMed] [Google Scholar]
- 83.Menon S, et al. The role of an iron-sulfur cluster in an enzymatic methylation reaction: methylation of CO dehydrogenase/acetyl-CoA synthase by the methylated corrinoid iron-sulfur protein. J Biol Chem. 1999;274:11513–11518. doi: 10.1074/jbc.274.17.11513. [DOI] [PubMed] [Google Scholar]
- 84.Menon S, et al. Role of the [4Fe-4S] cluster in reductive activation of the cobalt center of the corrinoid iron-sulfur protein from Clostridium thermoaceticum during acetyl-CoA synthesis. Biochemistry. 1998;37:5689–5698. doi: 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- 85.Evans JC, et al. Structures of the N-terminal modules imply large domain motions during catalysis by methionine synthase. Proc Natl Acad Sci USA. 2004;101:3729–3736. doi: 10.1073/pnas.0308082100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taurog RE, et al. Synergistic, random sequential binding of substrates in cobalamin-independent methionine synthase. Biochemistry. 2006;45:5083–5091. doi: 10.1021/bi060051u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peters JW, et al. X-ray crystal structure of the Fe-only hydrogenase (Cpl) from Clostridium pasteurianum to 1.8 angstrom resolution. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 88.Nicolet Y, et al. A novel FeS cluster in Fe-only hydrogenases. Trends Biochem Sci. 2000;25:138–143. doi: 10.1016/s0968-0004(99)01536-4. [DOI] [PubMed] [Google Scholar]